
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A polymer acceptor with an optimal LUMO energy level for all-polymer solar cells†
Zicheng
Ding‡
a,
Xiaojing
Long‡
ab,
Chuandong
Dou
*a,
Jun
Liu
*a and
Lixiang
Wang
a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, People's Republic of China. E-mail: liujun@ciac.ac.cn; chuandong.dou@ciac.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100864, People's Republic of China
First published on 14th June 2016
Abstract
A key parameter for polymer electron acceptors is the lowest unoccupied molecular orbital (LUMO) energy level (ELUMO). For state-of-the-art polymer electron acceptors based on the naphthalene diimide (NDI) unit, their ELUMO are low-lying and cannot be tuned, leading to a low open-circuit voltage (Voc) of the resulting all-polymer solar cells (all-PSCs). We report that polymer electron acceptors based on the double B←N bridged bipyridine (BNBP) unit exhibit tunable ELUMO because of their delocalized LUMOs over polymer backbones. The ELUMO of the copolymer of the BNBP unit and selenophene unit (P-BNBP-Se) is lower by 0.16 eV than that of the copolymer of the BNBP unit and thiophene unit (P-BNBP-T). As a result, the energy levels of P-BNBP-Se match well with the widely-used polymer donor, poly[(ethylhexyl-thiophenyl)-benzodithiophene-(ethylhexyl)-thienothiophene] (PTB7-Th). The electron mobility of P-BNBP-Se (μe = 2.07 × 10−4 cm2 V−1 s−1) is also higher than that of P-BNBP-T (μe = 7.16 × 10−5 cm2 V−1 s−1). While the all-PSC device based on the PTB7-Th:P-BNBP-T blend shows a moderate power conversion efficiency (PCE) of 2.27%, the corresponding device with P-BNBP-Se as the acceptor exhibits a PCE as high as 4.26%. Moreover, owing to the suitable ELUMO of P-BNBP-Se, the all-PSC device of P-BNBP-Se shows a Voc up to 1.03 V, which is higher by 0.22 V than that with the conventional NDI-based polymer acceptor. These results indicate that BNBP-based polymers can give all-PSCs with high PCEs, remarkably high Voc values and small photon energy losses.
Introduction
All-polymer solar cells (all-PSCs), which utilize polymers as both the electron donor and electron acceptor, have attracted much attention recently because of their great advantages over conventional polymer/fullerene PSCs.1 These advantages include enhanced light absorption of polymer acceptors, low cost, and improved mechanical/thermal stability. Great progress in all-PSCs has been made by using absorption-complementary polymer donor/acceptors, optimizing the blend morphologies, or developing new polymer acceptors.2 However, the further development of all-PSCs is severely limited by the lack of excellent polymer acceptors.3 To date, only several specific polymer acceptors based on the naphthalene diimide (NDI) unit, perylenediimide (PDI) unit and B←N bridged thienylthiazole (BNTT) unit can work as polymer acceptors for efficient all-PSCs with power conversion efficiencies (PCEs) exceeding 4%.4,5A key parameter for polymer acceptors is the lowest unoccupied molecular orbital (LUMO) energy level (ELUMO). In all-PSCs, the ELUMO difference between the acceptor and donor (ΔELUMO) is regarded as the driving force for the charge separation.6 The difference between the ELUMO of the acceptor and the highest occupied molecular orbital (HOMO) energy level (EHOMO) of the donor is related to the open-circuit voltage (Voc) of all-PSCs.6 Therefore, to get a large ΔELUMO for effective charge separation and to maximize Voc, the ELUMO of the polymer acceptor must be carefully optimized. The state-of-the-art polymer acceptors are the NDI-based conjugated polymers.4 Unfortunately, the ELUMO of these polymers are fixed at ca. −3.85 eV and cannot be effectively tuned, leading to a low Voc of the resulting all-PSCs. According to a study by Takimiya et al.,7 the fixed ELUMO of NDI-based polymers are due to the localized LUMOs on the NDI units. The ELUMO of the NDI-based conjugated polymers are determined by the NDI unit and are not affected by the copolymerization units. Thus, it is important but challenging to develop polymer acceptors with tunable ELUMO.
Following our strategy to develop polymer acceptors using the B←N unit,5 we have reported a new electron-deficient building block based on the B←N unit, double B←N bridged bipyridine (BNBP), to develop a polymer acceptor.8 In this manuscript, we report that BNBP-based polymer acceptors show tunable ELUMO because of their delocalized LUMOs over the polymer backbones. The ELUMO of the copolymer of the BNBP unit and selenophene unit (P-BNBP-Se) is lower by 0.16 eV than that of the copolymer of the BNBP unit and thiophene unit (P-BNBP-T) (Fig. 1). As a result, the energy levels of P-BNBP-Se match well with the widely-used polymer donor, poly[(ethylhexyl-thiophenyl)-benzodithiophene-(ethylhexyl)-thienothiophene] (PTB7-Th).9 While the all-PSC device based on the PTB7-Th:P-BNBP-T blend shows a moderate PCE of 2.27%, the corresponding device with P-BNBP-Se as the acceptor exhibits a PCE as high as 4.26% with a remarkably high Voc of 1.03 V. These results indicate that BNBP-based polymer acceptors have different electronic structures from those of the classical NDI-based polymer acceptors and that they can give all-PSCs with remarkably high Voc values and high PCEs.
