
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Click-chemistry approaches to π-conjugated polymers for organic electronics applications
Assunta
Marrocchi
 *a,
Antonio
Facchetti
bcd,
Daniela
Lanari
e,
Stefano
Santoro
a and
Luigi
Vaccaro
*a
*a,
Antonio
Facchetti
bcd,
Daniela
Lanari
e,
Stefano
Santoro
a and
Luigi
Vaccaro
*a
aLaboratory of Green Synthetic Organic Chemistry, CEMIN – Dipartimento di Chimica, Biologia e Biotecnologie, Università di Perugia, Via Elce di Sotto, 8, 06123 Perugia, Italy. E-mail: assunta.marrocchi@unipg.it; luigi.vaccaro@unipg.it
bPolyera Corporation, 8045 Lamon Avenue, Skokie, IL 60077, USA
cCenter of Excellence for Advanced Materials Research (CEAMR), King Abdulaziz University, Jeddah, Saudi Arabia
dNorthwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
eDipartimento di Scienze Farmaceutiche, Università di Perugia, Via del Liceo, 1, 06123 Perugia, Italy
First published on 27th June 2016
Abstract
Given the wide utility of click-chemistry reactions for the preparation of simple moieties within large architecturally complex materials, this minireview article aims at surveying papers exploring their scope in the area of π-conjugated polymers for application in organic electronics to enable advanced functional properties.
 Assunta Marrocchi | Assunta Marrocchi is currently Associate Professor at the Department of Chemistry, Biology and Biotechnology of the University of Perugia. She earned a PhD in Chemical Sciences at the same institution, working on a research project dealing with the design and synthesis of helically-shaped polycyclic aromatics. Her research interests include the design, sustainable synthesis and characterization of π-conjugated semiconductors for organic electronics, with a special emphasis on the preparation of semiconducting materials for organic solar cells. |
 Antonio Facchetti | Antonio Facchetti is a co-founder and currently the Chief Scientific Officer of Polyera Corporation. He is also an Adjunct Professor at Northwestern U. and KAU. He has published more than 360 research articles, 12 books/book chapters and holds more than 110 patents. He received the ACS Award for Creative Invention, the Italian Chemical Society Research Prize, the team IdTechEx Printed Electronics Europe Award, and the corporate FlexTech Award. He is a NAI, Kavli, AAAS, MRS, and RSC Fellow. He was selected among the “Top 100 Materials Scientists of the decade (2000-2010)” and recognized as a Highly Cited Scientist by Thomson Reuters (2014). |
 Daniela Lanari | Daniela Lanari obtained her PhD in Chemical Science from the University of Perugia in 2005. In the same year she joined the research group of Professor James Fraser Stoddart at the University of California Los Angeles (UCLA) where she worked as postdoctoral fellow on the templated synthesis of mechanically interlocked molecules. She is currently Assistant Professor (Associate Professorship qualification) at the University of Perugia (Dipartimento di Chimica e Tecnologia del Farmaco). Her main interests include the optimization of synthetic procedures and synthesis of solid catalysts for the sustainable preparation of target compounds including pharmaceuticals and organic semiconductors. |
 Stefano Santoro | Stefano Santoro received his Ph.D. degree from the University of Perugia in 2009 (with Prof. C. Santi and Prof. M. Tiecco). Between 2006 and 2007 he was a visiting Ph.D. student at Aarhus University, in the group of Prof. K. A. Jørgensen. In 2010 he moved to Stockholm University as a postdoctoral researcher with Prof. F. Himo, where he stayed until 2014. Since 2014 he has been an assistant professor at the University of Perugia. His main research interests are in the areas of homogeneous and heterogeneous catalysis, C–H functionalizations and mechanistic investigations by means of DFT calculations. |
 Luigi Vaccaro | Luigi Vaccaro is currently leading the Green S.O.C. group at the University of Perugia and was habilitated as Full Professor in 2013. He is Associate Editor of RSC Advances and Beilstein Journal of Organic Chemistry. He has received the Europa Medal from Society of Chemical Industry, London (2001), the ADP Award from Merck’s Chemistry Council for “Creative work in organic chemistry” (2006 and 2007), the Ciamician Medal from Società Chimica Italiana (2007) and the Vigevani Visiting Professorship (2014). His research is currently focused on the development of heterogeneous catalysis, safer media and flow chemistry to define novel green/sustainable processes. |
Since the realization that π-conjugated polymers can be successfully implemented in several electronic and photonic devices the field of organic opto-electronics has grown exponentially.1 The most investigated devices are light emitting diodes (OLEDs), field-effect transistors (OFETs), sensors, integrated circuits, solar energy storage, photovoltaic cells (OPVs), laser diodes, and RF-ID tags.1 Organic non-volatile memory is another key area of application that exploits the advantages of organic materials.2 Typical methodologies to achieve photo/electro active polymers in organic electronics are based on traditional transition metal (TM) catalyzed cross-coupling reactions such as Stille, Suzuki, and Heck-type polymerizations (Scheme 1A and 1B).3
 | ||
| Scheme 1 A general representation of polymerization using Stille, Suzuki, Heck and direct arylation cross-coupling reactions. | ||
Despite their great versatility and wide substrate scope, these reactions may present drawbacks including the number of steps to synthesize the proper monomers, instability of organometallic reagents, poor conversion related to unreactive monomers, difficulties in controlling the polymer architecture, and poor atom economy, due to the formation of stoichiometric amounts of (toxic) byproducts. Moreover, electronic grade organic materials generally require extensive purification processes to remove undesired traces of byproducts and/or metals.4
Recently, the direct arylation methodologies (Scheme 1C) have received increasing attention appearing to be a simple and environmentally benign alternative to traditional cross-coupling reactions.3,5
In this context, efforts for implementing efficacious and green novel approaches to organic semiconductors which minimize the use of solvents and reagents, as well as the number of workup procedures,6 undoubtedly represent important contributions for the development of the field.
Click chemistry7 shares with green chemistry some of the most fundamental principles, by means of which more efficient and environmentally benign synthetic protocols can be designed and implemented. The concept of “click chemistry” was first introduced by Sharpless8 in 2001. A set of stringent criteria must be met by a chemical process to be classified as “click-type”. Among the most important, the process should be (i) able to generate inoffensive by-products removable by non-chromatographic methods, (ii) carried out under simple reaction conditions, (iii) “spring-loaded” for a single trajectory, i.e. characterized by a high thermodynamic driving force that drives it quickly and irreversibly to high yield of a single reaction product, with high reaction specificity (iv) based on readily available starting materials and (v) without the use of an additional reaction medium or only using benign solvents.8 The copper(I)-catalyzed cycloaddition of azides and alkynes to give 1,2,3-triazoles (CuAAC) is the flagship of click chemistry. The uncatalyzed reaction, i.e. the Huisgen 1,3-dipolar cycloaddition of azides and alkynes,9 proceeds very slowly even at high temperatures and gives a mixture of 1,4- and 1,5-substituted 1,2,3-triazoles (Scheme 2A).
 | ||
| Scheme 2 Synthesis of 1,2,3-triazoles via 1,3-dipolar cycloaddition of azides and terminal alkynes. | ||
However, in the presence of a source of catalytically active copper(I) species7b the reaction is markedly accelerated as shown independently for the first time in 2002 by the research groups of Sharpless10 and Meldal.11 Moreover, the Cu(I)-catalyzed process occurs regioselectively, affording exclusively the 1,4-disubstituted isomer (Scheme 2B). It should be also noted that in alternative to Cu(I)-catalyzed process, some classes of 1,2,3-triazoles can be also prepared via very efficient organocatalytic methods.12
Several studies have been reported13 to elucidate the mechanistic pathway for CuAAC, on the basis of experimental evidence and density functional theory (DFT) calculations. The most recent experimental mechanistic study by Fokin and coworkers13a outlined in Scheme 3 demonstrated unambiguously the participation of a dinuclear copper intermediate.
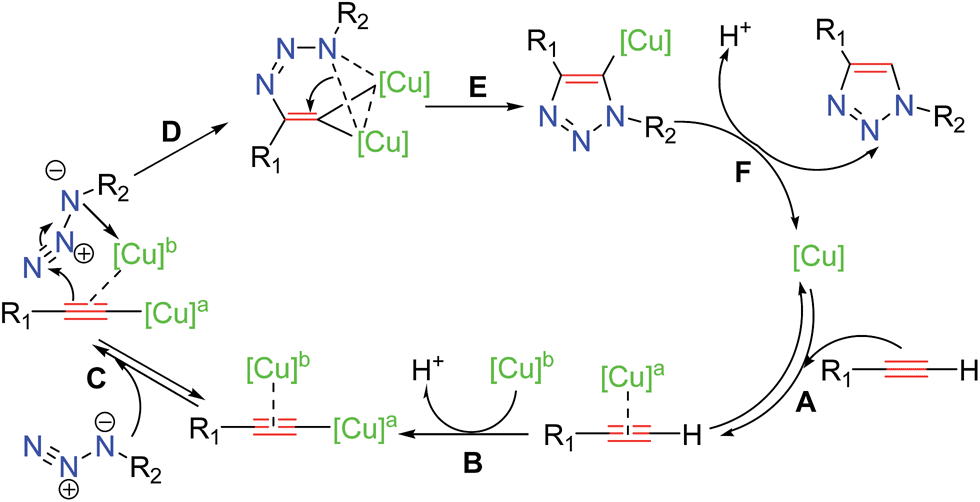 | ||
| Scheme 3 Most recent CuAAC mechanism proposed by Fokin and co-workers. | ||
Since then, click chemistry have found widespread application in various research areas, including the syntheses of dendrimers14 and rotaxanes,15 drug discovery,7,16 biochemistry,7,16b–d and for the chemical modification of surfaces and nanostructures.7
The utility of click reactions in polymer chemistry has also been explored. Although most studies reported post-functionalization of pre-formed polymers,7,17 recent efforts employed these reactions as efficient polymerization techniques (click polymerization).7d,e,g,18 The azide–alkyne click reaction, in particular, holds the promise to become a powerful synthetic tool to develop unprecedented functional materials which are expected to open new opportunities for organic electronics/photonics applications.
For instance, the triazole ring may serve as electron-accepting unit to impart optical nonlinearity to a given system.19
Triazole derivatives may also display the unique characteristic of aggregation-induced emission (AIE),18f,g,20 that is, their emission intensity may be enhanced in the solid/aggregate states, making them particularly desirable for applications as advanced photonic materials.
Although several studies indicate that the conjugation in triazole derived structures is limited through the triazole moiety,21 in the last decade a number of interesting examples demonstrated extended π-electron systems incorporating the 1,2,3-triazole moiety into the conjugation path.
Triazole-containing molecular systems may function as electron or even ambipolar22 transporting materials in the fabrication of organic light-emitting diodes. Moreover, Ratner, Mirkin and coworkers23 recently demonstrated that the triazole ring maintained the conjugation required for electronic transport in molecular transport junctions to bridge nanogaps, thereby enabling the creation of nanoelectronic devices with diverse functions and applications. Finally, Guldi and co-workers24 clearly demonstrated that aromatic 1,2,3-triazoles may represent excellent conjugating π-linkers for rapid and efficient photoinduced electron transfer between remote electron donor and electron acceptor moieties, namely zinc porphyrin and C60, respectively. This is an important factor for applications of organic materials in solar energy storage and photovoltaic devices.25 Similar conclusions have been drawn independently by several other groups,19b,20,26 when exploring the use of 1,2,3-triazoles to bridge donor–acceptor groups featuring different substitution patterns.
In this review article, we have surveyed seminal papers exploring the utility of the click reaction in the area of π-conjugated polymers for organic electronics. We have mainly focused the discussion on conjugated hetero-structures from azide–alkyne precursors. We have also briefly surveyed reactions other than CuAAC featuring the essential “click” attributes and that have been applied for the preparation of π-conjugated macromolecules. The key physical and morphological properties of the resulting materials as well as their performances in organic electronic devices have been discussed.
Several interesting studies27 have been carried out on the use of azide–alkyne click reaction as an effective strategy for tuning the associated self-assembly properties in the post-functionalization of pre-formed conjugated polymers. However, a survey of these methods is beyond the scope of this review.
The first report on the synthesis of conjugated polymers by the Cu(I)-catalysed 1,3-dipolar “click” reaction was published in 2005 by van Maarseveen, Reek and coworkers28 who synthesized linear poly(triazole)s P1–P3via polymerization of 2,7-diazidofluorene 1 and aromatic diynes 2–4 (Scheme 4).
 | ||
| Scheme 4 Synthesis of “click” polymers P1–P3. | ||
The reaction between 1 and 2,5-diethynylpyridine 2 was performed in THF/CH3CN, by employing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-monomer molar ratio, Cu (∼14 mol%)/Cu(OAc)2 (∼2 mol%) as the catalyst and tris-(benzyltriazolylmethyl)amine (∼0.8 mol%) as the ligand. Copolymer P1 was obtained with a Mn up to ∼25 kDa (PDI = 1.9). Under the same conditions, copolymers P2 and P3 were obtained with Mn up to 327 kDa (PDI = 1.21) and Mn = 8 kDa (PDI = 1.61), respectively.
:1 co-monomer molar ratio, Cu (∼14 mol%)/Cu(OAc)2 (∼2 mol%) as the catalyst and tris-(benzyltriazolylmethyl)amine (∼0.8 mol%) as the ligand. Copolymer P1 was obtained with a Mn up to ∼25 kDa (PDI = 1.9). Under the same conditions, copolymers P2 and P3 were obtained with Mn up to 327 kDa (PDI = 1.21) and Mn = 8 kDa (PDI = 1.61), respectively.
Interestingly, when the reaction between 1 and 2 was carried out at −10 °C for 65 h, P1 molecular weight was higher (Mn = 20.6 kDa, PDI = 2.86) than that of the polymer obtained at 25 °C for 170 h, thus suggesting an exothermic polymerization reaction. Unfortunately, no reaction yields were reported. The fluorene-containing copolymer absorption spectra were found to be the superposition of those of the monomers. Furthermore, the authors observed that P1–P3 were highly emissive in THF, with photoluminescence peaks located at 360–380, i.e. the emission was dominated by the fluorene units. The highest quantum yield (ΦF = 55%) was achieved for P3. These findings indicate a poor electronic communication between the polymers aromatic building blocks. However, cyclic voltammetry measurements on a model compound consisting of two pyridyl-triazole units bridged by a fluorene unit (i.e. the product between 1 and 2) showed one two-electron reduction (−1.86 V) but no oxidation, suggesting that the present materials might be useful semiconductors.
Shortly thereafter, Bunz and coworkers29 prepared the conjugated poly(triazole)s P4–P9 (Scheme 5) by reacting 2,7-diazidofluorenes 1 (or 5), and 4,4′-diazido-3,3-dimethoxy-biphenyl 6 with 2,5-dialkyl-1,4-diethynyl benzenes 7 and 8, in the presence of CuSO4 (5 mol%)/sodium ascorbate (SA).
 | ||
| Scheme 5 Synthesis of “click” polymers P4–P9. | ||
These copolymers were obtained in high yields (80–92%), with a maximum Mn value of 8.7 kDa achieved for P7 (PDI = 5.8). According to 1H- and 13C-NMR spectroscopy, all poly(triazole)s P4–P9 exhibited regioregular 1,4-substitution of the triazole unit and showed blue fluorescence in solution, with the highest quantum yields (ΦF ∼ 40%) achieved for P7 and P9. On the other hand, no fluorescence was observed in the solid state, which was attributed to aggregation phenomena.30 The optical properties were independent from P4–P9 molecular weights, thereby suggesting that the used building blocks led to species with localized HOMO/LUMO orbitals. This was also corroborated by quantum mechanical calculations on the model compound P8. Interestingly, fluorescence measurements and theoretical calculations suggested that29 protonation at the 3-position of the triazole group in P8 resulted in the decrease of HOMO–LUMO gap and more delocalized frontier molecular orbitals. On these basis, the use of alkyne and azide precursors substituted with electron-donating and electron-withdrawing groups, respectively, is expected to further lower the band-gap of P4–P9, which may become of interest as semiconductors.
Finally, by using a heated tip of an AFM cantilever (∼225 °C) the authors29 succeeded in writing crisp nanoscale features into P7 thin-films. The absence of tackiness and ripping led the authors to the conclusion that these materials were attractive as novel semiconductors that could have been easily thermally structured.
Lee, Jin and coworkers31 reported the preparation of π-conjugated soluble poly(triazole)s P10–P12 (Scheme 6) by click polymerization (∼90% yields) of 2,7-diazido-9,9-dioctylfluorene (9) with 2,7-diethynyl-9,9-dioctylfluorene (10), 4,7-diethynylbenzothiadiazole (11), and 2,7-diethynylcarbazole (12), respectively, employing a 1:1 co-monomer molar ratio and CuSO4·5H2O/sodium ascorbate/triethylamine as the catalytic system.
 | ||
| Scheme 6 Synthetic routes for the “click” polymers P10–P12. | ||
An average Mn as high as ∼8.3 kDa was obtained for P10 (PDI = 1.92) whereas P11 and P12 both exhibited lower Mn ∼ 6 kDa (PDI = 1.38 and 1.92, respectively). Polymers P10–P12 were found to be stable up to 300 °C (via TGA) and exhibited optical absorption maxima in CHCl3 in the range of 328–350 nm, with P10 being the most red-shifted. Similar UV-vis absorption spectra were recorded in the solid state. The well-structured PL spectra of P10–P12 in solution revealed a blue light emission between 370 and 406 nm, whereas their emission spectra in the solid state were slightly red-shifted (15–40 nm). The HOMO and LUMO energy levels of P10 are −5.23 eV and −3.25 eV, respectively, whereas a HOMO of −5.39 (5.35) eV and a LUMO of −2.44 (3.14) eV were found for P12, on the basis of cyclic voltammetry and optical absorption data.
More recently, Nesterov and coworkers32 reported an interesting stepwise methodology based on surface-initiated Cu(I)-catalyzed click polymerization to synthesize “brush” polymer P13 (Scheme 7). It is noteworthy to highlight that this approach allows the access to surface-grafted (“brush”) polymers which are an interesting alternative for high-performance, long-term operation organic electronics, possibly helping charge injection and charge transport processes which are crucial for many devices.33
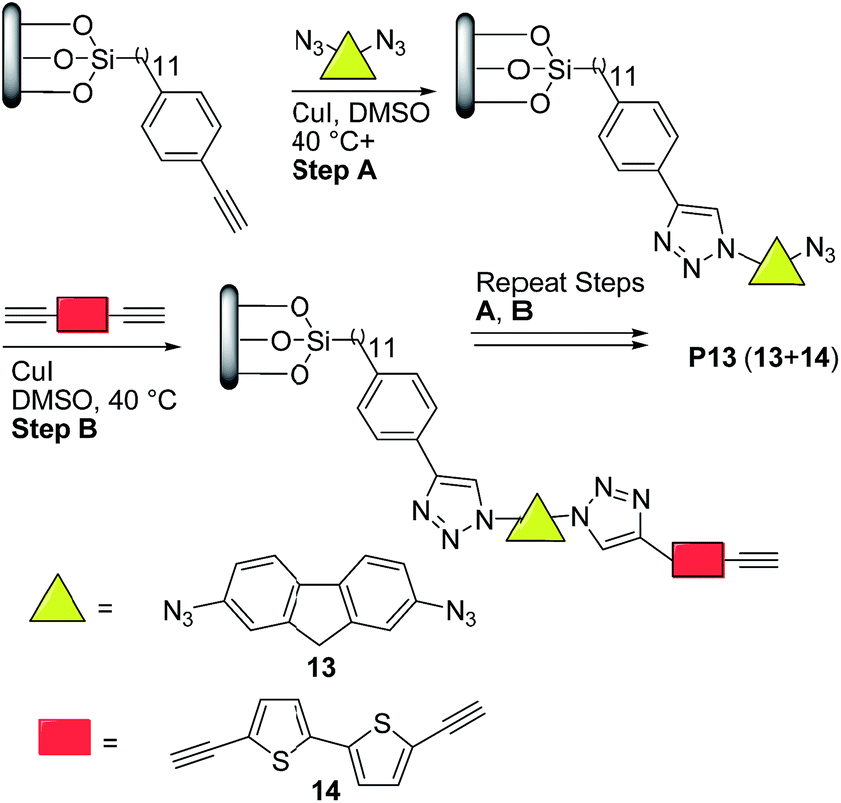 | ||
| Scheme 7 Preparation of semiconducting polymer thin films via surface-initiated stepwise “click” polymerization. | ||
Thus, the authors32 functionalized a quartz substrate with a monolayer of trimethylsilyl-acetylene initiator. Next, the activated surface was immersed in a dimethyl sulfoxide (DMSO) solution of the bis-azide monomer 13 (Scheme 7) in the presence of 10 mol% CuI, followed by rinsing. Finally, the substrate was placed into a DMSO solution of the bisacetylene monomer 14 and 10 mol% CuI, followed by rinsing. This sequence was repeated for 34 times. Note here that the initially prepared monomer solutions could last for the entire duration of polymerization, thereby avoiding wasting monomers. Moreover, no oligomer/polymer formation was detected in these solutions, which indicated that a controlled stepwise polymerization occurred only on the substrate surface.
Atomic force microscopy (AFM) data revealed that P13 films exhibited a thickness of ∼28 nm and a surface morphology (rms ∼ 6 nm) featuring uniform cylindrical domains of ∼90 nm in diameter with tight packing density (Fig. 1). The domain morphologies were investigated by polarization-dependent ultraviolet photoemission spectroscopy (UPS) and found to be oriented normally to the substrate protruding throughout the film. Furthermore, the authors claimed that each domain likely featured a uniform, well-ordered packing of the macromolecules. UV-vis thin-film absorption spectrum exhibited a maximum at ∼350 nm, with a broad band spanning to almost 700 nm, as well as an optical band gap of 2.52 eV. The HOMO and LUMO energy levels for P13 were found to be −5.28 eV and −2.76 eV, respectively, on the basis of cyclic voltammetry experiments and optical absorption data.
 | ||
| Fig. 1 Surface morphology imaged by contact mode AFM. (a) Topograph of the film of P13 prepared on a quartz surface (roughness, rms, ∼6 nm); (b) corresponding lateral force image. Centre area has a trench in the film made by ‘‘nanoshaving’’ to show continuity of the cylindrical domains. Reproduced with permission.32 Copyright 2011, The Royal Society of Chemistry. | ||
The potential of ‘click’ polymers from azide–alkyne precursors as conducting molecular wires has been demonstrated by Luo et al.34 More specifically, the authors prepared oligophenylenetriazole wires O1 and O2 (Scheme 8) having systematically varied lengths up to 10 nm. The O1 and O2 wires were built from Au surface by the general route exemplified for the synthesis of O2 in Scheme 8.
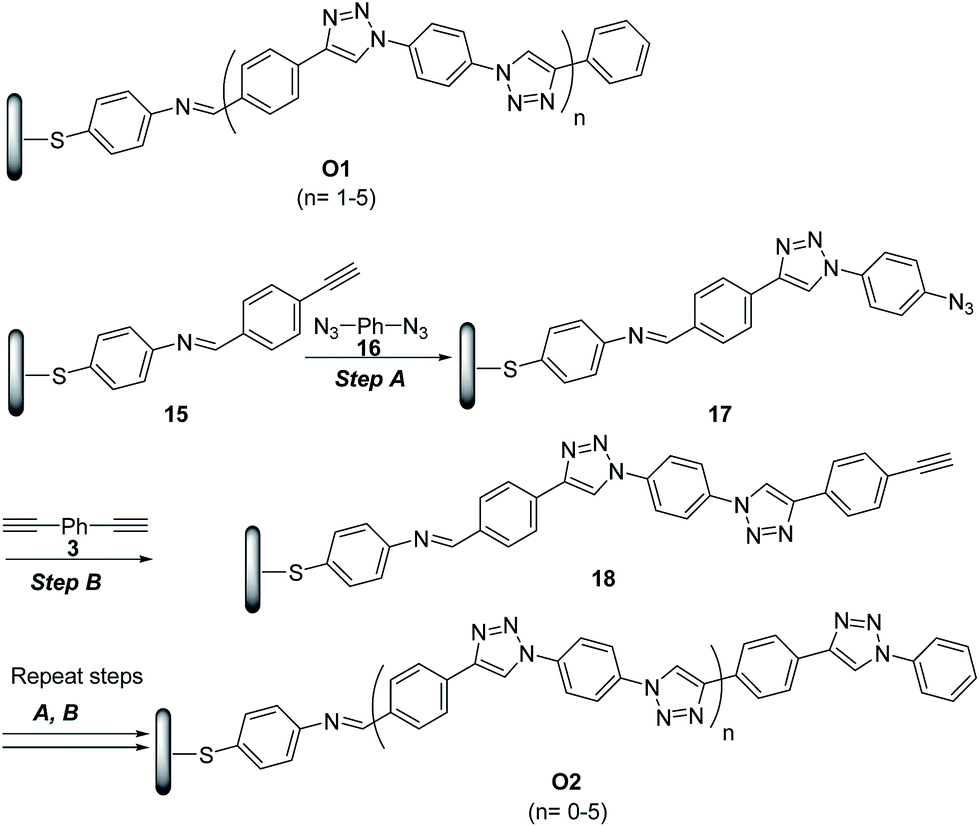 | ||
| Scheme 8 Synthetic route to oligophenylenetriazoles O1 and O2. (A) CuSO4·5H2O (2 mmol%), SA (15 mmol%), EtOH/H2O (2.5:1), rt; (B) CuSO4·5H2O (2 mmol%), SA (15 mmol%), CH3CN/H2O (2.5:1), rt. | ||
Electrical measurements of oligophenylenetriazole wires were performed by conducting probe atomic force microscopy (CP-AFM) and, interestingly, their current–voltage characteristics were reported to be similar to other conjugated wire molecules,35 with a transition from tunnelling to hopping transport as wire length increased.
Bäuerle and co-workers36 successfully employed the click chemistry approach to synthesize a range of thiophene-based oligomers of a donor–acceptor type O3, in which the thiophene moiety is the donor (electron-rich) moiety and 1,2,3-triazoles are the acceptor (electron-poor) units. These systems were designed bearing in mind that a 1,2,3-triazole can act as a weak electron-acceptor.19b Excellent yields up to 99% were obtained by reacting an equimolar mixture of the corresponding terminal acetylenes and in situ generated organic azide. Optimal conditions involved the use of copper(I) iodide (10 mol%)/sodium ascorbate (10 mol%)/N,N′-dimethylethylenediamine (DMEDA, 20 mol%) as the catalytic system in ethanol–water (50 °C, 20 h) (Scheme 9). Notably, a conjugation through the triazole ring was generally operative, as indicated by the spectroscopic and redox properties of the above systems. These data points out again the importance of designing suitable substrates in such approach to limit the charge trapping characteristics of the triazole ring.
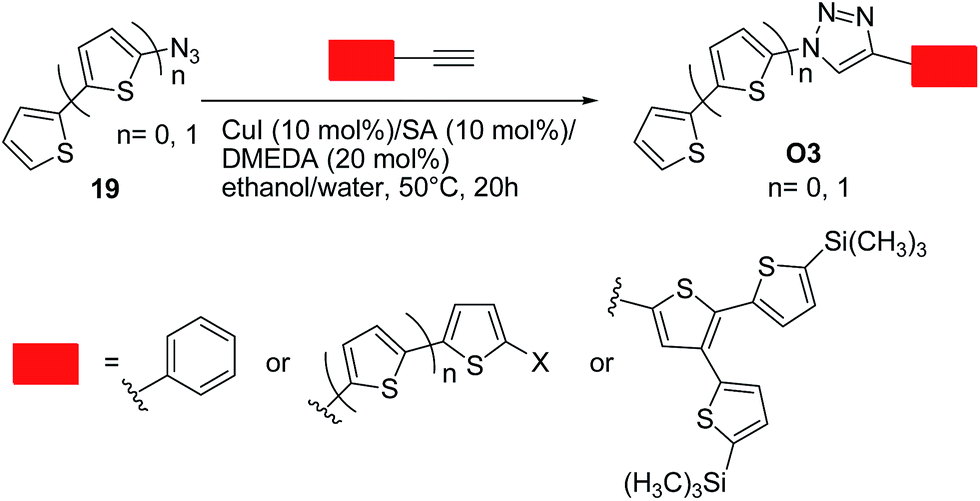 | ||
| Scheme 9 Synthesis of 1,4-disubstituted 1,2,3-triazoles O3 from azides 19 and corresponding terminal acetylenes. | ||
Very recently Song et al.37 demonstrated that triazole rings are advantageous units to high performance electrical memory polymers, in view of the high hole affinity of the three nitrogens with lone-pair electrons. A series of brush polystyrenyl (PS) derivatives P14–P16 (Scheme 10) bearing the triazole moiety in their bristle were prepared in high yield (up to 98%) employing as a key step the click reaction of a poly(4-azidomethylstyrene) 20 with 4-[(4-ethynylphenyl)ethynyl]-N,N-dihexadecylaniline 21. Next, the authors investigated the triazole containing polymers electrical memory characteristics by incorporating them as active layer into devices with aluminium top and bottom electrodes (Fig. 2).
 | ||
| Scheme 10 Synthetic route to P14–P16. TCNE and TCNQ stand for tetracyanoethylene and 7,7,8,8-tetracyanoquinodimethane, respectively. | ||
 | ||
| Fig. 2 (A) Device structure and (B) representative I–V curves of the devices incorporating (a) P14, (b) P15, and (c) P16 30 nm thick films [adapted from ref. 37]. | ||
Interestingly, P14–P16 revealed to be highly suitable for the production of unipolar permanent memory devices that can be operated with very low power consumption, a high ON/OFF current ratio and high stability and reliability. Furthermore, the memory type could be tuned from p-type (P14) to n-type (P15, P16) by the incorporation of a strong electron accepting TCNE or TCNQ moiety into the ethynylphenyl unit linked to the triazole moiety, as a result of a reduced LUMO level with regard to the electrode work function.
Apart from the combination of the azide–alkyne functionalities, other reactions based on “spring-loaded” reactants and featuring the essential “click-chemistry” attributes have been exploited to synthesize π-conjugated macromolecules for organic electronics applications.
For instance, Jørgensen and Krebs38 and, more recently, Eichen and coworkers39 developed a stepwise directional synthetic route (Fig. 3) to oligo-arylene-vinylenes.
 | ||
| Fig. 3 Schematic illustration of Jørgensen38 and Eichen39 “click” approach to oligo-arylene-vinylenes. Adapted with permission.39b Copyright 2011, Wiley-VCH. | ||
Oligomers of arylene-vinylenes are of interest in many areas of materials research, in some cases as models for the corresponding poly-p-phenylene-vinylenes (PPVs) that are used in organic light emitting diodes, field effect transistors and photovoltaics.1
Most remarkable the development of such alternative reaction scheme by the Jørgensen group38 was prompted by the observation that when palladium catalyzed reactions were used for polymerization, incorporation of small palladium nanoparticles was inevitable. As a consequence these metal residues were very difficult or impossible to remove, substantially affecting the device performance.4
Thus, the authors employed only one monomer featuring two different terminal functionalities. On a stilbene core, it was introduced a phosphonate ester group at one extremity and an acetal-protected aldehyde at the other (Fig. 3A). The oligomerization then started with the Horner–Wadsworth–Emmons (HWE) reaction between an aldehyde and the monomer in the presence of potassium t-butoxide.
The product of this HWE reaction was an end-capped “monomer” that was subsequently subjected to the deprotection of the aldehyde in the presence of dilute hydrochloric acid, as depicted in Fig. 3A. Oligomerization then proceeded by alternating reaction of the previous aldehyde-terminated p-phenylenevinylene fragment with the monomer and deprotection of the acetal.
A series of oligomers O4 (Fig. 4) featuring up to 11 phenylene-vinylene units were successfully prepared in good to high yields (57–96%). Interestingly the authors demonstrated that the proposed synthetic scheme gave easy access to oligomers of high purity.
 | ||
| Fig. 4 Examples of π-conjugated “click” oligo-arylene-vinylenes reported by Jørgensen et al.38 and Eichen et al.39 | ||
Subsequently, the oligomers could be further derivatized easily at the aldehyde position to create a series of systems with a range of electron-accepting or electron-donating substituents. Photovoltaic cells using the structure of ITO/PEDOT:PSS/O4/Al were assembled. Illuminated under simulated sunlight (AM1.5) gave short circuit currents (Isc) in the range of 0.015–0.5 mA cm−2 and typical open circuit voltage (Voc) of 0.4–0.8 V. The maximum efficiency obtained was ∼0.1%.
Eichen and coworkers39 applied “click”-type approaches depicted in Fig. 3 to produce a wide range of structurally controlled functionalized oligo arylenevinylene systems, including O5. The general process used bifunctional monomers consisting of a π-conjugated backbone bearing two functional groups, i.e. a phosphine/phosphonate group and an acetal-protected aldehyde. The first step in the arylene-vinylene formation was a Wittig–Horner reaction between a functionalized aldehyde (so-called start unit) with the in situ generated ylide of a bifunctional monomer (using t-BuOK). Next, the product was deprotected to release the aldehyde group of the di-arylenevinylenes for the subsequent step. This sequence has been repeated several times, using the different bifunctional monomers to construct the oligomer with the desired sequence. Additionally, at a certain stage, the deprotected oligomer has been coupled with a bifunctional system bearing two phosphine/phosphonate groups, producing symmetrical oligo-phenylene-vinylenes (Fig. 3B).
Optical absorption/fluorescence spectroscopy and cyclic voltammetry were extensively used to characterize the electronic structure of the arylenevinylene systems, thereby highlighting the tuneability of the optical and HOMO/LUMO band positions and, ultimately, showing the potential inherent to such approach.
Next, organic field-effect transistors and light emitting diodes were prepared from selected materials, and field-effect mobilities (μ) up to ∼1 × 10−3 cm2 V−1 s−1 as well as CIE chroma coordinates (x, y) = (0.6354, 0.3625) were achieved.
Moreover, very recently, Demissie et al.40 and Smith et al.41 reported the successful synthesis of π-conjugated molecular wires on Au surfaces using imine click-like (condensation) reaction to ensure high yields (∼99%) in a simple sequential monomer addition process. The reaction schematic in Scheme 11 demonstrates the alternate addition of benzene-1,4-dicarbaldehyde 22 (ref. 40) (or thiophene-2,5-carboxyaldehyde 23)41 and 1,4-diaminobenzene 24 monomers to give O6 and O7-type oligomers. The wires ranged in length from 0.6 to 5.5 nm. Electrical transport measurements were carried out for O7-type oligomers,41 revealing that charge transport in short oligomers was temperature-dependent whereas for longer ones it is activated, consistent with a crossover from tunneling to hopping transport. Interestingly, optical and electrochemical measurements indicated that for wires featuring more than three repeating units charge is not delocalized across the entire wire length.
 | ||
| Scheme 11 Reaction schematics for the molecular wires O6 and O7 self-assembled on Au surface. | ||
Research efforts have been also devoted to exploit thiol-yne click42 reaction to synthesize electronically active π-conjugated polymers. Indeed, since the thiol-yne reaction allows simple addition of two thiol groups to an alkyne, it appears to be perfectly suitable to enable multifunctional conjugated structures. For instance, Tang and co-workers developed the first example of Rh-catalyzed thiol-yne click polymerization for the synthesis of a series of linear poly(vinylenesulphide)s (PVSs), including P17a and P17b (Scheme 12).43 The Rh(PPh)3Cl catalyzed click polymerization of dithiol 25 and diynes 26a,b was carried out under mild conditions at room temperature, leading to the corresponding sulphur rich polymers 17a and 17b in high yields (∼85–92%) and good-to-high molecular weight (Mw = 13.3 kDa, PDI = 3.4 and Mw = 7 kDa, PDI = 3.2, respectively). Polymer 17a featured mixed isomers with E/Z ratio of 50:50 whereas when the ferrocene-containing dyine 26b was employed, a PVS with high E content (∼90%) was obtained. In a further contribution44 the same group reported a non-metallic catalyst-mediated click polymerization of dithiol 25 and a series of dipropiolates, including 26c (Scheme 12). The reaction was carried out at room temperature (24 h), and readily provided the corresponding PVS 17c with high molecular weight (Mw = 21 kDa, PDI = 2.9) and a predominant Z configuration (Z/E = 78%) in a satisfactory yield (73.5%). Tang and co-workers succeeded45,46 also in establishing a catalyst-free thiol-yne click polymerization and preparing conjugated linear and hyperbranched PVSs (e.g.P17d–f, Scheme 12, and P18, Scheme 13, respectively). The polymerizations of the aromatic dithiol 25 and diynes 26d–f or 27 in equimolar ratio could be performed under very mild conditions (THF, 30 °C), without any additive. Importantly, this polymerization was quite efficient (78–97% yield) and PVSs with Mw up to 61 kDa (P18, PDI = 4.96) could be obtained. The E/Z ratio generally resulted to be ∼50/50. Interestingly, the AIE active20a,d tetraphenylethylene moiety containing PVSs, i.e.P17c, P17f and P18 were also found to possess the AIE feature.
 | ||
| Scheme 12 Syntheses of poly(vinylsulfide)s P17via thiol-yne click polymerization reactions of monomers 25 and 26. | ||
 | ||
| Scheme 13 Synthesis of hyperbranched π-conjugated polymer P18 by catalyst-free thiol-yne reaction between monomers 25 and 27. | ||
Conclusions
In conclusion, click chemistry may provide an efficient and versatile way for the synthesis of structurally diverse π-conjugated polymers/oligomers. Particularly, the CuAAC reaction continues to confirm its role as a powerful synthetic tool contributing to unaccountable applications. It also proves to be very effective for the synthesis of opto-electronic materials opening new opportunities for organic electronics applications. As inferred from the number of contributions emerged in the last few years, the “click” polymerization is yet far to be fully exploited in this field. Nevertheless, the examples of conjugated triazole-based materials reported to date have highlighted the great potential for new directions, identifying a new family within organic materials. The ultimate scope of this review is to anticipate the need of future research in this area. A major issue that need to be addressed in the near future is to improve the experimental conditions to allow a (CuAAC) click polymerization reaction to proceed efficiently without catalyst or in such a way that copper can be removed completely. Indeed, although there are many reports7d,h,20b,42c on metal- or catalyst-free azide–alkyne cycloadditions and other click reactions, only few papers44–46 have addressed this issue for the preparation of opto-electronic polymers. Additionally, there are examples7a,b reporting the utilization of supported heterogeneous Cu(I) catalysis for CuAAC reactions but only for the preparation of non-conjugated systems. It is well known6a,7a,b that carrying out a reaction using a heterogeneous catalyst, product purification can be simplified because of the facile recovery from the reaction mixture by filtration/centrifugation. In addition, in most cases the need of chromatography for metal removal may be avoided, thus reducing the operation cost and the waste associated with the process. Heterogeneous catalysts are also easy to handle and are usually recyclable and safer to be stored/discarded. Another fundamental question is the control over selectivity in the click polymerization when using a broader range of monomers to access different conjugated polymers. Further efforts in this direction are certainly needed to achieve a more mature and broader scope synthetic methodology. Among the possible routes for optimization, the proper design of monomers, catalysts, and/or process conditions appear to be promising strategies. Expanding the range of azide/alkyne monomer substituents to broaden the development of CuAAC click-polymerization towards conjugated polymers is also desirable.Acknowledgements
We gratefully acknowledge the Università degli Studi di Perugia and the “Fondazione Cassa di Risparmio di Terni e Narni“ for financial support. AF thanks Polyera Corporation, KAU (grant # 80-130-35-HiCi), and AFOSR (grant # 9550-15-1-0044) for support.Notes and references
- For recent representative reviews see: (a) Y. Yao, H. Dong and W. Hu, Adv. Mater., 2016, 28, 4513–4523 CrossRef CAS PubMed; (b) C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed; (c) L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed; (d) K. Muellen and W. Pisula, J. Am. Chem. Soc., 2015, 137, 9503–9505 CrossRef CAS PubMed; (e) Z. Yi, S. Wang and Y. Liu, Adv. Mater., 2015, 27, 3589–3606 CrossRef CAS PubMed; (f) S. Savagatrup, A. D. Printz, T. F. O'Connor, A. V. Zaretski and D. J. Lipomi, Chem. Mater., 2014, 26, 3028–3041 CrossRef CAS; (g) S. J. Benight, C. Wang, J. B. H. Tok and Z. Bao, Prog. Polym. Sci., 2013, 38, 1961–1977 CrossRef CAS; (h) A. Marrocchi, D. Lanari, A. Facchetti and L. Vaccaro, Energy Environ. Sci., 2012, 5, 8457–8474 RSC.
- (a) V. Khikhlovskyi, A. J. J. Van Bremen, R. A. J. Janssen, G. H. Gelink and M. Kemerink, Org. Electron., 2016, 31, 56–62 CrossRef CAS; (b) S. Nam, J. Seo, H. Kim and Y. Kim, Appl. Phys. Lett., 2015, 107, 153302 CrossRef; (c) J. S. Meena, S. M. Sze, U. Chand and T.-Y. Tseng, Nanoscale Res. Lett., 2014, 9, 526–559 CrossRef PubMed; (d) S.-T. Han, Y. Zhou and V. A. L. Roy, Adv. Mater., 2013, 25, 5425–5449 CrossRef CAS PubMed.
- Metal catalyzed cross-coupling reactions and more, ed. A. de Meijere, S. Braese and M. Oestreich, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- For representative examples see: (a) G. Strappaveccia, E. Ismalaj, C. Petrucci, D. Lanari, A. Marrocchi, M. Drees, A. Facchetti and L. Vaccaro, Green Chem., 2015, 17, 365–372 RSC; (b) T. P. Osedach, T. L. Andrew and V. Bulović, Energy Environ. Sci., 2013, 6, 711–718 RSC; (c) W. L. Leong, G. C. Welch, L. G. Kaake, C. J. Takacs, Y. Sun, G. C. Bazan and A. J. Heeger, Chem. Sci., 2012, 3, 2103–2109 RSC.
- For representative examples see: (a) P.-O. Morin, T. Bura and M. Leclerc, Mater. Horiz., 2016, 3, 11–20 RSC; (b) S.-L. Suraru, J. A. Lee and C. Luscombe, ACS Macro Lett., 2016, 5, 724–729 CrossRef CAS; (c) A. E. Rudenko and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 135–147 CrossRef CAS; (d) L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed; (e) A. Facchetti, A. Marrocchi and L. Vaccaro, Angew. Chem., Int. Ed., 2012, 51, 3520–3523 CrossRef CAS PubMed.
- (a) A. Marrocchi, A. Facchetti, D. Lanari, C. Petrucci and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763–786 RSC; (b) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- For recent representative examples see: (a) S. Chassaing, V. Beneteau and P. Pale, Catal. Sci. Technol., 2016, 6, 923–957 RSC; (b) E. Haldon, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC; (c) F. Alonso, Y. Moglie and G. Radivoy, Acc. Chem. Res., 2015, 48, 2516–2528 CrossRef CAS PubMed; (d) A. Qin, Y. Liu and B. Z. Tang, Macromol. Chem. Phys., 2015, 216, 818–828 CrossRef CAS; (e) H. Wu, H. Li, R. T. K. Kwok, E. Zhao, J. Z. Sun, A. Qin and B. Z. Tang, Sci. Rep., 2014, 4, 5107–5112 Search PubMed; (f) M. Juríček, P. H. J. Kouwer and A. E. Rowen, Chem. Commun., 2011, 47, 8740–8749 RSC; (g) Themed issue (4) Click chemistry: function follows form, ed. M. G. Finn and V. V. Folkin, Chem. Soc. Rev., 2010, 39, 1221–1408 Search PubMed; (h) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., 2001, 113, 2056–2075 ( Angew. Chem., Int. Ed. , 2001 , 40 , 2004–2021 ) CrossRef.
- R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565–598 ( Angew. Chem. , 1963 , 75 , 604–637 ) CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- For recent results on N-substituted 1,2,3-triazoles see: (a) J. Thomas, S. Jana, J. John, S. Liekensb and W. Dehaen, Chem. Commun., 2016, 52, 2885–2888 RSC; (b) J. Thomas, J. John, N. Parekh and W. Dehaen, Angew. Chem., Int. Ed., 2014, 53, 10155–10159 CrossRef CAS PubMed. For representative papers on 1H-1,2,3-triazoles see: (c) D. Amantini, F. Fringuelli, O. Piermatti, F. Pizzo, E. Zunino and L. Vaccaro, J. Org. Chem., 2005, 70, 6526–6529 CrossRef CAS PubMed; (d) G. D'Ambrosio, F. Fringuelli, F. Pizzo and L. Vaccaro, Green Chem., 2005, 7, 874–877 RSC.
- For representative examples see: (a) T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef PubMed; (b) G.-C. Kuang, P. M. Guha, W. S. Brotherton, J. T. Simmons, L. A. Stankee, B. T. Nguyen, R. J. Clark and L. Zhu, J. Am. Chem. Soc., 2011, 133, 13984–14001 CrossRef CAS PubMed; (c) B. F. Straub, Chem. Commun., 2007, 3868–3870 RSC; (d) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed; (e) V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210 CrossRef CAS PubMed.
- For representative examples see: (a) S. Mongkhontreerat, M. V. Walter, O. C. J. Andren, Y. Cai and M. Malkoch, Adv. Funct. Mater., 2015, 25, 4837–4843 CrossRef CAS; (b) C. Deraedt, N. Pinaud and A. Didier, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS PubMed; (c) A. Didier, L. Liang, A. Rapakousiou and J. Ruiz, Acc. Chem. Res., 2012, 45, 630–640 CrossRef PubMed; (d) P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3932 CrossRef CAS PubMed.
- (a) X. Hou, C. Ke and J. F. Stoddart, Chem. Soc. Rev., 2016 10.1039/c6cs00055j; (b) H. Li, Z. Zhu, A. C. Fahrenbach, B. M. Savoie, C. Ke, J. C. Barnes, J. Lei, Y.-L. Zhao, L. M. Lilley, T. J. Marks, M. A. Ratner and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 456–467 CrossRef CAS PubMed; (c) O. S. Miljanic, W. R. Dichtel, I. Aprahamian, R. D. Rohde, H. D. Agnew, J. R. Heath and J. F. Stoddart, QSAR Comb. Sci., 2007, 26, 1165–1174 CrossRef CAS; (d) W. R. Dichtel, O. S. Miljanic, J. M. Spruell, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2006, 128, 10388–10390 CrossRef CAS PubMed.
- (a) X. Wang, B. Huang, X. Liu and P. Zhan, Drug Discovery Today, 2016, 21, 118–132 CrossRef CAS PubMed; (b) T. T. Trinh, C. Laure and J.-F. Lutz, Macromol. Chem. Phys., 2015, 216, 1498–1506 CrossRef CAS; (c) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed; (d) D. M. Beal and L. H. Jones, Angew. Chem., Int. Ed., 2012, 51, 6320–6326 CrossRef CAS PubMed.
- For recent representative reviews see: (a) G. Delaitre, N. K. Guinard and C. Barner-Kwollik, Acc. Chem. Res., 2015, 48, 1296–1307 CrossRef PubMed; (b) P. Espeel and F. E. Du Prez, Macromolecules, 2015, 48, 2–14 CrossRef CAS; (c) T. Michinobu, Macromol. Chem. Phys., 2015, 216, 1387–1395 CrossRef CAS; (d) T. Michinobu, Chem. Soc. Rev., 2011, 40, 2306–2316 RSC.
- (a) Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC; (b) A. B. Lowe, Polymer, 2014, 55, 5517–5549 CrossRef CAS; (c) W. H. Binder and R. Zirbs, Click Chemistry in macromolecular synthesis in, Encyclopaedia of Polymer Science and Technology, ed. H. F. Mark, J. Wiley & Sons, 4th edn, 2014, vol. 3, pp. 186–230 Search PubMed; (d) C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed; (e) P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC; (f) A. Qin and B.-Z. Tang, Chem. Soc. Rev., 2010, 39, 2522–2544 RSC; (g) A. Qin, J. W. Y. Lam and B.-Z. Tang, Macromolecules, 2010, 43, 8693–8702 CrossRef CAS.
- (a) A. Qin, C. K. W. Jim, W. Lu, J. W. Y. Lam, M. Haussler, Y. Dong, H. H. Y. Sung, I. D. Williams, G. K. L. Wong and B. Z. Tang, Macromolecules, 2007, 2308–2317 CrossRef CAS; (b) M. Parent, O. Mongin, K. Kamada, C. Katan and M. Blanchard-Desche, Chem. Commun., 2005, 2029–2031 RSC.
- (a) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC; (b) Q. Wang, H. K. Li, Q. Wei, J. Z. Sun, J. Wang, X. A. Zhang, A. J. Qin and B. Z. Tang, Polym. Chem., 2013, 4, 1396–1401 RSC; (c) Q. Wang, M. Chen, B. Yao, J. Wang, J. Mei, J. Z. Sun, A. J. Qin and B. Z. Tang, Macromol. Rapid Commun., 2013, 34, 796–802 CrossRef CAS PubMed; (d) A. Qin, J. W. Y. Lam and B. Z. Tang, Prog. Polym. Sci., 2012, 37, 182–209 CrossRef CAS; (e) J. Wang, J. Mei, E. Zhao, Z. Song, A. Qin, J. Z. Sun and B. Z. Tang, Macromolecules, 2012, 45, 7692–7703 CrossRef CAS.
- P. D. Jarowski, Y.-L. Wu, B. Schweizer and F. Diederich, Org. Lett., 2008, 10, 3347–3350 CrossRef CAS PubMed.
- (a) C.-H. Cheng, C.-A. Wu, F.-I. Y. Wu and C.-H. Shih, m-Terphenyl compound derivatives and application for organic light emitting diode, US Pat., 8475939B2, 2011; (b) Y. Tao, Q. Wang, L. Ao, C. Zhong, C. Yang, J. Qin and D. Ma, J. Phys. Chem. C, 2010, 114, 601–609 CrossRef CAS; (c) M.-K. Kim, J. Kwon, T.-H. Kwon and J.-I. Hon, New J. Chem., 2010, 34, 1317–1322 RSC.
- (a) C. A. Mirkin, X. Chen, A. B. Braunschweig, M. J. Wiester, X. Xu and W. L. Daniel, Click chemistry, molecular transport junction and colorimetric detection of copper, US Pat., 2011/0033940A1, 2011; (b) X. Chen, A. B. Braunschweig, M. J. Wiester, S. Yeganeh, M. A. Ratner and C. A. Mirkin, Angew. Chem., Int. Ed., 2009, 48, 5178–5181 CrossRef CAS PubMed.
- G. De Miguel, M. Wielopolski, D. I. Schuster, M. A. Fazio, O. P. Lee, C. K. Haley, A. L. Ortiz, L. Echegoyen, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2011, 133, 13036–13054 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- (a) K. Ladomenou, V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Coord. Chem. Rev., 2016, 306, 1–42 CrossRef CAS and references therein; (b) S. Ast, T. Fischer, H. Mueller, W. Mickler, M. Schwichtenberg, K. Rurack and H.-J. Holdt, Chem.–Eur. J., 2013, 19, 2990–3005 CrossRef CAS PubMed; (c) T. Duan, K. Fan, Y. Fu, C. Zhong, X. Chen, T. Peng and J. Qin, Dyes Pigm., 2012, 94, 28–33 CrossRef CAS; (d) M. A. Fazio, O. P. Lee and D. I. Schuster, Org. Lett., 2008, 10, 4979–4982 CrossRef CAS PubMed.
- For recent representative examples see: (a) P. Kumari, K. Khawas, S. Hazra and B. K. Kuila, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1785–1794 CrossRef CAS; (b) A.-R. Davies and K. R. Carter, Macromolecules, 2015, 48, 1711–1722 CrossRef; (c) A. R. Davis and K. R. Carter, Langmuir, 2014, 30, 4427–4433 CrossRef CAS PubMed; (d) K. Kim, T. K. An, J. Kim, J. Y. Jeong, J. Jang, H. Kim, Y. J. Baek, Y.-H. Kim, S. H. Kim and C. E. Park, Chem. Mater., 2014, 26, 6467–6476 CrossRef CAS; (e) A. S. Lang, M.-A. Muth, C. David Heinrich, M. Carassco-Orozco and M. Thelakkat, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1480–1486 CrossRef CAS.
- D. J. V. C. van Steenis, O. R. P. David, G. P. F. van Strikdonck, J. H. van Maarseveen and J. N. H. Reek, Chem. Commun., 2005, 4333–4335 RSC.
- S. Bakbak, P. J. Leech, B. E. Carson, S. Saxena, W. P. King and U. H. F. Bunz, Macromolecules, 2006, 39, 6793–6795 CrossRef CAS.
- (a) J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS; (b) S. Chen, A. Su, Y. Huang, C. Su, G. Peng and S. Chen, Macromolecules, 2002, 35, 4229–4232 CrossRef CAS; (c) S. Hecht and J. M. J. Frechet, Angew. Chem., Int. Ed., 2001, 40, 74–91 CrossRef CAS; (d) R. Deans, J. Kim, M. R. Machacek and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 8565–8566 CrossRef CAS.
- (a) M. A. Karim, Y.-R. Cho, J. S. Park, S. C. Kim, J. W. Lee, Y.-S. Gal and S.-H. Jin, Chem. Commun., 2008, 1929–1931 RSC; (b) M. A. Karim, Y.-R. Cho, J. S. Park, Y. I. Ryu, M. J. Lee, M. Song, S. H. Jin, J. W. Lee and Y. S. Gal, Macromol. Chem. Phys., 2008, 209, 1967–1975 CrossRef CAS.
- E. Hwang, K. L. Lusker, J. C. Garno, Y. Losovyj and E. E. Nesterov, Chem. Commun., 2011, 11990–11992 RSC.
- (a) M. Alonzi, D. Lanari, A. Marrocchi, C. Petrucci and L. Vaccaro, RSC Adv., 2013, 3, 23909–23923 RSC; (b) N. Marshall, S. K. Sontag and J. Locklin, Chem. Commun., 2011, 47, 5681–5689 RSC.
- L. Luo and C. D. Frisbie, J. Am. Chem. Soc., 2010, 132, 8854–8855 CrossRef CAS PubMed.
- (a) S. H. Choi, C. Risko, M. C. Ruiz Delgado, B. Kim, J.-L. Bredas and C. D. Frisbie, J. Am. Chem. Soc., 2010, 132, 4358–4368 CrossRef CAS PubMed; (b) S. H. Choi, B. Kim and C. D. Frisbie, Science, 2008, 320, 1482–1486 CrossRef CAS PubMed.
- S. Potratz, A. Mishra and P. Bäuerle, Beilstein J. Org. Chem., 2012, 8, 683–691 CrossRef CAS PubMed.
- S. Song, Y.-G. Ko, H. Lee, D. Wi, B. J. Ree, Y. Li, T. Michinobu and M. Ree, NPG Asia Mater., 2015, 7, e228, DOI:10.1038/am.2015.130.
- M. Jørgensen and F. C. Krebs, J. Org. Chem., 2004, 69, 6688–6696 CrossRef PubMed.
- (a) K. N. Shivananda, I. Cohen, E. Borzin, Y. Gerchikov, M. Firstenberg, O. Solomeshch, N. Tessler and Y. Eichen, Adv. Funct. Mater., 2012, 22, 1489–1501 CrossRef CAS; (b) M. Firstenberg, K. N. Shivananda, I. Cohen, O. Solomeshich, V. Medvedev, N. Tessler and Y. Eichen, Adv. Funct. Mater., 2011, 21, 634–643 CrossRef CAS.
- A. T. Demissie, G. Haugstad and C. D. Frisbie, J. Am. Chem. Soc., 2015, 137, 8819–8828 CrossRef CAS PubMed.
- C. E. Smith, S. O. Odoh, S. Ghosh, L. Gagliardi, C. J. Cramer and C. D. Frisbie, J. Am. Chem. Soc., 2015, 137, 15732–15741 CrossRef CAS PubMed.
- For representative reviews on this topic see: (a) A. B. Lowe, Polymer, 2014, 55, 5517–5549 CrossRef CAS; (b) R. Hogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CrossRef PubMed; (c) B. Yao, J. Sun, A. Qin and B. Z. Tang, Chin. Sci. Bull., 2013, 58, 2711–2718 CrossRef CAS; (d) C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- J. Liu, J. W. Y. Lam, C. K. W. Jim, J. C. Y. Ng, J. Shi, H. Su, K. F. Yeung, Y. Hong, M. Faisal, Y. Yu, K. S. Wong and B. Z. Tang, Macromolecules, 2011, 44, 68–79 CrossRef CAS.
- C. K. W. Jim, A. Qin, J. W. Y. Lam, F. Mahtab, Y. Yu and B. Z. Tang, Adv. Funct. Mater., 2010, 20, 1319–1328 CrossRef CAS.
- B. Yao, J. Mei, J. Li, J. Wang, H. Wu, J. Z. Sun, A. Qin and B. Z. Tang, Macromolecules, 2014, 47, 1325–1333 CrossRef CAS.
- B. Yao, T. Hu, H. Zhang, J. Li, J. Z. Sun, A. Qin and B. Z. Tang, Macromolecules, 2015, 48, 7782–7791 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |