
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ferrocenyl chiral bisphosphorus ligands for highly enantioselective asymmetric hydrogenation via noncovalent ion pair interaction†
Caiyou
Chen
a,
Heng
Wang
a,
Zhefan
Zhang
a,
Shicheng
Jin
a,
Songwei
Wen
a,
Jianjian
Ji
a,
Lung Wa
Chung
b,
Xiu-Qin
Dong
*a and
Xumu
Zhang
*ab
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: xumu@whu.edu.cn; xiuqindong@whu.edu.cn
bDepartment of Chemistry South University of Science and Technology of China, Shenzhen, 518055, P.R. China
First published on 30th June 2016
Abstract
A new class of ferrocenyl chiral bisphosphorus ligand, Wudaphos, was developed, and exhibits excellent ee and activity (ee up to 99%, TON up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) for the asymmetric hydrogenation of both 2-aryl and 2-alkyl acrylic acids through ion pair noncovalent interaction under base free and mild reaction conditions. Well-known anti-inflammatory drugs such as naproxen and ibuprofen together with the intermediate for the preparation of Roche ester and some bioactive compounds were also efficiently obtained with excellent ee. Control experiments were conducted and revealed that the ion pair noncovalent interaction and chain length played important roles.
000) for the asymmetric hydrogenation of both 2-aryl and 2-alkyl acrylic acids through ion pair noncovalent interaction under base free and mild reaction conditions. Well-known anti-inflammatory drugs such as naproxen and ibuprofen together with the intermediate for the preparation of Roche ester and some bioactive compounds were also efficiently obtained with excellent ee. Control experiments were conducted and revealed that the ion pair noncovalent interaction and chain length played important roles.
Introduction
Since the 1960s, the exploration of new efficient chiral phosphine ligands remains a continuous work for transition-metal catalyzed asymmetric reactions.1 The development of new asymmetric reactions largely relies on the exploration of new efficient chiral phosphine ligands.2 As a result, the research of developing more efficient and practical phosphine ligands in terms of excellent enantioselectivity and activity, good air stability, and ease of preparation remains highly desirable in asymmetric catalysis.Attractive noncovalent interactions play a key role in the design of catalysts. Similar to enzymatic catalysis, attractive noncovalent interactions are responsible for many of the remarkable rate acceleration and stereoselectivity improvements by lowering the kinetic barrows through stabilization of the transition state and suppressing the degree of freedom in the transition state.3 Consequently, intensive research efforts have been directed toward the development of a new efficient catalytic system utilizing attractive noncovalent interactions.4 As summarized by Jacobson et al. (Table 1),3 among the three representative noncovalent interactions, the steric repulsion is highly distance dependent (1/r12) wherein weak interaction was observed with long distance. H-Bond interaction (1/r2) and ion pair interaction (1/r) are relatively less distance dependent and the ion pair interaction showed the strongest interaction. We anticipated that the strong ion pair noncovalent interaction can be utilized in the development of a new efficient catalytic system for asymmetric hydrogenation (AH), wherein the substrates can interact with the catalyst strongly, accelerating the reaction rate to a large extent. However, only a few examples have been reported concerning of the strong ion pair noncovalent interaction and the substrate scope was limited.5
| Entry | Noncovalent interaction | Energy dependence on distance |
|---|---|---|
| 1 |
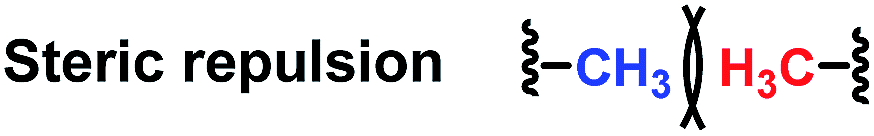
|
1/r12 |
| 2 |
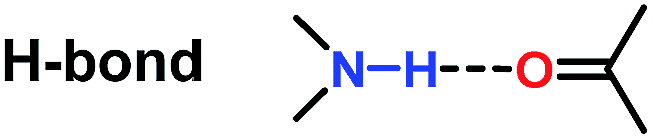
|
Complicated ∼1/r2 |
| 3 |
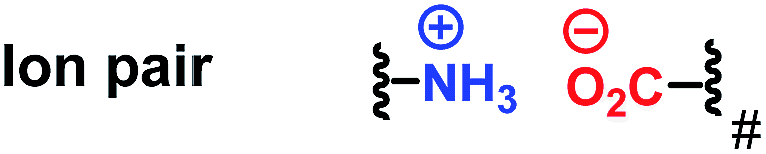
|
1/r |
With regard to the utilization of the strong ion pair noncovalent interaction in the development of new chiral catalysts for AH, we found that Ugi's amine is a privileged motif in which the dimethyl amine moiety can play as a proton acceptor which can interact with the acid substrates strongly through noncovalent ion pair interaction (Scheme 1). Moreover, the Ugi's amine motif can conveniently incorporate planar chirality, C-chirality and P-chirality into the catalytic system as exemplified by many efficient chiral ligands such as Josiphos,6 Walphos,7 Taniaphos,8 Bophoz,9 Mandyphos,10 TRAP,11 Trifer5a and Chenphos.5b Herein, we report a new class of bisphosphorus ligands incorporating the Ugi's amine motif (Scheme 1). This type of bisphosphorus ligand possesses one chiral phosphine and the chiral Ugi amine moiety. Two large substituents of the non-chiral phosphine together with a ferrocene backbone block three quadrants, and the small substituent of the chiral phosphine makes the remaining quadrant open. This three blocked quadrant model is believed to have good chiral induction.12 Furthermore, the dimethyl amine unit in the ligand can be a proton acceptor and thus can interact with the acid substrates through noncovalent ion pair interaction. According to the above hypothesis, this catalytic system is believed to exhibit excellent enantioselectivity and activity in the AH of unsaturated acid substrates.
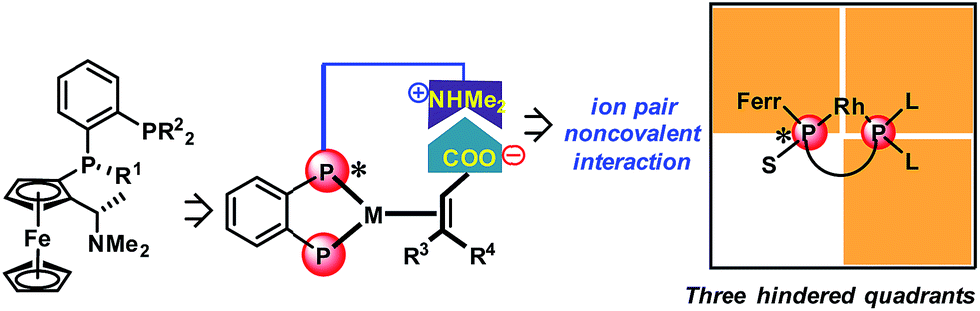 | ||
| Scheme 1 The new ferrocenyl ligands and the corresponding model of the three hindered quadrants. L = large substituent, S = small substituent, and Ferr = ferrocene. | ||
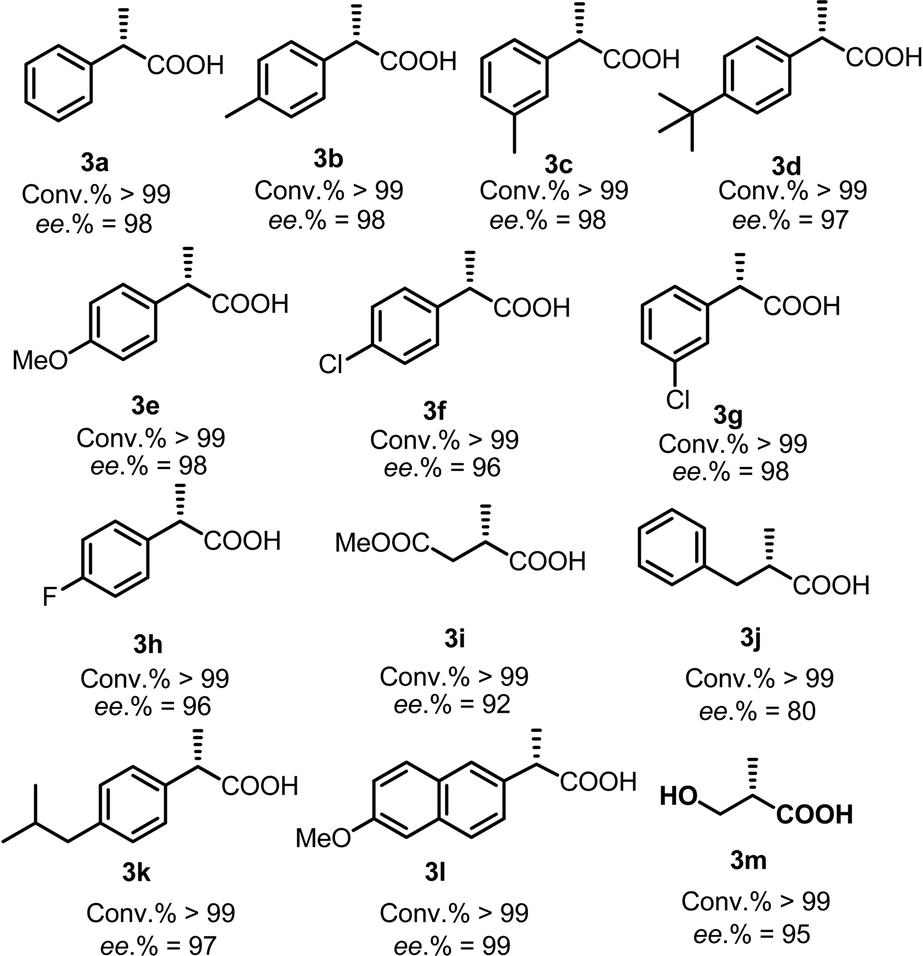
AH of 2-substituted acrylic acids will generate chiral α-substituted propanoic acids which are important units that widely exist in pharmaceuticals and fine chemicals (Scheme 2). Good examples are the well-known non-steroid anti-inflammatory and analgesic drugs such as naproxen, ibuprofen and flurbiprofen,13 the esterification potent inhibitors 4 against the inflammatory phenotype of the cystic fibrosis (CF) lung disease,14 the bioactive natural product 5 isolated from the Fusarium oxysporum which shows cytotoxicity against three human cancer cell lines PC-3, PANC-1 and A549,15 and artemisnin16 which is a famous drug against Plasmodium falciparum malaria that won the 2015 Nobel Prize in medicine. Furthermore, α-substituted propanoic acid can also be used to prepare the synthetically important Roche ester, a well-known synthon in total synthesis.17 Although AH of 2-substituted acrylic acids was previously realized using chiral Ru catalysts,18 the reactions were limited by the generally required high pressure. Chiral Rh and Ir catalysts have also recently been reported to realize this valuable transformation.19 However, most of these catalytic systems need an equivalent base to neutralize the free acid.19 This method is non-direct and needs additional procedure to remove the base. Herein we report a ferrocenyl catalytic system that can finely utilize the free acid via the attractive noncovalent ion pair interaction to realize this valuable transformation in a direct and concise way without any base with ee (enantiomeric excess) up to 99% and TON up to 20000 (Scheme 3).
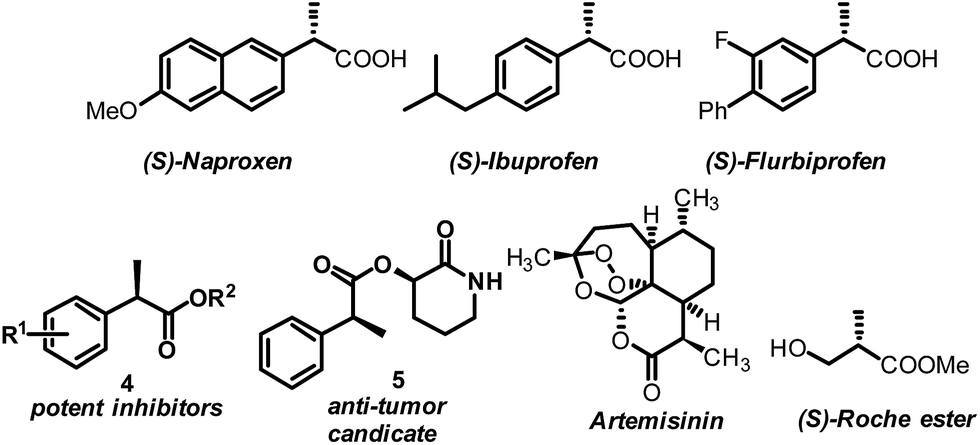 | ||
| Scheme 2 Representative drugs and chemicals featuring the α-substituted propanoic acid moiety. | ||
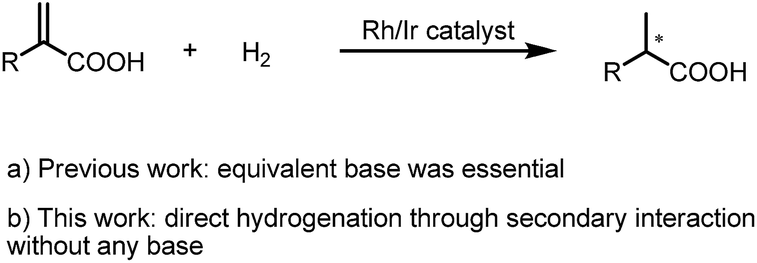 | ||
| Scheme 3 AH of 2-substituted acrylic acids. | ||
Results and discussion
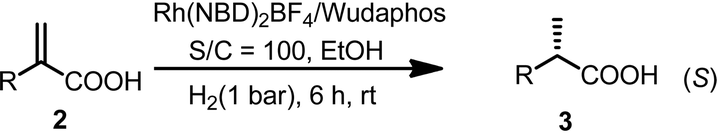
The new ferrocenyl bisphosphorus ligands we report herein can be easily synthesized in two-pot with very high diastereoselectivity (dr > 99:1) (Scheme 4). Starting from (S)-Ugi amine, a one pot sequential reaction gave 1 efficiently. Importantly, compound 1 was obtained as a single diastereomer as determined by NMR which makes the synthesis to be very simple and practical. The subsequent lithiation followed by treating with different chlorophosphines afforded the desired ligands L1–L5 in good yields. Moreover, the absolute configuration of L1 was determined using X-ray spectrum analysis as (Sc, RFC, Sp).20 Importantly, ligands L1–L5 are all highly air stable even when stored under air for more than one year.
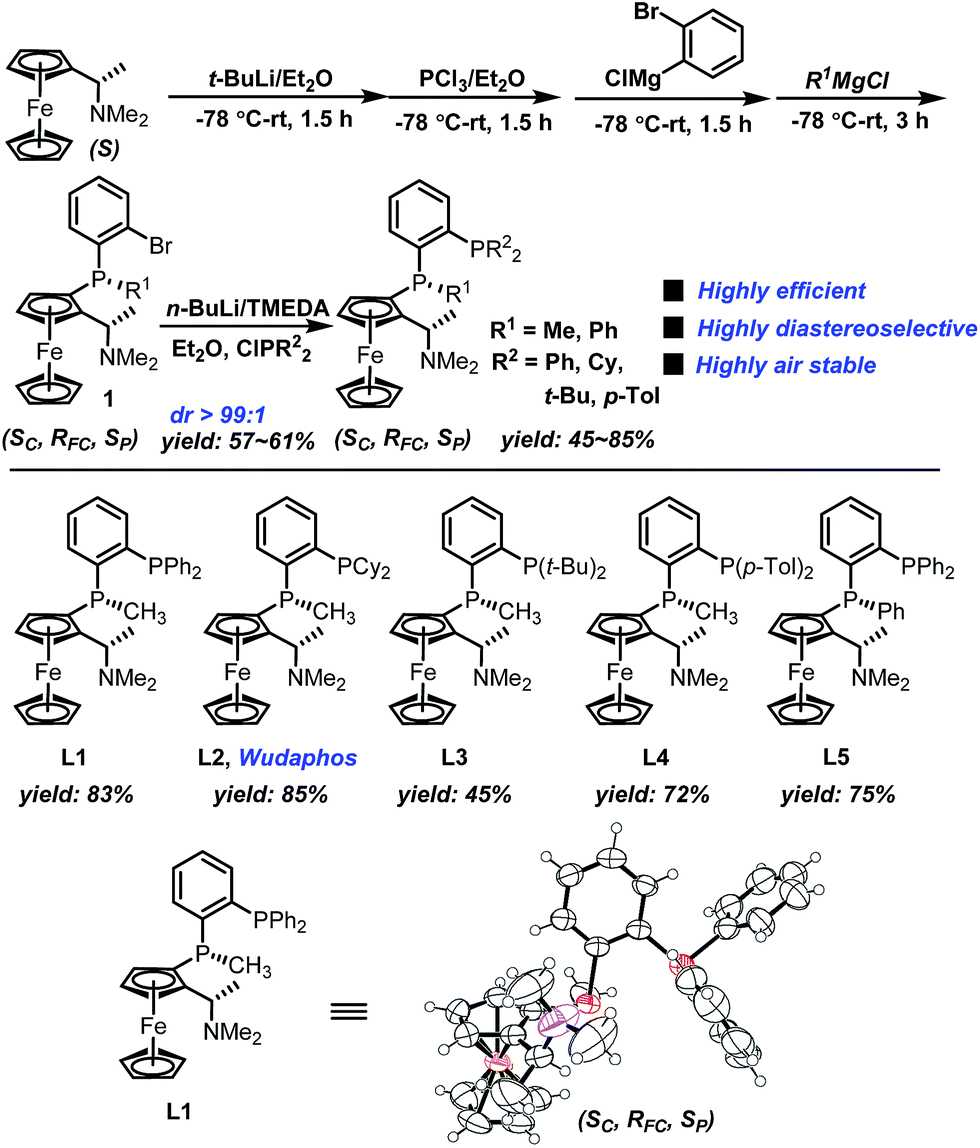 | ||
| Scheme 4 Synthesis of the ferrocenyl new bisphosphorus ligands. | ||
With ligands L1–L5 in hand, AH of 2-substituted acrylic acids was initiated by evaluating ligand effects using 2-phenyl acrylic acid 2a as a model substrate (Table 2). We were pleased to find that the substrate was all smoothly converted except when using L5 as the ligand (Table 2, entry 5) in the absence of any base. The hydrogenation results highly depended on the ligand structure. With L1, a good ee was obtained (Table 2, entry 1). Changing the phenyl group in L1 into a cyclohexyl group (L2, Table 2, entry 2), the ee significantly increased to 98%. However, to our surprise, further changing the phenyl group into the steric bulky t-butyl group resulted in a very low ee and the product configuration was changed from (S) to (R) (L3, Table 2, entry 3). This is probably due to the much too big steric hindrance of the t-butyl group which influences the noncovalent ion pair interaction between the ligand and substrate. Further changing the phenyl group into the p-tolyl group resulted in a decreased ee (L4, Table 2, entry 4). Using L5 as a ligand with a phenyl group instead of a methyl group attached to the chiral phosphine, the ee dropped significantly (L5, Table 2, entry 5). This result corresponds to the hypothesis that we made that the ligand should have three blocked quadrants and make the remaining quadrant open in order to get good enantioselectivities (Scheme 1). In terms of reactivity and enantioselectivity, L2 was selected as the optimum ligand for further investigation. Due to the remarkable performance, herein we name L2 Wudaphos.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Conv.b% | ee % | Configurationd |
|
a The reaction was conducted in a 0.1 mmol scale in 1 mL of EtOH, [Rh(NBD)2]BF4 (NBD = norbornadiene) was used as metal precursor, S/C = 100, L/Rh = 1.1:1, temperature = rt, H2 pressure = 1 bar, reaction time = 6 h.
b Substrate conversion, determined using 1H NMR.
c Enantiomeric excess of 3a, determined using chiral HPLC after treating 3a with CH2N2.
d Configuration of 3a, determined by comparing the optical rotation data with those reported in the literature.
|
||||
| 1 | L1 | >99 | 84 | (S) |
| 2 | Wudaphos | >99 | 98 | (S) |
| 3 | L3 | >99 | 33 | (R) |
| 4 | L4 | >99 | 74 | (S) |
| 5 | L5 | 67 | 57 | (S) |
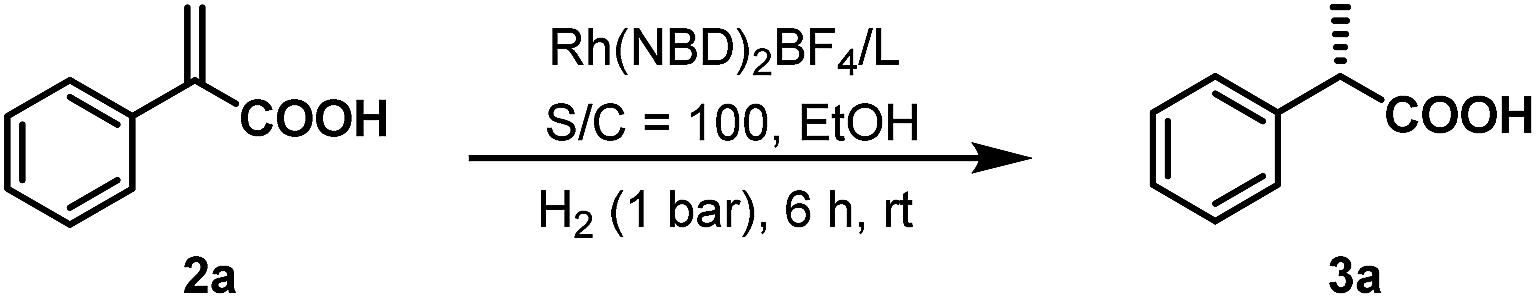
Subsequently, the substrate scope of the AH of 2-substituted acrylic acids was investigated using the optimum ligand Wudaphos under the best reaction conditions (for the screening of the reaction conditions and the discussion of the solvent effects, see ESI†). As listed in Table 3, 2-aryl acrylic acids were efficiently hydrogenated with excellent enantioselectivities under mild reaction conditions in the absence of any base regardless of whether the substituents on the phenyl ring were electron donating (Table 3, 3a–3e), electron withdrawing (Table 3, 3h), or halogens (Table 3, 3f–3h). 2-Alkyl acrylic acids were also smoothly hydrogenated with high ee (Table 3, 3i, 3j, 3m). Thus, the intermediate for the preparation of Roche ester was conveniently obtained in high ee (Table 3, 3m). Furthermore, the well-known anti-inflammatory drugs ibuprofen and naproxen were also easily obtained in excellent ee under mild conditions (Table 3, 3k, 3l). Our catalytic system shows a clear advance compared with the previous systems owing to its high enantioselectivity and base free conditions.
|
|
|---|
|
a The reaction was conducted in a 0.1 mmol scale in 1 mL of EtOH, [Rh(NBD)2]BF4 (NBD = norbornadiene) was used as the metal precursor, Wudaphos was used as the ligand, S/C = 100, L/Rh = 1.1:1, temperature = rt, H2 pressure = 1 bar, reaction time = 6 h, the configuration of all the product was determined as (S) by comparing the optical rotation data with those reported by the literature, the ee was determined via chiral HPLC after esterification with CH2N2, and the conversion of the substrates was determined using 1H NMR.
|
|
The asymmetric hydrogenation of 2-substituted acrylic acids was also conducted with low catalyst loading using 2a as a model substrate under 50 bar H2 atmosphere. Satisfyingly, our catalyst system showed excellent activity under very mild conditions in the absence of any base when the catalyst loading was 0.02 mol% (S/C = 5000) without any decrease of the ee (Table 4, entry 1). Moreover, the hydrogenation also proceeded smoothly with full conversion and high ee when lowering the catalyst loading to 0.01 mol% (S/C = 10000, Table 4, entry 2) or 0.005 mol% (S/C = 20000, Table 4, entry 3) albeit with a slight drop of ee. As shown in Scheme 5, the potent inhibitors 4 (ref. 14) and the bioactive natural product 5 (ref. 15) can be readily synthesized following literature procedures starting from the hydrogenation product 3a.
|
|
||||
|---|---|---|---|---|
| Entry | S/C | Time (h) | Conv.b% | ee % |
|
a The reaction was conducted in EtOH, [Rh(NBD)2]BF4 (NBD = norbornadiene) was used as the metal precursor, Wudaphos was used as the ligand, L/Rh = 1.1:1, temperature = rt, and H2 pressure = 50 bar.
b Substrate conversion, determined via1H NMR.
c Enantiomeric excess of 3a, determined using chiral HPLC after esterification with CH2N2.
|
||||
| 1 | 5000 | 6 | >99 | 98 |
| 2 | 10000 |
12 | >99 | 97 |
| 3 |
20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 000 000
|
24 | >99 | 96 |
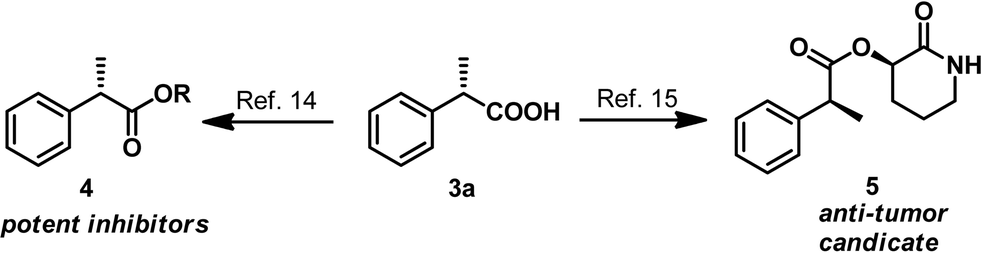 | ||
| Scheme 5 Synthesis of the potent inhibitors 4 and the bioactive natural product 5. | ||
In order to gain more insights into this catalytic system, several control experiments were also conducted. The effect of the chain length between the olefin and the acid moiety was first investigated. It was found that the chain length played an important role in determining the ee. Although the hydrogenation reactions of compounds 6, 7 and 2a all proceeded smoothly, the ee obtained in the hydrogenation of 6 and 7 was very low (Scheme 6), which indicated that the excellent enantiomeric control was based on a matched chain length. Subsequently, the effect of the ion pair noncovalent interaction was also investigated. Hydrogenation of the ester substrate 8 did not occur at all. The ee of the hydrogenation of 2a also dropped evidently when adding 0.5 equivalent of Cs2CO3. Moreover, only a racemic product was observed when one equivalent amount of triethylamine was added. It is probably due to the reason that the ion pair interaction between the ligand and substrate was interrupted by the additional base. These results suggested that the ion pair noncovalent interaction between the ligand and the acid substrates is critical (Scheme 6).
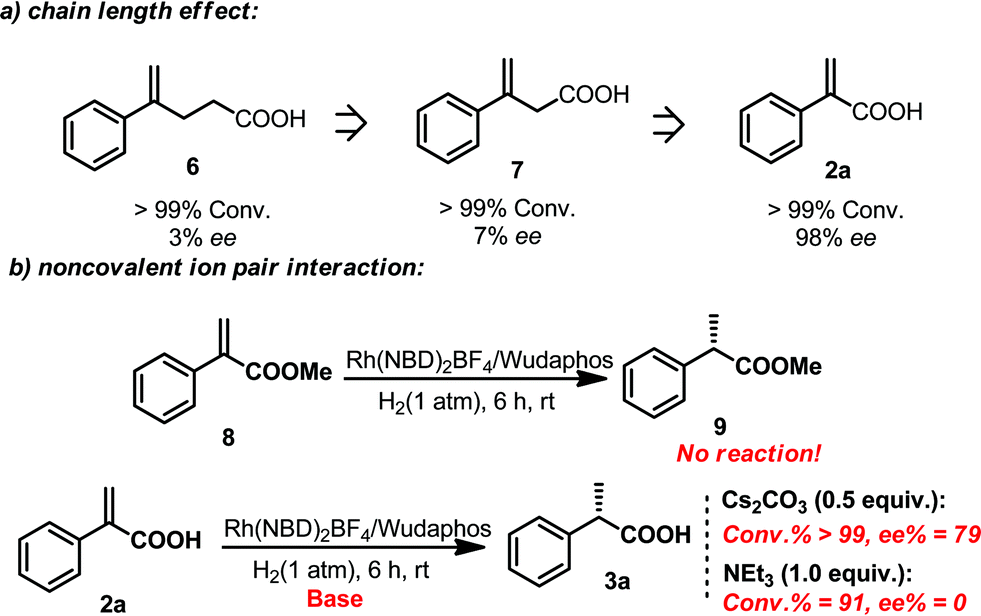 | ||
| Scheme 6 Control experiments for the investigation of the chain length effect and the ion pair noncovalent interaction effect. | ||
On the basis of the observed (S)-enantioselectivity, the X-ray crystal structure of ligand L1 (Scheme 1) and previous computational studies,21 3D models were built to account for the important roles of the ion pair interaction and the small substituent on the phosphine ligand (Scheme 7). The favorable ionic pair interaction is present and the phenyl group on the substrate experiences less repulsion with the phosphine ligand for (S)-hydrogenation. In contrast, such an ionic pair interaction cannot form and the phenyl group on the substrate has larger repulsion with the ferrocenyl group for the (R)-hydrogenation. These 3D models also correspond to the hypothesis that we made that the good enantiomeric control of the Wudaphos is based on the three hindered quadrant model.
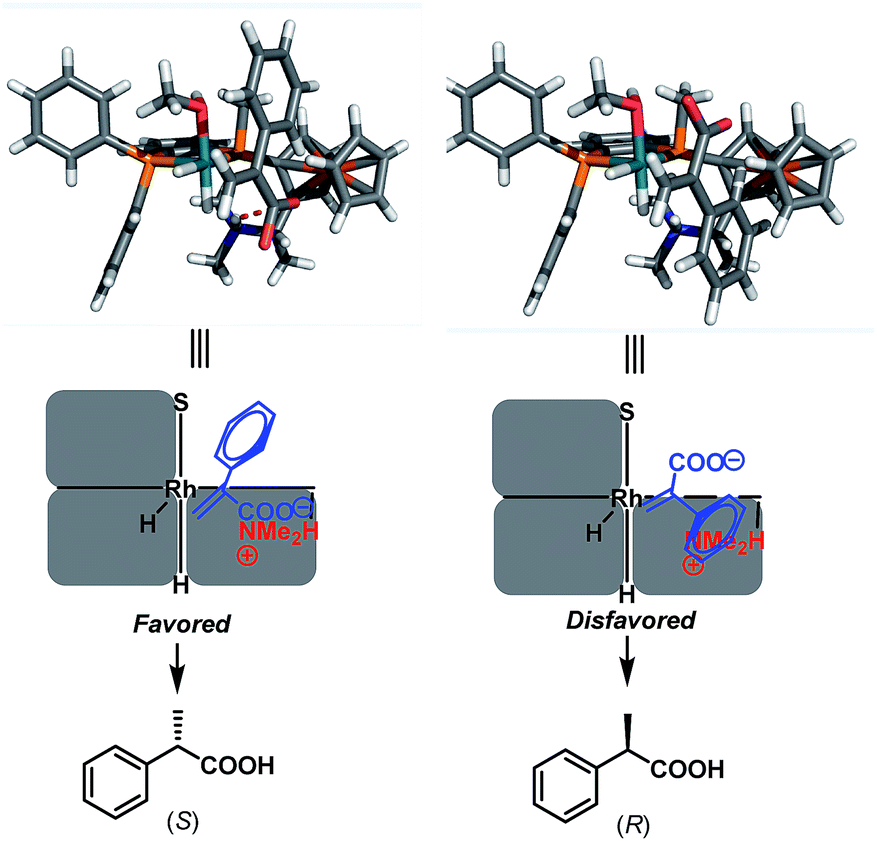 | ||
| Scheme 7 3D models and the predicted enantiomeric control. | ||
Conclusions
In summary, a new class of ferrocenyl chiral bisphosphorus ligand, Wudaphos, was developed. The Wudaphos type ligands are highly air stable and exhibit excellent ee and activity (ee up to 99%, TON up to 20000) for the asymmetric hydrogenation of both 2-aryl and 2-alkyl acrylic acids. Importantly, the hydrogenation reaction was efficiently realized through the attractive ion pair noncovalent interaction in base free and mild reaction conditions, which shows a clear advance compared with the previous catalytic system. Well-known anti-inflammatory drugs such as naproxen and ibuprofen together with the intermediate for the preparation of Roche ester and some bioactive compounds were efficiently obtained with an excellent ee. Control experiments were conducted and revealed the ion pair noncovalent interaction played an important role and the excellent enantiomeric control was based on the matched chain length between the olefin and the acid moiety.
Acknowledgements
We are grateful for the financial support by the grant from Wuhan University (203273463, 203410100064), “111” Project of the Ministry of the Education of China and the National Natural Science Foundation of China (Grant No. 21372179, 21432007, 21502145).Notes and references
- (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (b) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999 Search PubMed.
- (a) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2010, 111, 1713 CrossRef PubMed; (b) Comprehensive Chirality, ed. H. Yamamoto and E. Carreira, Elsevier, 2012 Search PubMed; (c) Stereoselective Synthesis, ed. R. S. Atkinson, Wiley, 1995 Search PubMed.
- R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678 CrossRef CAS PubMed.
- M. Sawamura and Y. Ito, Chem. Rev., 1992, 92, 857 CrossRef CAS.
- (a) W. Chen, P. J. McCormack, K. Mohammed, W. Mbafor, S. M. Roberts and J. Whittall, Angew. Chem., Int. Ed., 2007, 46, 4141 CrossRef CAS PubMed; (b) W. Chen, F. Spindler, B. Pugin and U. Nettekoven, Angew. Chem., Int. Ed., 2013, 52, 8652 CrossRef CAS PubMed.
- A. Togni, C. Breutel, A. Schnyder, F. Spindler, H. Landert and A. Tijani, J. Am. Chem. Soc., 1994, 116, 4062 CrossRef CAS.
- W. Weissensteiner, T. Sturm and F. Spindler, Adv. Synth. Catal., 2003, 345, 160 CrossRef.
- M. Lotz, K. Polborn and P. Knochel, Angew. Chem., Int. Ed., 2002, 41, 4708 CrossRef CAS PubMed.
- N. W. Boaz, S. D. Debenham, E. B. Mackenzie and S. E. Large, Org. Lett., 2002, 4, 2421 CrossRef CAS PubMed.
- A. J. J. Perea, A. Borner and P. Knochel, Tetrahedron Lett., 1998, 39, 8073 CrossRef.
- M. Sawamura, H. Hamashima, M. Sugawara, N. Kuwano and Y. Ito, Organometallics, 1995, 14, 4549 CrossRef CAS.
- (a) G. Hoge, H. Wu, W. S. Kissel, D. A. Pflum, D. J. Greene and J. Bao, J. Am. Chem. Soc., 2004, 126, 5966 CrossRef CAS PubMed; (b) K. Huang, X. Zhang, T. J. Emge, G. Hou, B. Cao and X. Zhang, Chem. Commun., 2010, 46, 8555 RSC.
- (a) D. Lednicer and L. A. Mitscher, The Organic Chemistry of Drug Synthesis, Wiley, New York, 1977, vol. 1 Search PubMed D. Lednicer and L. A. Mitscher, The Organic Chemistry of Drug Synthesis, Wiley, New York, 1980, vol. 2; Search PubMed (b) T. Y. Shen, Angew. Chem., Int. Ed., 1972, 11, 460 CrossRef CAS PubMed; (c) P.-J. Harrington and E. Lodewijk, Org. Process Res. Dev., 1997, 1, 72 CrossRef CAS.
- S. Tchilibon, J. Zhang, Q.-F. Yang, O. Eidelman, H. Kim, H. Caohuy, K. A. Jacobson, B. S. Pollard and H. B. Pollard, Biochem. Pharmacol., 2005, 70, 381 CrossRef CAS PubMed.
- P. R. Krishna, P. V. A. Kumar, V. S. Mallula and K. V. S. Ramakrishna, Tetrahedron, 2013, 69, 2319 CrossRef CAS.
- (a) Y. Tu, M. Ni, Y. Zhong, L. Li, S. Cui, M. Zhang, X. Wang and X. Liang, Yaoxue Xuebao, 1981, 16, 366 CAS; (b) R. W. Burg, B. M. Miller, E. E. Baker, J. Birnbaum, S. A. Currie, R. Hartman, Y. R. Kong, L. Monaghan and G. Olson, Antimicrob. Agents Chemother., 1979, 15, 361 CrossRef CAS PubMed; (c) J. R. Egerton, D. A. Ostlind, L. S. Blair, C. H. Eary, D. Suhayda, S. Cifelli, R. F. Riek and W. C. Campbell, Antimicrob. Agents Chemother., 1979, 15, 372 CrossRef CAS PubMed.
- (a) S. J. Mickel, G. H. Sedelmeier, D. Niederer, F. Schuerch, G. Koch, E. Kuesters, R. Daeffler, A. Osmani, S. Weibel, M. E. Schmid, A. Hirni, K. Schaer and R. Gamboni, Org. Process Res. Dev., 2004, 8, 107 CrossRef CAS; (b) I. Paterson, O. Delgado, G. J. Florence, I. Lyothier, M. O. Brien, J. P. Scott and N. Sereinig, J. Org. Chem., 2005, 70, 150 CrossRef CAS PubMed.
- (a) T. Ohta, H. Takaya, M. Kitamura, K. Nagai and R. Noyori, J. Org. Chem., 1987, 52, 3174 CrossRef CAS; (b) Q.-H. Fan, C.-Y. Ren, C.-H. Yeung, W.-H. Hu and A. S. C. Chan, J. Am. Chem. Soc., 1999, 121, 7407 CrossRef CAS; (c) C.-C. Pai, C.-W. Lin, C.-C. Lin, C.-C. Chen and A. S. C. Chan, J. Am. Chem. Soc., 2000, 122, 11513 CrossRef CAS; (d) Q.-H. Fan, Y.-M. Chen, X.-M. Chen, D.-Z. Jiang, F. Xi and A. S. C. Chan, Chem. Commun., 2000, 789 RSC; (e) R. A. Brown, P. Pollet, E. McKoon, C. A. Eckert, C. L. Liotta and P. G. Jessop, J. Am. Chem. Soc., 2001, 123, 1254 CrossRef CAS PubMed; (f) G.-J. Deng, B. Yi, Y.-Y. Huang, W.-J. Tang, Y.-M. He and Q.-H. Fan, Adv. Synth. Catal., 2004, 346, 1440 CrossRef CAS; (g) L. Qiu, F. Y. Kwong, J. Wu, W. H. Lam, S. Chan, W.-Y. Yu, Y.-M. Li, R. Guo, Z. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2006, 128, 5955 CrossRef CAS PubMed; (h) L. Qiu, Y.-M. Li, F. Y. Kwong, W.-Y. Yu, Q.-H. Fan and A. S. C. Chan, Adv. Synth. Catal., 2007, 349, 517 CrossRef CAS.
- (a) F. Robin, F. Mercier, L. Ricard, F. Mathey and M. Spagnol, Chem.–Eur. J., 1997, 3, 1365 CrossRef CAS; (b) B. Zupančič, B. Mohar and M. Stephan, Adv. Synth. Catal., 2008, 350, 2024 CrossRef; (c) M. S. Tephan, D. Šterk and B. Mohar, Adv. Synth. Catal., 2009, 351, 2779 CrossRef; (d) B. Zupančič, B. Mohar and M. Stephan, Org. Lett., 2010, 12, 1296 CrossRef PubMed; (e) B. Zupančič, B. Mohar and M. Stephan, Org. Lett., 2010, 12, 3022 CrossRef PubMed; (f) Y. Zhang, Z.-B. Han, F.-Y. Li, K.-L. Ding and A. Zhang, Chem. Commun., 2010, 46, 156 RSC; (g) S.-F. Zhu, Y. Yu, S. Li, L. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2012, 51, 8872 CrossRef CAS PubMed; (h) K. Dong, Y. Li, Z. Wang and K. Ding, Org. Chem. Front., 2014, 1, 155 RSC.
- The X-ray crystal data of L1 has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1456993.†.
- I. D. Gridnev and T. Imamoto, ACS Catal., 2015, 5, 2911 CrossRef CAS , and references therein.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1456993. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01845a |
| This journal is © The Royal Society of Chemistry 2016 |