Results and discussion
Scheme 1 shows the synthetic route of P-BNBP-Se and P-BNBP-T. The three monomers were prepared following literature methods and the two polymers were synthesized in Stille-polymerization conditions.8 Their chemical structures are confirmed by 1H NMR and elemental analysis. According to gel permeation chromatography (GPC), with 1,2,4-trichlorobenzene as the eluent at 150 °C, the number-average molecular weight (Mn) and polydispersity (PDI) are 26.3 kDa and 1.93 for P-BNBP-Se and 46.2 kDa and 1.81 for P-BNBP-T, respectively. According to the thermogravimetric analysis (TGA), P-BNBP-T and P-BNBP-Se show a good thermal stability with thermal decomposition temperatures (Td) of over 350 °C (ESI†). In addition, the two polymers show a good solubility in common organic solvents, including chlorobenzene (CB), chloroform (CHCl3) and o-dichlorobenzene (o-DCB).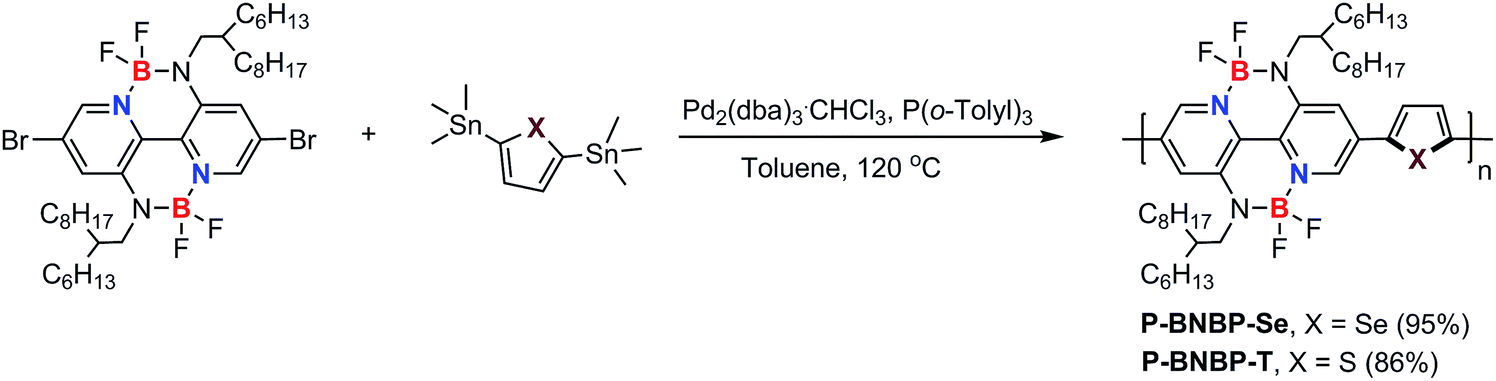 | ||
| Scheme 1 Synthetic route of P-BNBP-Se and P-BNBP-T. | ||
To elucidate the molecular orbitals of the two polymers, density functional theory (DFT) calculations at the B3LYP/6-31G* level of theory were performed with the model compounds containing six repeating units with the long alkyl chains replaced by methyl groups.10 For comparison, we also show the DFT calculation result of the state-of-the-art polymer acceptor, (poly((N,N′-bis(2-octyldodecyl)-1,4,5,8-naphthalenedicarboximide-2,6-diyl)-alt-5,5′-(2,2-bithiophene))) (N2200 or P(NDI2ODT2)) (Fig. 1a).11 As shown in Fig. 2, the calculated LUMO of the model compound of N2200 is localized on the NDI units, indicating that its ELUMO is determined by the NDI unit and cannot be effectively tuned by changing the co-monomer units. This is consistent with the DFT calculation and experimental results of NDI-based conjugated polymers in the literature.4,11 In contrast, the calculated LUMOs of the model compounds of P-BNBP-Se/P-BNBP-T are delocalized over the BNBP units and the selenophene/thiophene units. Therefore, the LUMO levels of BNBP-based polymers are determined by both the BNBP unit and the co-monomer unit. The LUMO levels of BNBP-based polymers should be effectively tuned by changing the co-monomer units.
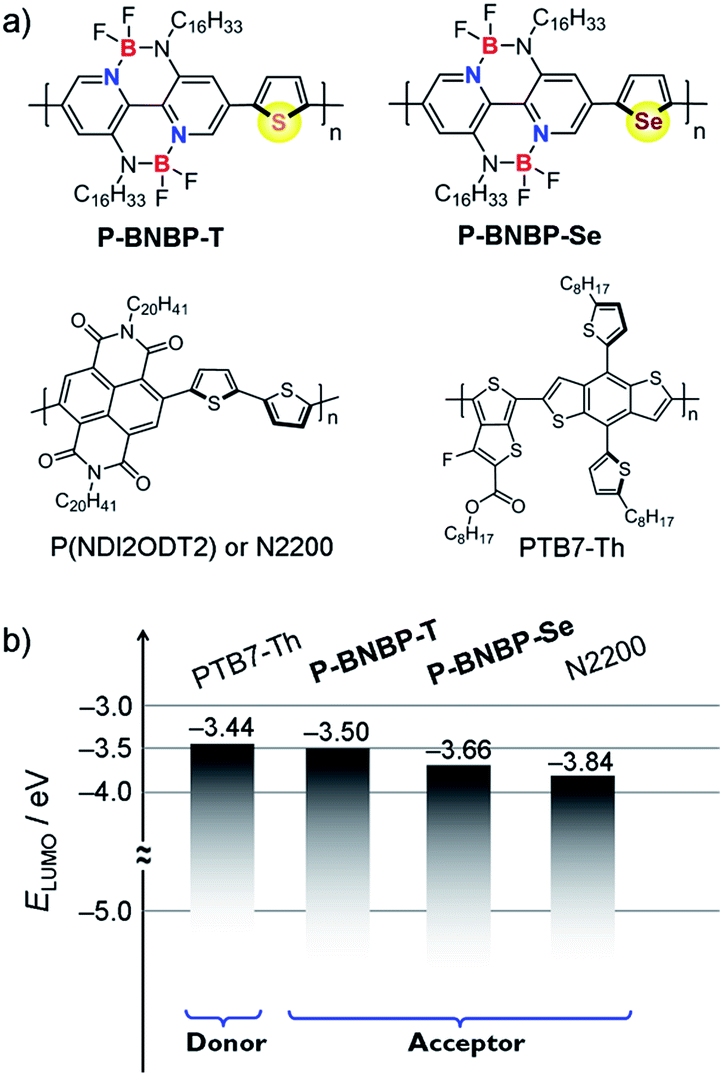 | ||
| Fig. 1 (a) Chemical structures of P-BNBP-Se, P-BNBP-T, N2200 and PTB7-Th and (b) their LUMO energy level alignments. | ||
 | ||
| Fig. 2 Kohn–Sham LUMOs of model compounds of P-BNBP-Se, P-BNBP-T and N2200, based on calculations at the B3LYP/6-31G* level. | ||
Cyclic voltammetry was employed to estimate the LUMO/HOMO energy levels of the two polymers (ESI†).12 As shown in Fig. 3a, P-BNBP-Se exhibits irreversible reduction and oxidation waves with onset potentials of Eredonset = −1.14 V and Eoxonset = +1.04 V, respectively. Accordingly, the ELUMO/HOMO of P-BNBP-Se are estimated to be −3.66 eV/−5.84 eV (Table 1). Similarly, the ELUMO/HOMO of P-BNBP-T are estimated to be −3.50 eV/−5.77 eV (Table 1). As reported previously, the model compound of the BNBP unit itself has an ELUMO of −3.19 eV. The ELUMO of the two BNBP-based polymers are much lower than that of the BNBP unit. Moreover, the ELUMO of P-BNBP-Se is lower than that of P-BNBP-T by 0.16 eV. These results confirm that the LUMO levels of BNBP-based polymers can be effectively tuned by changing the co-monomer units. This is consistent with the delocalized LUMOs in the DFT calculation results. The lower-lying ELUMO of P-BNBP-Se is attributed to the lower electronegativity of the Se atom (2.4) than the S atom (2.5) and the empty orbital of the Se atom.13
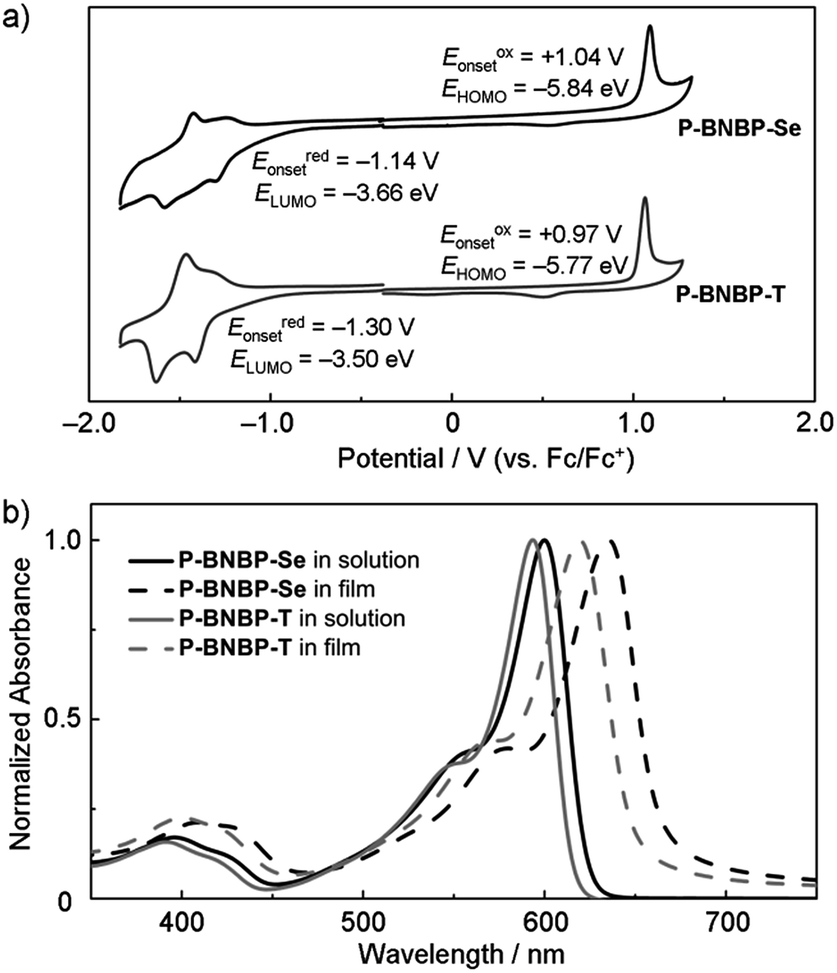 | ||
| Fig. 3 (a) Cyclic voltammogram of P-BNBP-Se and P-BNBP-T in thin films using a Ag/AgCl reference electrode, Fc = ferrocene; (b) UV/Vis absorption spectra of P-BNBP-Se and P-BNBP-T in o-DCB solutions and in thin films. | ||
| Polymer | M n (kDa) | PDI | λ abs (nm) | λ abs (nm) | ε (cm−1) | E optg (eV) | E oxonset (V) | E redonset (V) | E HOMO (eV) | E LUMO (eV) | μ e (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Measured in o-DCB solution. b Measured in thin film. c Onset potential vs. Fc/Fc+. d E HOMO/LUMO = −(4.80 + Eoxonset/Eredonset) eV. | |||||||||||
| P-BNBP-Se | 26.3 | 1.93 | 600 | 635 | 1.49 × 105 | 1.87 | +1.04 | −1.14 | −5.84 | −3.66 | 2.07 × 10−4 |
| P-BNBP-T | 46.0 | 2.01 | 593 | 622 | 1.45 × 105 | 1.92 | +0.97 | −1.30 | −5.77 | −3.50 | 7.16 × 10−5 |
Fig. 3b shows the absorption spectra of P-BNBP-Se and P-BNBP-T in dilute o-DCB solutions and in thin films. Both of the two polymers in solutions show broad absorption bands around λ = 580 nm. The absorption spectrum is slightly redshifted for P-BNBP-Se compared to P-BNBP-T. In thin film, P-BNBP-Se exhibits a maximum absorption at 635 nm, while P-BNBP-T shows the absorption peak at 622 nm. Both of the two films show high absorption coefficients (ε), suggesting their intense light absorption. According to the onset absorption wavelength in thin films, the optical band gaps (Eg) of P-BNBP-Se and P-BNBP-T are estimated to be 1.87 eV and 1.92 eV, respectively. The electron mobilities (μe) of P-BNBP-Se and P-BNBP-T were estimated using the space-charge-limited current (SCLC) method with the current density-voltage curves of the electron-only devices (device structure: ITO/PEIE/polymer/Ca/Al).14 The electron mobility of P-BNBP-Se (μe = 2.07 × 10−4 cm2 V−1 s−1) is higher than that of P-BNBP-T (μe = 7.16 × 10−5 cm2 V−1 s−1) (ESI†). The higher electron mobility of P-BNBP-Se is due to the stronger intermolecular interactions in Se-containing polymers because of the larger and more polarizable radii of the selenium atom than the sulfur atom. This is confirmed by the smaller π–π stacking distance of P-BNBP-Se (dπ–π = 3.77 Å) than that of P-BNBP-T (dπ–π = 3.81 Å) (ESI†). The electron mobility of P-BNBP-Se is comparable to the hole mobilities of typical polymer electron donors, which is very favourable for its application as a polymer electron acceptor in all-PSCs.
To investigate the application of P-BNBP-Se and P-BNBP-T as electron acceptors in all-PSCs, we select a widely-used polymer donor, PTB7-Th. All-PSC devices were fabricated with a configuration of ITO/PEDOT:PSS/PTB7-Th:P-BNBP-Se or P-BNBP-T/Ca/Al (ESI†). The active layer was spin-coated from the blend in o-DCB solution without any additives. Fig. 4 shows the current density–voltage (J–V) curves under AM 1.5G illumination (100 mW cm−2) and the external quantum efficiency (EQE) spectra of the optimal devices. The photovoltaic parameters are summarized in Table 2. The PTB7-Th![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P-BNBP-T (3:1, w:w) device shows a PCE of 2.27% with a Voc of 1.12 V, a short-circuit current density (Jsc) of 5.24 mA cm−2 and a fill factor (FF) of 0.39. The device based on the PTB7-Th:P-BNBP-Se (2:1, w:w) blend exhibits a PCE of 4.26% with a Voc of 1.03 V, a Jsc of 10.02 mA cm−2 and a FF of 0.42. This PCE value is comparable to that of the reference all-PSC device based on the PTB7-Th:N2200 (1:1, w:w) blend from the chloroform solution (PCE = 4.57%), indicating that P-BNBP-Se is an excellent polymer acceptor. Compared with the device of P-BNBP-T, the device of P-BNBP-Se shows a slightly decreased Voc and much increased Jsc. The slightly decreased Voc is attributed to the lower ELUMO of P-BNBP-Se than that of P-BNBP-T. On the other hand, the Voc of the P-BNBP-Se device is higher than that of the N2200 device by 0.22 V (Table 2) because the ELUMO of P-BNBP-Se is higher than that of N2200. The much increased Jsc of the P-BNBP-Se device than that of the P-BNBP-T device is in accordance with their EQE values (EQEmax = 0.47 for P-BNBP-Se and EQEmax = 0.25 for P-BNBP-T) (Fig. 4b). The Jsc calculated from the integration of the EQE spectra agrees well with the Jsc values obtained from the J–V scans within an error of 5%.
:P-BNBP-T (3:1, w:w) device shows a PCE of 2.27% with a Voc of 1.12 V, a short-circuit current density (Jsc) of 5.24 mA cm−2 and a fill factor (FF) of 0.39. The device based on the PTB7-Th:P-BNBP-Se (2:1, w:w) blend exhibits a PCE of 4.26% with a Voc of 1.03 V, a Jsc of 10.02 mA cm−2 and a FF of 0.42. This PCE value is comparable to that of the reference all-PSC device based on the PTB7-Th:N2200 (1:1, w:w) blend from the chloroform solution (PCE = 4.57%), indicating that P-BNBP-Se is an excellent polymer acceptor. Compared with the device of P-BNBP-T, the device of P-BNBP-Se shows a slightly decreased Voc and much increased Jsc. The slightly decreased Voc is attributed to the lower ELUMO of P-BNBP-Se than that of P-BNBP-T. On the other hand, the Voc of the P-BNBP-Se device is higher than that of the N2200 device by 0.22 V (Table 2) because the ELUMO of P-BNBP-Se is higher than that of N2200. The much increased Jsc of the P-BNBP-Se device than that of the P-BNBP-T device is in accordance with their EQE values (EQEmax = 0.47 for P-BNBP-Se and EQEmax = 0.25 for P-BNBP-T) (Fig. 4b). The Jsc calculated from the integration of the EQE spectra agrees well with the Jsc values obtained from the J–V scans within an error of 5%.
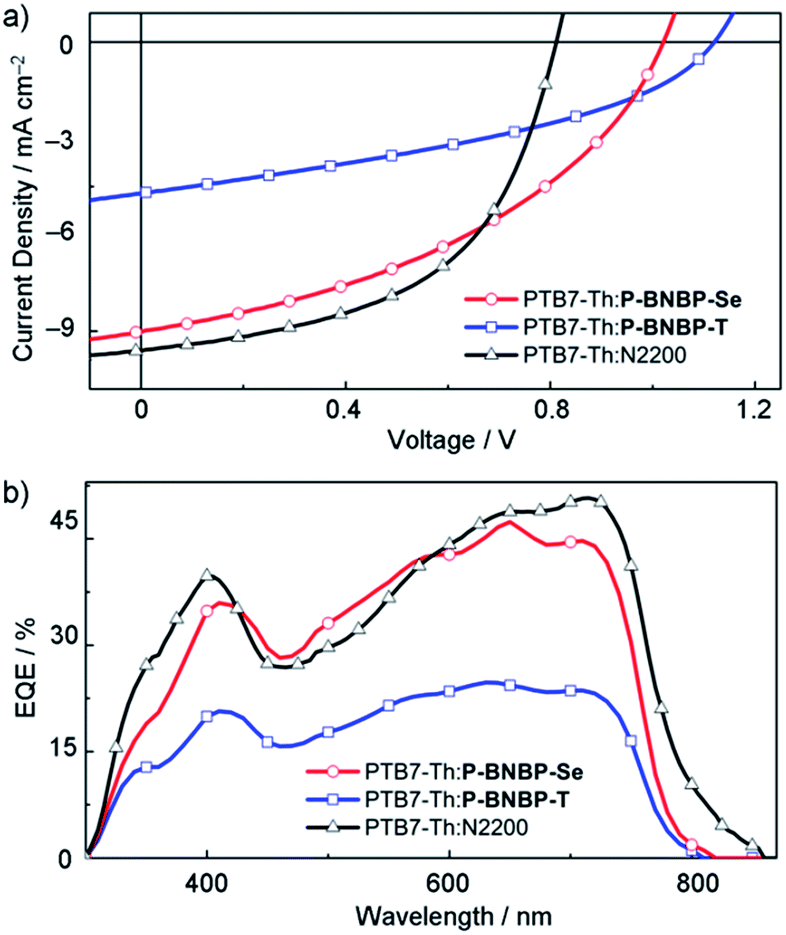 | ||
| Fig. 4 (a) J–V curves and (b) EQE spectra of the all-PSC devices based on the PTB7-Th:P-BNBP-Se, PTB7-Th:P-BNBP-T and PTB7-Th:N2200 blends, respectively. | ||
The charge carrier mobilities of the two blends were investigated using the SCLC method with the electron-only and hole-only devices (ESI†).14 The electron mobility and hole mobility (μh) of the PTB7-Th:P-BNBP-Se blend are estimated to be 3.34 × 10−5 cm2 V−1 s−1 and 2.38 × 10−4 cm2 V−1 s−1, respectively. In comparison, the PTB7-Th:P-BNBP-T blend exhibits a μe = 5.96 × 10−6 cm2 V−1 s−1 and μh = 7.28 × 10−4 cm2 V−1 s−1, respectively. The higher electron mobility and the balanced electron/hole mobilities of the PTB7-Th:P-BNBP-Se blend are due to the enhanced electron mobility of P-BNBP-Se. We also investigated the bimolecular charge recombination in the all-PSC devices using the light-intensity dependence of the J–V curves (Fig. 5). The Jsc follows a power-law dependence on the illumination intensity (Jsc ∝ Plightα), where Plight is light intensity and α is the calculated power-law exponent. The α values are 0.93 for the PTB7-Th:P-BNBP-Se device and 0.94 for the PTB7-Th:P-BNBP-T device, which are close to unity, suggesting that the bimolecular charge recombination is weak in the two devices at a short circuit condition.15 Both the weak bimolecular recombination and the high and balanced electron/hole mobilities of PTB7-Th:P-BNBP-Se can explain its excellent device performance.
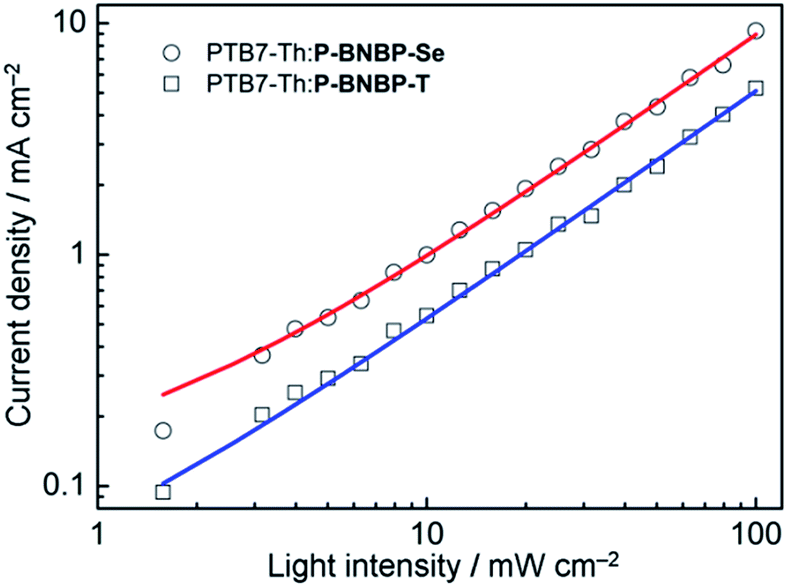 | ||
| Fig. 5 Short-circuit current density (Jsc) versus light intensity (Plight) data and power-law (Jsc ∝ Plightα) fittings for the all-PSC devices. | ||
The morphologies of the PTB7-Th:P-BNBP-Se and PTB7-Th:P-BNBP-T blends were characterized by transmission electron microscopy (TEM) and atomic force microscopy (AFM). As shown in Fig. 6, the TEM images exhibit similar nano/microstructures without large-size aggregation. The AFM images of the two blends similarly reveal smooth surface morphologies with the same root-mean-square (RMS) roughness of 1.47 nm and domain sizes of around 20–40 nm. The phase separation morphologies of the two blends are beneficial for good all-PSC devices.
 | ||
| Fig. 6 The TEM images and the AFM height images of ((a) and (b)) the PTB7-Th:P-BNBP-Se blend and ((c) and (d)) the PTB7-Th:P-BNBP-T blend, respectively. | ||
In organic photovoltaics (OPVs), the ΔELUMO of the donor and acceptor is regarded as the driving force for charge separation. The ΔELUMO should be larger than a specific value for efficient charge separation. If ΔELUMO is too large, there is a lot of energy loss in the charge separation process, leading to a low Voc because the Voc of OPVs is related to the difference between the EHOMO of the donor and ELUMO of the acceptor.6 In our previous report, an all-PSC device based on the PTB7:P-BNBP-T blend (ΔELUMO = 0.19 eV) showed a good PCE of 3.38%.8 As shown in Fig. 1b, the ΔELUMO is only 0.06 eV for PTB7-Th:P-BNBP-T, and thus the all-PSC device shows a high Voc but produces a low PCE due to the insufficient charge separation.4g As the ELUMO of P-BNBP-Se is lower than that of P-BNBP-T, the ΔELUMO for PTB7-Th:P-BNBP-Se is increased to 0.22 eV and ensures an efficient charge separation, resulting in higher Jsc and PCE values. Moreover, due to the suitable ELUMO of P-BNBP-Se, the PTB7-Th:P-BNBP-Se device produces a high Voc of 1.03 V, which is higher than that of the PTB7-Th:N2200 device by 0.22 V. These results indicate that the suitable ELUMO of P-BNBP-Se plays an important role in enhancing the all-PSCs device performance.
It is worthy to note the remarkably low photon energy losses (Eloss) of the all-PSCs based on P-BNBP-Se and P-BNBP-T. Eloss is defined as the difference between the lowest optical bandgap of the donor/acceptor and the eVoc of the organic photovoltaic (OPV) device (Eloss = Eg − eVoc).16 Typically, OPVs have large Eloss values of 0.7–1.0 eV. It has been proposed that the lowest attainable Eloss of OPVs is 0.6 eV, despite several exceptional examples.17 As listed in Table 2, the Eloss for the device of PTB7-Th:P-BNBP-Se and PTB7-Th:P-BNBP-T is 0.56 eV and 0.47 eV, respectively. To our best knowledge, the Eloss of 0.47 eV is the lowest one for OPVs reported so far. A small Eloss is always observed for all-PSCs with BNBP-based polymers as electron acceptors and the exact reason is as yet unknown. We speculate that the small Eloss is related to the high-lying LUMO levels of the BNBP-based polymers.
Conclusions
In summary, we have developed a polymer acceptor based on the BNBP unit and selenophene unit with an optimal ELUMO to simultaneously enable charge separation and maximize Voc. BNBP-based polymers have delocalized LUMOs over the polymer backbones, so their ELUMO can be tuned by changing the comonomer unit. The ELUMO of P-BNBP-Se is lower by 0.16 eV than that of P-BNBP-T and consequently matches well with that of the polymer donor, PTB7-Th. While the all-PSC device based on PTB7-Th:P-BNBP-T shows a moderate PCE of 2.27%, the corresponding device with P-BNBP-Se as the acceptor exhibits a PCE as high as 4.26%. Moreover, the device of P-BNBP-Se shows a Voc of up to 1.03 V and Eloss as small as 0.56 eV. These results indicate that BNBP-based polymer acceptors have different electronic structures from those of classical NDI-based polymer acceptors and that they can give all-PSCs with remarkably high Voc values and high PCEs.Experimental section
Synthesis of P-BNBP-Se
The dibromo-substituted BNBP monomer was synthesized according to the previous report.8 The starting materials of the dibromo-substituted BNBP monomer (220 mg, 0.248 mmol), 2,5-bis(trimethylstannyl)selenophene (114.1 mg, 0.248 mmol), Pd2(dba)3·CHCl3 (4.5 mg, 0.0050 mmol) and P(o-tolyl) (12.1 mg, 0.04 mmol) were placed in a two-necked flask under argon, and then dried toluene (11 mL) was added. After the mixture was stirred at 115 °C for 48 h, an end-capping reaction was carried out by adding 2,5-bis(trimethylstannyl)selenophene (3 mg) and then bromobenzene (200 mg). After cooling, the resulting organic phase was extracted with CHCl3 (150 mL) and washed with water. After the solvents were removed, the residue was dispersed in methanol and the precipitate was collected. The obtained dark solid was dispersed in acetonitrile, and was collected and dried in a vacuum overnight. Yield: 213 mg (95%). 1H NMR (400 MHz, CDCl3, 25 °C): δ 8.41 (s, 1H), 7.76 (s, 1H), 7.66 (s, 1H), 3.63 (br, 2H), 1.85 (br, 1H), 1.42–1.27 (br, 24H), 0.85 (br, 6H). GPC (TCB, polystyrene standard, 150 °C): Mn = 26300, PDI = 1.93. Anal. calcd for C46H72B2F4N4Se: C, 64.42; H, 8.46; B, 2.52; F, 8.86; N, 6.53; Se, 9.21. Found: C, 64.25; H, 8.58; N, 6.65; Se, 9.05.
Synthesis of P-BNBP-T
The starting materials of the dibromo-substituted BNBP monomer (150 mg, 0.166 mmol), 2,5-bis(trimethylstannyl)thiophene (68.5 mg, 0.166 mmol), Pd2(dba)3·CHCl3 (3.5 mg, 0.0033 mmol) and P(o-tolyl) (8.1 mg, 0.027 mmol) were placed in a two-necked flask under argon, and then dried toluene (4 mL) was added. After the mixture was stirred at 115 °C for 50 h, an end-capping reaction was carried out by adding 2,5-bis(trimethylstannyl)thiophene (3 mg) and then bromobenzene (200 mg). After cooling, the resulting organic phase was extracted with CHCl3 (150 mL) and washed with water. After the solvents were removed, the residue was dispersed in methanol and the precipitate was collected. The obtained dark solid was dispersed in acetonitrile, and was collected and dried in a vacuum overnight. Yield: 116.5 mg (86%). 1H NMR (400 MHz, CDCl3, 25 °C): δ 8.47 (s, 1H), 7.74 (s, 1H), 7.60 (s, 1H), 3.64 (br, 2H), 1.87 (br, 1H), 1.43–1.25 (br, 24H), 0.83 (br, 6H). GPC (TCB, polystyrene standard, 150 °C): Mn = 46200, PDI = 1.81. Anal. calcd for C46H72B2F4N4S: C, 68.14; H, 8.95; B, 2.67; F, 7.37; N, 6.91; S, 3.95. Found: C, 67.83; H, 8.82; N, 6.75; S, 4.05.
Acknowledgements
The authors are grateful for the financial support by the 973 Project (No. 2014CB643504), the Nature Science Foundation of China (No. 51373165, No. 21404099, No. 21574129), the “Thousand Talents Program” of China, the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB12010200), and the State Key Laboratory of Supramolecular Structure and Materials in Jilin University (No. sklssm201608).Notes and references
- (a) T. Kim, J.-H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T.-S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS PubMed; (b) A. Facchetti, Mater. Today, 2013, 16, 123 CrossRef CAS; (c) G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CrossRef CAS.
- (a) L. Gao, Z.-G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884 CrossRef CAS PubMed; (b) Y.-J. Hwang, B. A. E. Courtright, A. S. Ferreira, S. H. Tolbert and S. A. Jenekhe, Adv. Mater., 2015, 27, 4578 CrossRef CAS PubMed; (c) Y.-J. Hwang, T. Earmme, B. A. E. Courtright, F. N. Eberle and S. A. Jenekhe, J. Am. Chem. Soc., 2015, 137, 4424 CrossRef CAS PubMed; (d) T. Earmme, Y.-J. Hwang, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2014, 26, 6080 CrossRef CAS PubMed; (e) T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960 CrossRef CAS PubMed.
- (a) H. Benten, D. Mori, H. Ohkita and S. Ito, J. Mater. Chem. A, 2016, 4, 5340 RSC; (b) L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666 CrossRef CAS PubMed.
- (a) N. Zhou, A. S. Dudnik, T. I. N. G. Li, E. F. Manley, T. J. Aldrich, P. Guo, H.-C. Liao, Z. Chen, L. X. Chen, R. P. H. Chang, A. Facchetti, M. O. de la Cruz and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 1240 CrossRef CAS PubMed; (b) C. Lee, H. Kang, W. Lee, T. Kim, K.-H. Kim, H. Y. Woo, C. Wang and B. J. Kim, Adv. Mater., 2015, 27, 2466 CrossRef CAS PubMed; (c) H. Kang, M. A. Uddin, C. Lee, K.-H. Kim, T. L. Nguyen, W. Lee, Y. X. Li, C. Wang, H. Y. Woo and B. J. Kim, J. Am. Chem. Soc., 2015, 137, 2359 CrossRef CAS PubMed; (d) J. W. Jung, J. W. Jo, C.-C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K.-Y. Jen, Adv. Mater., 2015, 27, 3310 CrossRef CAS PubMed; (e) L. Ye, X. Jiao, M. Zhou, S. Zhang, H. Yao, W. Zhao, A. Xia, H. Ade and J. Hou, Adv. Mater., 2015, 27, 6046 CrossRef CAS PubMed; (f) P. Cheng, L. Ye, X. Zhao, J. Hou, Y. Li and X. Zhan, Energy Environ. Sci., 2014, 7, 1351 RSC; (g) Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan, J. Reinspach, J. Mei, A. L. Appleton, G. I. Koleilat, Y. Gao, S. C. B. Mannsfeld, A. Salleo, H. Ade, D. Zhao and Z. Bao, Adv. Mater., 2014, 26, 3767 CrossRef CAS PubMed; (h) I. H. Jung, W.-Y. Lo, J. Jang, W. Chen, D. L. Zhao, E. S. Landry, L. Y. Lu, D. V. Talapin and L. Yu, Chem. Mater., 2014, 26, 3450 CrossRef CAS; (i) C. Mu, P. Liu, W. Ma, K. Jiang, J. B. Zhao, K. Zhang, Z. H. Chen, Z. H. Wei, Y. Yi, J. N. Wang, S. H. Yang, F. Huang, A. Faccheti, H. Ade and H. Yan, Adv. Mater., 2014, 26, 7224 CrossRef CAS PubMed; (j) D. Mori, H. Benten, I. Okada, H. Ohkita and S. Ito, Energy Environ. Sci., 2014, 7, 2939 RSC; (k) M. Schubert, B. A. Collins, H. Mangold, I. A. Howard, W. Schindler, K. Vandewal, S. Roland, J. Behrends, F. Kraffert, R. Steyrleuthner, Z. Chen, K. Fostiropoulos, R. Bittl, A. Salleo, A. Facchetti, F. Laquai, H. W. Ade and D. Neher, Adv. Funct. Mater., 2014, 24, 4068 CrossRef CAS; (l) E. J. Zhou, J. Z. Cong, K. Hashimoto and K. Tajima, Adv. Mater., 2013, 25, 6991 CrossRef CAS PubMed; (m) L. Xue, Y. Yang, Z.-G. Zhang, X. Dong, L. Gao, H. Bin, J. Zhang, Y. Yang and Y. Li, J. Mater. Chem. A, 2016, 4, 5810 RSC.
- (a) R. Zhao, C. Dou, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 5313 CrossRef CAS PubMed; (b) C. Dou, Z. Ding, Z. Zhang, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 3648 CrossRef CAS PubMed.
- R. A. Street, Adv. Mater., 2016, 28, 3814 CrossRef CAS PubMed.
- K. Takimiya, I. Osaka and M. Nakano, Chem. Mater., 2014, 26, 587 CrossRef CAS.
- (a) C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436 CrossRef CAS PubMed; (b) X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016 DOI:10.1002/adam.201601205.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591 CrossRef.
- DFT calculations were performed using Gaussian 09 program: M. J. Frisch, et al., Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009. For details, see ESI.†.
- R. Steyrleuthner, M. Schubert, I. Howard, B. Klaumünzer, K. Schilling, Z. Chen, P. Saalfrank, F. Laquai, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2012, 134, 18303 CrossRef CAS PubMed.
- Z. Ding, Z. Miao, Z. Xie and J. Liu, J. Mater. Chem. A, 2016, 4, 2413 RSC.
- (a) D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375 CrossRef CAS PubMed; (b) R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314 CrossRef CAS PubMed.
- P. W. M. Blom, M. J. M. de Jong and M. G. van Munster, Phys. Rev. B: Solid State, 1997, 55, R656 CrossRef CAS.
- (a) A. K. K. Kywa, D. H. Wang, V. Gupta, W. L. Leong, L. Ke, G. C. Bazan and A. J. Heeger, ACS Nano, 2013, 7, 4569 CrossRef PubMed; (b) S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
- (a) R. A. J. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847 CrossRef CAS PubMed; (b) D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939 CrossRef CAS.
- (a) K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085 CrossRef CAS PubMed; (b) W. Li, K. H. Hendriks, A. Furlan, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 2231 CrossRef CAS PubMed; (c) M. Wang, H. Wang, T. Yokoyama, X. Liu, Y. Huang, Y. Zhang, T.-Q. Nguyen, S. Aramaki and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 12576 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, thermal property, theoretical calculations, as well as all-PSC device fabrications and characterizations. See DOI: 10.1039/c6sc01756h |
| ‡ Z. Ding and X. Long contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |