
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metallacarboranes as tunable redox potential electrochemical indicators for screening of gene mutation†
Tania
García-Mendiola
ab,
Victoria
Bayon-Pizarro
a,
Adnana
Zaulet
c,
Isabel
Fuentes
c,
Félix
Pariente
ab,
Francesc
Teixidor
c,
Clara
Viñas
*c and
Encarnación
Lorenzo
*ab
aDepartamento Química Analítica y Análisis Instrumental, Universidad Autónoma de Madrid, Spain. E-mail: encarnacion.lorenzo@uam.es
bInstituto Madrileño de Estudios Avanzados (IMDEA) Nanociencia, Spain
cInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Barcelona, Spain. E-mail: clara@icmab.es
First published on 8th June 2016
Abstract
The substitution of hydrogen with chlorine in the metallacarborane [3,3-Fe(1,2-closo-C2B9H11)2]− cluster modulates the formal potential of the Fe3+/Fe2+ redox couple, shifting it to a more positive value. Hence, very similar redox probes with a wide range of formal potentials, ranging from negative to positive values, are available. Thus, we have achieved the synthesis and studied the electrochemical behaviour of the sodium salt of [3,3-Fe(8,9,12-Cl3-1,2-closo-C2B9H8)2]− in aqueous media. This strategy allows tuning of the redox potential of the [3,3-Fe(1,2-closo-C2B9H11)2]− framework with a minor change in its shape and dimensions. We also describe the interaction of the prepared [3,3-Fe(8,9,12-Cl3-1,2-closo-C2B9H8)2]− and the pristine [3,3-Fe(1,2-closo-C2B9H11)2]− with DNA. These studies have been carried out not only with DNA in solution but also with DNA immobilized on screen-printed gold electrodes. The results obtained point to a strong interaction between the metallacarboranes and DNA, to a different extent with single stranded DNA (ssDNA) compared to double stranded DNA (dsDNA). This property makes them selective and wide-ranging potential electrochemical indicators of hybridization. The suitability of these new redox indicators for selective DNA biosensor development has been probed by the direct detection of two different mutations associated with cystic fibrosis in PCR amplicons extracted from blood cells.
Introduction
The detection of specific DNA sequences provides the fundamental basis for monitoring a wide variety of viral infections, as well as genetic and infectious diseases. DNA biosensors based on nucleic acid recognition processes have received considerable attention in rapid and inexpensive DNA assays.1,2 Different techniques including fluorescence, surface plasmon resonance, quartz crystal microbalance measurements and electrochemistry have been employed to detect DNA hybridization. Among them, electrochemical transducers are powerful tools for interfacing DNA recognition at the molecular level and converting the hybridization event into an analytical signal.3–6 They offer attractive advantages such as high sensitivity, low cost and minimal power requirements. Thus, electrochemical DNA biosensors represent a dynamic research area focused on the development of point-of-care tests.7 These devices rely on the conversion of DNA base pair recognition events into a useful electrical signal, using either a label-free or label-based method.8 The label free method based on the use of an electroactive hybridization indicator is one of the most attractive owing to its simplicity. In this case, the conversion of the hybridization/recognition event into a measurable electrochemical signal is achieved by using an electroactive compound that interacts with single and double stranded DNA to different extents.2,4 Recently, small molecules and transition metal complexes, generally cations,9–13 have been used as electrochemical hybridization indicators. However, not all those proposed are sensitive enough for direct single-nucleotide polymorphism (SNP) genotyping. Hence, the study of the interaction of new molecules with DNA in order to design new drugs and diagnostic reagents still remains a formidable task.Metallacarbaboranes (or metallacarboranes) [3,3′-M(1,2-closo-C2B9H11)2]z− are a class of inorganic polyhedral clusters containing carbon, boron, hydrogen, and metal atoms in various combinations.14 A typical metallacarborane is a sandwich of two [C2B9H11]2− (dicarbollide) clusters with a metal ion in the center. Fig. 1 displays the [3,3′-Fe(1,2-closo-C2B9H11)2]− and [3,3′-Fe(8,9,12-Cl3-1,2-closo-C2B9H8)2]− platforms with their cluster vertex numbering. They are metal sandwich complexes similar to ferrocene (see Fig. 1) and have a good reversible electroactive couple, Fe3+/Fe2+, but unlike ferrocene they are water-soluble. Metallacarboranes are becoming a subject of growing interest to the broad chemical community owing to their unique combination of features and properties, including the rigidity of the cages and their relative rotary motion, their hydrophobicity, and their chemical and thermal stability due to delocalized charge. The inclusion of carbon atoms in the boron framework causes a differential reactivity of the cluster with either acidic (carbon-bound) or hydridic (boron-bound) hydrogen atoms. Moreover, changing the metals further increases the variability, influencing, for instance, the charge or redox properties. Altogether, a broad range of interactions can be formed between metallacarboranes and their neighboring molecules. For example, the B–X bond (X = halogen) in boron clusters is quite stable to reduction and this could be used as an advantage for the redox tuning of boron clusters. Indeed, it has been demonstrated that the incorporation of Cl or I in [3,3′-Co(1,2-closo-C2B9H11)2]− gradually and systematically shifts the formal potential of Co3+/Co2+ to positive values.15 Thus, the redox potential of a platform can be tuned with a minor change in its shape and dimensions so as to adjust it to a specific purpose.
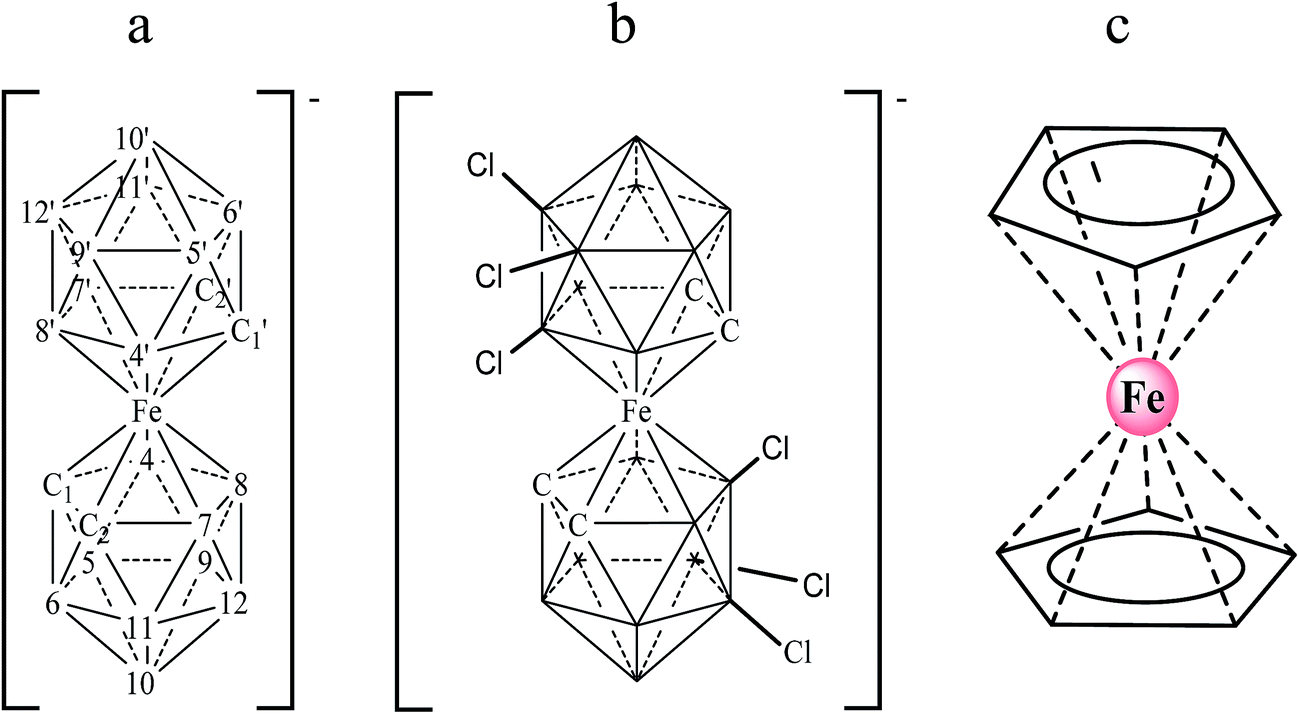 | ||
| Fig. 1 Chemical structures of the anionic metallacarboranes [3,3′-Fe(1,2-closo-C2B9H11)2]− (a) and [3,3′-Fe(8,9,12-Cl3-1,2-closo-C2B9H8)2]− (b) with their cluster vertex numbering; neutral ferrocene (c). | ||
In addition, [3,3′-Co(1,2-closo-C2B9H11)2]− and [3,3′-Co(8-I-1,2-closo-C2B9H10)2]− have proved to have an extraordinary capacity for self-assembly in water.16,17 This has been demonstrated by the formation of vesicles and micelles for [3,3′-Co(1,2-closo-C2B9H11)2]− or vesicles and lamellae for [3,3′-Co(8-I-1,2-closo-C2B9H10)2]−, in this order, on increasing the concentration of the cobaltabisdicarbollide. Therefore, it was expected that (i) due to the great tendency to adhere to surfaces through hydrogen or dihydrogen bonds,18 and (ii) due to the self assembly capacity, a large multiplier effect could occur at defined potentials.
In spite of these properties, few studies related to DNA and metallacarboranes can be found in the literature. Lesnikowski et al. have reported the use of metallacarborane units covalently linked to an oligonucleotide DNA strand as an active label method for the electrochemical detection of DNA hybridization,19–21 but to date no one has studied their interaction with DNA and their use as free redox hybridization indicators. These compounds may exhibit interesting interactions with DNA; they are anions, and interactions with DNA have usually been described for cations.
In this work, we describe the interaction of two ferrabisdicarbollide species, [Na·2.5H2O][3,3′-Fe(1,2-closo-C2B9H11)2] (denoted Na[FESANE] in the text) and its hexachlorinated derivative [Na·2.5H2O][3,3′-Fe(8,9,12-Cl3-1,2-closo-C2B9H8)2] (denoted Na[Cl6-FESANE]), with DNA, and their application as negative and positive potential redox indicators for direct gene mutation detection in PCR amplicons extracted from blood cells.
Results and discussion
Synthesis and electrochemical properties of the metallacarboranes
Ferrocene, with its high stability, is the archetype of sandwich compounds in which a metal is bound in a hapto manner to two arene ligands.22 Ferrocene and the metallacarborane sandwich clusters [3,3′-M(1,2-closo-C2B9H11)2]z− (Fig. 1) are air stable.23 The substitution of some hydrogen atoms for halogens leads to the halogenated derivatives of pristine [3,3′-M(1,2-closo-C2B9H11)2]z−.15,24–26 Similarly to ferrocene, [3,3′-M(1,2-closo-C2B9H11)2]z− species are reversibly electroactive,27 but in contrast to ferrocene they are anions whose charge is delocalized through the large molecular volume.28 Dehydrochlorination and dehydroiodination on the [3,3′-Co(1,2-closo-C2B9H11)2]− platform causes an anodic shift of the formal potential (E0′) of the associated redox couple.15,25,26 The equivalent [3,3′-Fe(1,2-closo-C2B9H11)2]− platform, [FESANE]−, is also susceptible to dehydrochlorination, giving rise to [3,3-Fe(8,9,12-Cl3-1,2-closo-C2B9H8)2]−. We have studied the electrochemical behaviour of these two metallacarboranes. Fig. 2 shows cyclic voltammograms (CVs) at 100 mV s−1 for 1.0 mM Na[FESANE] (a) and Na[Cl6-FESANE] (b) at a bare gold screen-printed electrode (AuSPE) using 0.1 M phosphate buffer (PB) pH 7.0 solution as the supporting electrolyte (black curve). In the case of [FESANE]−, it can be seen that a pair of redox peaks, which we ascribe to the metal center (Fe), appear at an E0′ value of −0.283 V with a peak potential separation (ΔEp) of 74 mV. In the case of [Cl6-FESANE]−, a very similar CV response is obtained, but at an E0′ value of 0.348 V. This shift of about +0.6 V is due to the hydrogen cluster substitution with chlorine groups. The presence of B–Cl bonds in the cluster causes the shift in E0′, as was previously observed for [3,3′-Co(1,2-closo-C2B9H11)2]−.25 Hence, the dehydrochlorination of the cluster triggers the redox potential of the complex to be used for a specific purpose. In this work, the inclusion of six chlorine groups in the [FESANE]− framework allows the preparation of platforms where the redox couple Fe3+/Fe2+ has a positive or negative formal potential for [FESANE]− and [Cl6-FESANE]−, respectively.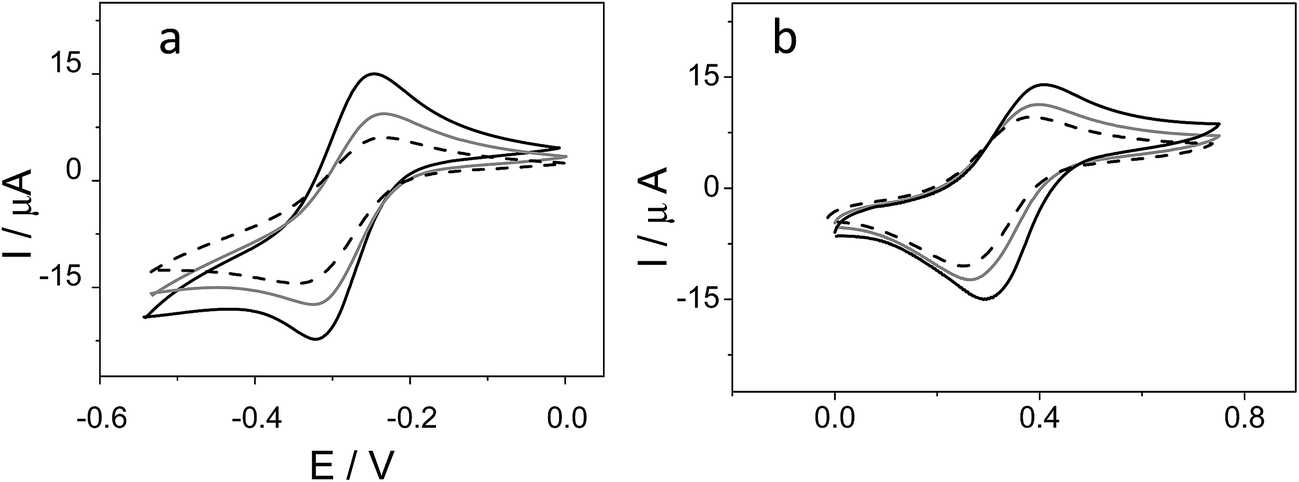 | ||
| Fig. 2 (a) Cyclic voltammograms of 1.0 mM Na[FESANE] in 0.1 M PB pH 7.0 solution at a bare AuSPE (black curve), a calf thymus double stranded DNA AuSPE (CT-dsDNA/AuSPE; grey curve) and a calf thymus single stranded DNA AuSPE (CT-ssDNA/AuSPE; dashed curve). (b) The same experiments for Na[Cl6-FESANE]. | ||
In the case of [FESANE]−, the ΔE value is close to the value of 60 mV expected for a freely diffusing one-electron reversible redox process. However, in the case of [Cl6-FESANE]−, the ΔE value is slightly higher, suggesting some limitations in the charge-transfer kinetics, probably due to the presence of the chlorine groups.
In both cases, at a higher ionic strength (0.1 M PB pH 7.0 solution + 0.4 M NaCl) a slightly more reversible shape but similar E0′ and ΔEp values were observed (see Fig. 1 of ESI†). Based on this result one would assume that there are not severe limitations in the charge–transfer kinetics due to the solution resistance. Moreover, the cathodic peak current was proportional to the square root of the scan rate (v1/2), as expected for a process controlled by diffusion.
Interaction with DNA
As we have previously mentioned these compounds may exhibit interesting interactions with DNA. Hence, we focused our attention on studying the interaction of [FESANE]− and [Cl6-FESANE]− with DNA either in solution or on the electrode surface.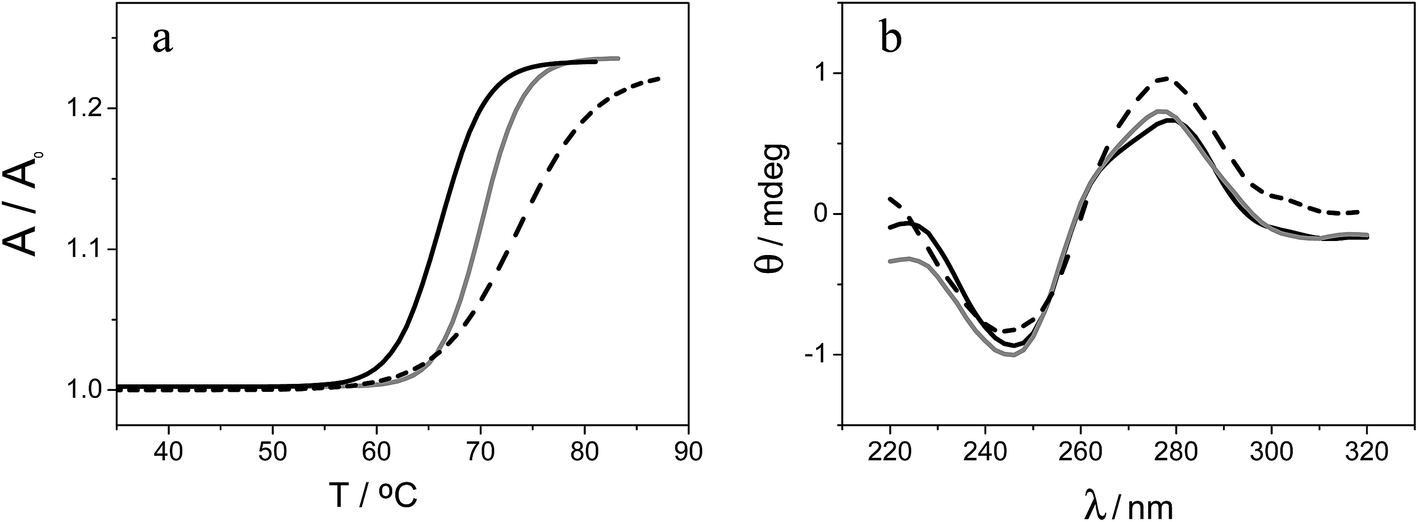 | ||
| Fig. 3 (a) Melting curves of CT-dsDNA (80 μM) in the absence (black curve) and in the presence of 8 μM Na[FESANE] (grey curve) and Na[Cl6-FESANE] (dashed curve). (b) Circular dichroism spectra of CT-dsDNA (80 μM) in the absence (black curve) and in the presence of 8 μM Na[FESANE] (grey curve) and Na[Cl6-FESANE] (dashed curve). | ||
In an effort to better understand the interactions of DNA and metallacarboranes Na[FESANE] and Na[Cl6-FESANE], the following experiments were conducted.
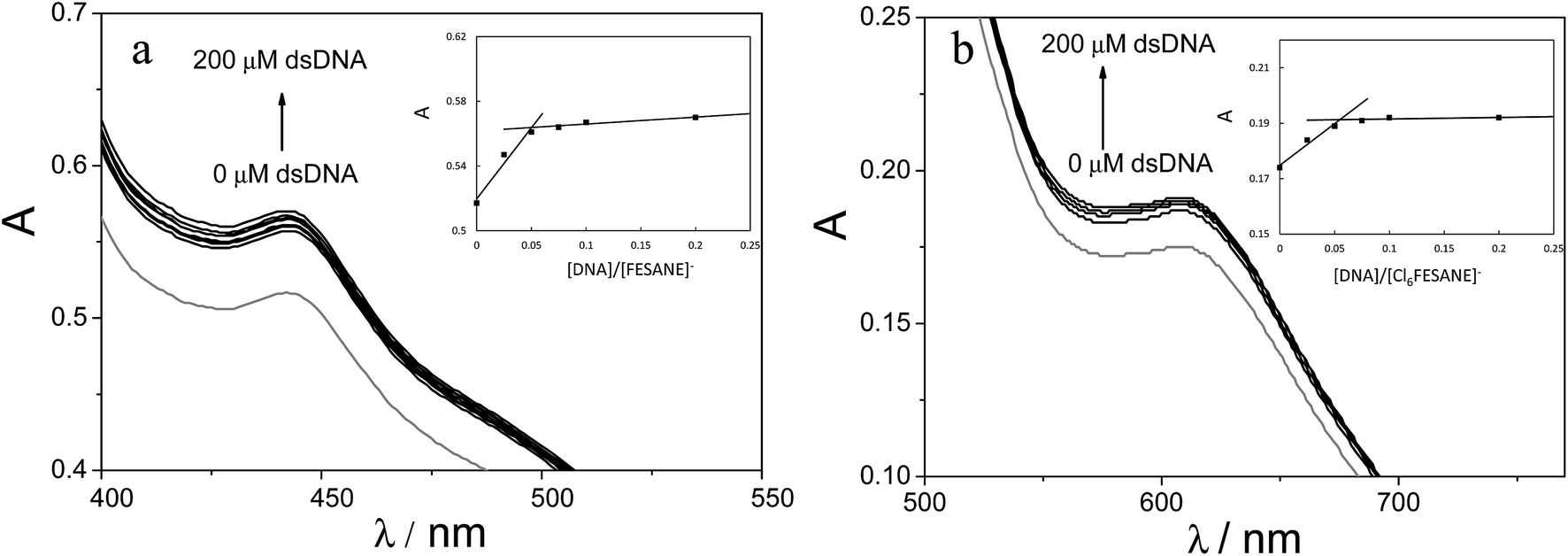 | ||
| Fig. 4 Absorption spectra of 1.0 mM (a) Na[FESANE] and (b) Na[Cl6-FESANE] in 0.1 M PB pH 7.0 solution in the absence (grey curve) and in the presence of increasing amounts of CT-dsDNA (from 0 to 200 μM). Inset: absorbance vs. [DNA]/Na[FESANE] and absorbance vs. [DNA]/Na[Cl6-FESANE] plots. | ||
It is well known that the use of pulse voltammetry, such as Differential Pulse Voltammetry (DPV), permits better discrimination between non-faradaic and faradaic current, which is convenient for studying the electrochemical behaviour of redox species that may interact with DNA present in the solution. For this reason, current titrations were performed by means of DPV by keeping the metallacarborane concentration constant while varying the concentration of CT-dsDNA. Fig. 5 shows differential pulse voltammograms for Na[FESANE] and Na[Cl6-FESANE]. From the Ipvs. [DNA]/[metallacarborane] plot (see inset Fig. 5a and b), it was found that about 20 molecules of metallacarborane are bound to a DNA base pair for both metallacarboranes. This high value, although it agrees well with that obtained from the spectrophotometric titrations, is quite surprising and it can be explained on the grounds of the peculiar structure of [FESANE]− and [Cl6-FESANE]−, which leads to the possibility of their self-assembly. They are composed of two bulky and highly hydrophobic dicarbollide semicages, each of them bearing two negative charges that “sandwich” a Fe(III) ion as the polar part. The remaining negative charge is delocalized over the entire [FESANE]− platform that can be represented by the Greek letter θ, as in the case of [3,3′-Co(1,2-closo-C2B9H11)2]− (denoted [COSANE]−).16 As can be seen in Fig. 1, because of the charge and θ shape of the [FESANE]− anion, counterions can neither approach it closely enough to reduce the Born energy, nor can the hydrophobic rigid ends deform to partition on one side of the head group, as in double-tailed surfactants.
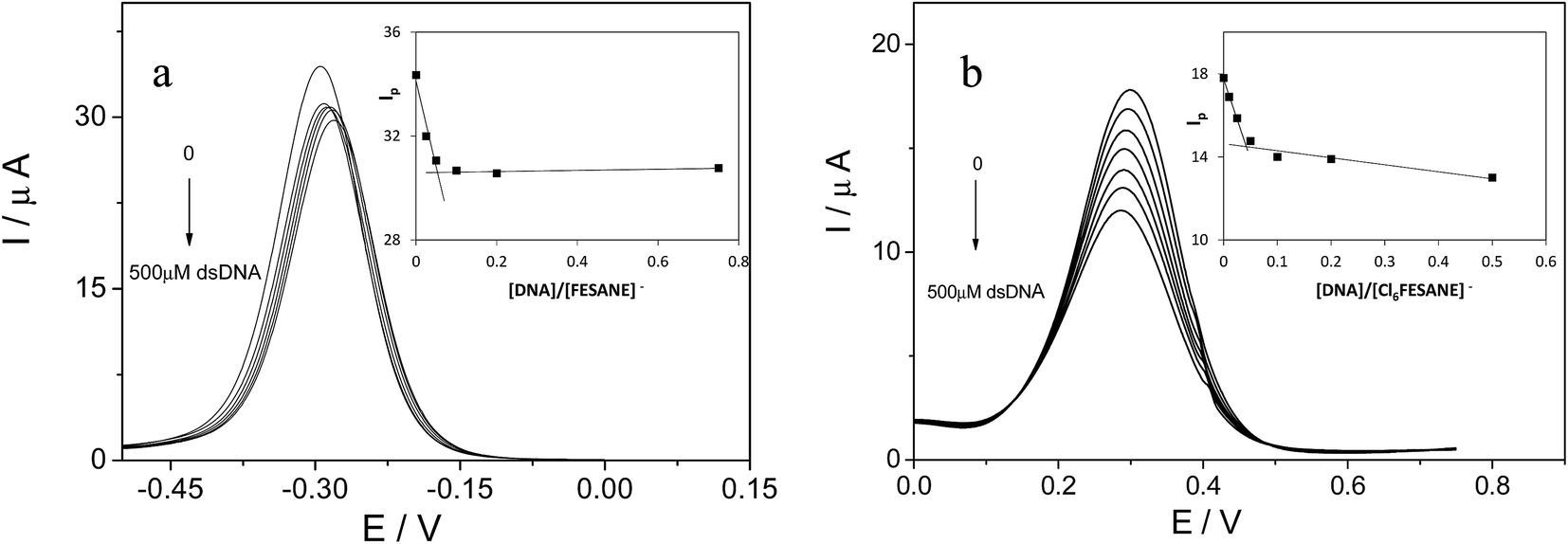 | ||
| Fig. 5 Differential pulse voltammograms of 1.0 mM of Na[FESANE] (a) and Na[Cl6-FESANE] (b) in the absence and in the presence of increasing amounts of CT-dsDNA (from 0 to 500 μM). Inset: Ipvs. [DNA]/Na[FESANE] and Ipvs. [DNA]/Na[Cl6-FESANE] plots. | ||
If [FESANE]− aggregates like [COSANE]−,16 vesicles with a radius of around ∼20 nm could be expected. In order to corroborate this hypothesis, Dynamic Light Scattering (DLS) experiments were carried out. In these experiments, the concentration of [FESANE]− in the solution was fixed (1.0 mM in water) and the concentration of CT-dsDNA increased from 0 to 900 μM. From the plot of particle size (nm) versus CT-dsDNA concentration (Fig. 3 of ESI†), it can be observed that the diameter of the [FESANE]− aggregates is about 39 nm. Upon addition of DNA, a linear increase in particle size up to 63 nm is observed at 150 μM DNA and then a plateau was reached. A new sudden size enlargement appears at a DNA concentration above 300 μM, reaching a size of 75 nm. These results seem to indicate that DNA is surrounding the [FESANE]− aggregates.
In the case of CT-dsDNA/AuSPE the peak current was directly proportional to the scan rate, v, which is characteristic of surface-confined redox processes. This seems to indicate that part of the metallacarborane present in the solution is bonding to the CT-dsDNA layer immobilized on the electrode surface. The general process can be described by Scheme 1, similar to the one previously proposed by Bard and Carter.40
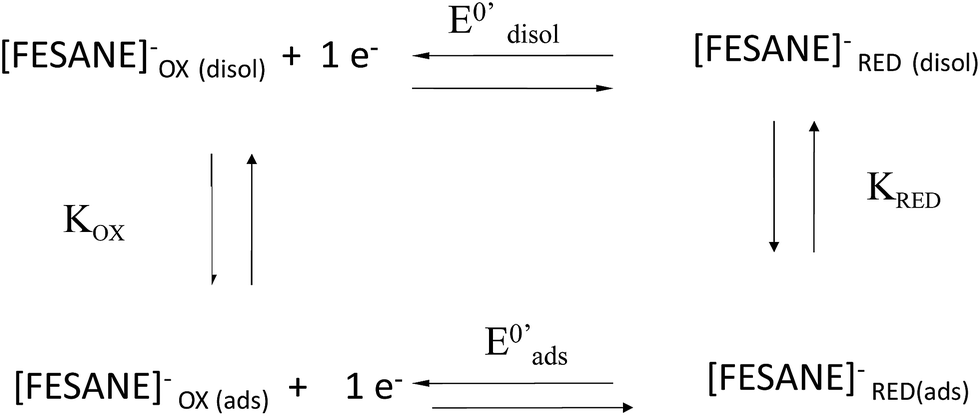 | ||
| Scheme 1 Square scheme of the general process. | ||
After repetitive potential cycles, there is an increase in the peak current with no change in E0, confirming the accumulation of the metallacarborane in the immobilized DNA layer (Fig. 4a of ESI†). If, after repetitive cycling, the electrode was removed from the cell, rinsed with water, and placed in an electrolyte solution containing no metallacarborane, a pair of voltammetric peaks, less defined but very similar to those previously obtained, were observed (Fig. 4b of ESI†). This indicates that the incorporated metallacarborane remained tightly bound to the DNA layer on the electrode surface.
If the ionic strength of the solution increases (0.1 M PB pH 7.0 solution + 0.4 M NaCl), either the shape or the formal potential of the voltammetric waves remain equal. These results confirm that the interaction between CT-dsDNA/AuSPE and the metallacarboranes is largely independent of the ionic strength, which is consistent with intercalation (Fig. 5 of ESI†).
Table 1 displays the E0′ values of [FESANE]− and [Cl6-FESANE]− using a bare AuSPE, a CT-dsDNA/AuSPE and a CT-ssDNA/AuSPE. E0′ shifted to less negative (in the case of [FESANE]−) or less positive (in the case of [Cl6-FESANE]−) values for a CT-dsDNA/AuSPE compared to those obtained at the bare AuSPE. On the other hand, for CT-ssDNA/AuSPE there is a shift of E0′ to more negative (in the case of [FESANE]−) or less positive (in the case of [Cl6-FESANE]−) values compared to those obtained at the bare AuSPE. From these shifts (ΔE0′) and using eqn (1) of the Experimental section, according to the model of Bard and Carter,40 the ratio of the equilibrium surface-binding constants (Kox/Kred) were estimated. For CT-dsDNA/AuSPE the values obtained were 0.76 and 1.73, for [FESANE]− and [Cl6-FESANE]−, respectively, while at a CT-ssDNA/AuSPE, the equilibrium surface-binding constants were estimated to be 1.14 and 3.35 for [FESANE]− and [Cl6-FESANE]−, respectively.
| AuSPE E0′ (V) | CT-dsDNA/AuSPE E0′ (V) | CT-ssDNA/AuSPE E0′ (V) | |
|---|---|---|---|
| [FESANE]− | −0.283 | −0.276 | −0.291 |
| [Cl6-FESANE]− | 0.348 | 0.334 | 0.317 |
The value of 0.76 for Kox/Kred obtained in the case of [FESANE]− suggests that the reduced form interacts with the CT-dsDNA backbone more strongly than the oxidized form, which is characteristic of intercalative interactions. However, in the case of [Cl6-FESANE]− a Kox/Kred value of 1.14 indicates that the oxidized form interacts more strongly than the reduced form, suggesting some electrostatic component in the interaction. This attests the first of the two possibilities given above concerning the role of the [Cl6-FESANE]−. Nevertheless, the effect of ionic strength is not relevant in the interaction of both clusters with DNA (Fig. 5 of ESI†), which points to the intercalation being predominant.
In the case of CT-ssDNA/AuSPEs, as one would expect, either for [FESANE]− or [Cl6-FESANE]− the values of Kox/Kred indicate that the oxidized form interacts with the CT-ssDNA backbone more strongly than the reduced form, which agrees well with an electrostatic interaction.
A unique 25-mer thiolated sequence of this bacterium (SH-HP1; 10 μl of 40 μM) was chemisorbed onto the AuSPE through the thiol group, with a surface coverage (σ) of around 90 pmol cm−2, and used as the probe. In the hybridization test, a complementary (HP2C), a non-complementary (HP2NC) and a Single Nucleotide Polymorphism (SNP) sequence (HP2SNP), were selected as the target DNA. Changes in the differential pulse voltammogram peak currents of the clusters accumulated at the probe modified electrode before and after hybridization (see Scheme 2) were obtained and are shown in Fig. 6. As can be seen, hybridization of the probe (SH-HP1) with the complementary HP2C chain in the biosensor recognition layer resulted in a dramatic increase in the DPV response (about 8 and 5 times for [FESANE]− and [Cl6-FESANE]−, respectively), while virtually no change in current was obtained for the non-complementary sequence (compare the black and grey curves in Fig. 5) either for [FESANE]− or [Cl6-FESANE]−. The selectivity of the system was probed by hybridization with a SNP target (HP2SNP) sequence. This target will give a distorted double-helix, which may interact with DNA in a different way. Based on this, one would anticipate a different biosensor response when compared to hybridization with a fully complementary target sequence (HP2C). As can be seen, there is a significant decrease (about 50%) in the peak current values compared to those obtained when the SH-HP1/AuSPE electrode was hybridized with its fully complementary sequence, HP2C, (Fig. 5, dashed curve). This diminution in the peak current shall be interpreted as a decrease in the binding constant of the metallacarborane with the distorted helix. However, the peak current obtained when the SNP was present was much higher (about 2 and 4 times for [FESANE]− and [Cl6-FESANE]−, respectively) than that obtained for a fully non-complementary sequence (see grey curve in Fig. 6). This allows clear discrimination between non-complementary, fully complementary and mismatched base pairs. To our knowledge this is the first time that metallacarboranes have been used for such a purpose.
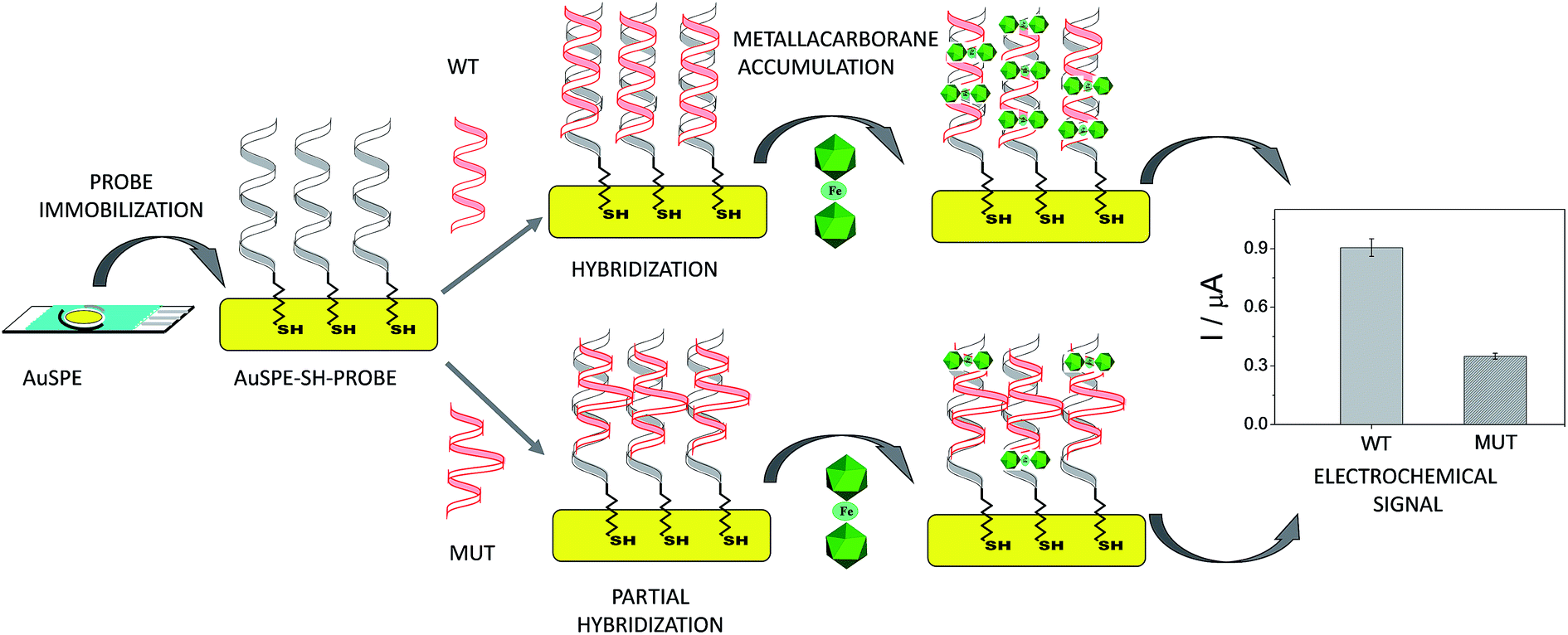 | ||
| Scheme 2 Scheme of the biosensor development. | ||
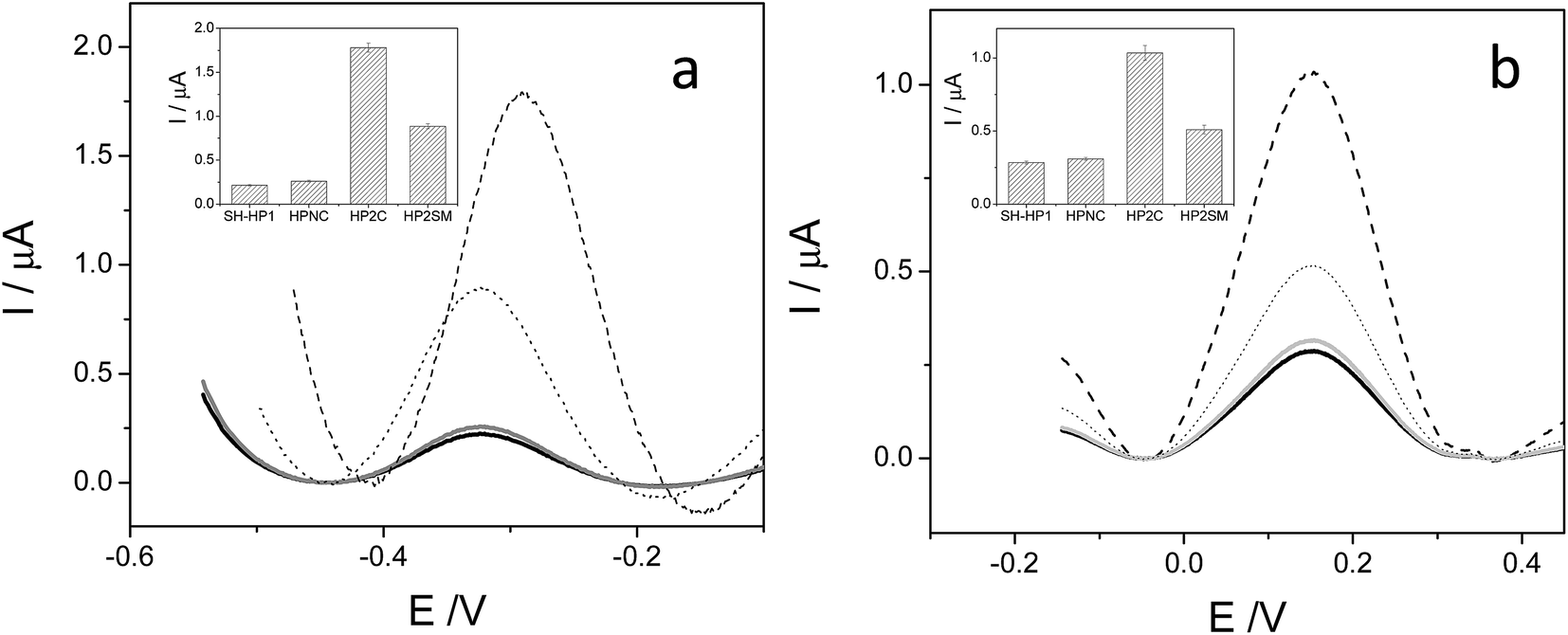 | ||
| Fig. 6 Differential pulse voltammograms of 1.0 mM Na[FESANE] (a) and Na[Cl6-FESANE] (b) accumulated on a SH-HP1/AuSPE before (black curve) and after hybridization with a complementary HP2C (dashed curve), non complementary HP2NC (grey curve) and Single Nucleotide Polymorphism (SNP) sequence HP2SNP (dotted curve). Inset: peak current and error bar diagrams of the biosensor response (N = 5, RSD less than 5% for all measurements). | ||
Gene mutation detection
Based on these promising results, we decided to go one step further and apply the DNA biosensor to diagnose genetic diseases through the detection of gene mutations associated to such diseases in real DNA PCR amplicons extracted from blood cells. Genetic diseases may be diagnosed by different test categories. Among them, mutation identification by sequencing of genes is the gold standard. However, these methods have serious drawbacks as routine diagnostic tools because of their labour intensity and cost. Hence, new, accurate and facile strategies for the detection of point mutations are therefore absolutely necessary. In this sense, the DNA biosensor could be an alternative to a classical gene assay due to its several advantages, in particular the low cost and simplicity.As a case study, two different mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR) were detected. In particular, we chose the F508del (MUTF508del)41 and p.Gly542Stop (SNPG542X) mutations, both associated with cystic fibrosis. The F508del mutation consists of a three-nucleotide deletion, which causes the loss of a phenylalanine residue of the CFTR protein,42 and the p.Gly542Stop mutation results in a truncated CFTR protein.
In the approach employed, no labelled oligonuclotides are required. Two different thiolated sequences from exons 11 and 12 (named SH-WT11 and SH-WT12; see Table 3 of the Experimental section), were assembled on the electrode surface, through the thiol group, and hybridized with the corresponding denaturated PCR target, which comprises around 300 bp PCR amplicons of exons 11 and 12 from the CFTR gene, carrying the mutations MUTF508del and SNPG542X associated to cystic fibrosis (see Table 3). As a control, two wild type amplicons, WT11 and WT12 from exons 11 and 12, respectively, were used. All samples were validated by the suppliers (Instituto de Genética Médica y Molecular de Madrid, Spain) by sequentiation methods.
The detection relies on the comparison of the voltammetric transduction of the hybridization reaction between the immobilized probe and the target DNA sequences (wild type or mutant) present in the sample. DPV of the metallacarborane accumulated on the double strand DNA layer, formed on the electrode surface after hybridization, was used to obtain the biosensor response. Fig. 7 shows peak current bar diagrams of the biosensor response before and after hybridization with a solution containing: the control sequence (WT) or mutated sequences, SNPG542X and MUTF508del, using [FESANE]− (a, b) or [Cl6-FESANE]− (c, d) as the redox indicator. The difference between the signals from the mutated and wild type target DNA allows unambiguous confirmation of the presence of a mutation without the need for the stringent conditions (i.e. formamide) usually employed for such a purpose. In addition, it is worth noting that the decrease in the current signal is a slightly higher for SNPG542X than for the MUTF508del samples. In this case the biosensor permits better discrimination between the mutated and wild type samples. This agrees well with the major distorted DNA helix formed after hybridization with the sequence carrying this mutation, which involves a three-nucleotide deletion.
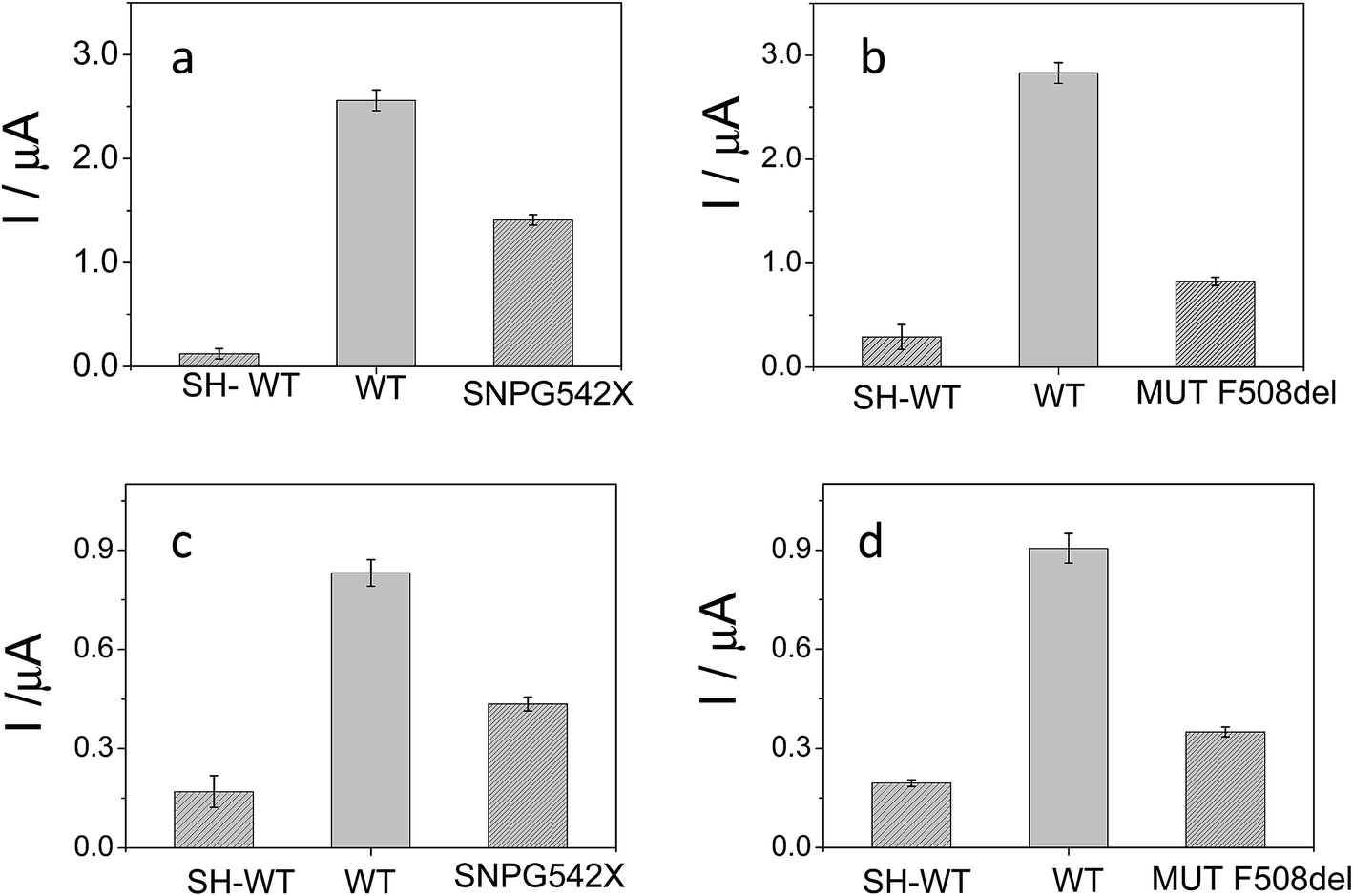 | ||
| Fig. 7 Peak current bar diagrams of the biosensor response before and after hybridization with a solution containing: the control sequence (WT) and mutated sequences, SNPG542X and MUTF508del, using [FESANE]− (a, b) or [Cl6-FESANE]− (c, d) as the redox indicator. | ||
The reproducibility of the developed methodology was evaluated from the response of 5 different biosensors, prepared in the same manner (see Experimental section), to either wild type or mutated target DNA. Reproducible signals were obtained with a relative standard deviation (RSD) of 5% or less in all cases. Hence, it can be concluded that the proposed screening method not only allows DNA detection but also discriminates between wild and mutated samples. These results compare well with those obtained with other more conventional (cationic) redox indicators as the ruthenium complex, pentaamine ruthenium [3-(2-phenanthren-9-yl-vinyl)-pyridine],12 or dyes such as Azure A or Safranine,43 but metallacarboranes are anionic species. This is the first time that an anion has been employed as the redox indicator of a hybridization event for the detection of gene mutations. Rapid and precise screening of small genetic variations, such as single nucleotide polymorphisms (SNPs) or point mutations as deletions, plays an important role in human diseases. As genetic markers, mutations can be used to trace the generational inheritance patterns associated with specific diseases. As diagnostic markers, point mutations can be used for early cancer44 or human disease detection, such as hereditary liver disorders or cystic fibrosis.
Experimental section
Chemicals
Potassium nitrate, sodium phosphate and sodium chloride were obtained from Scharlab Company. Double stranded calf thymus DNA (CT-dsDNA) was purchased from Sigma-Aldrich Co. CT-dsDNA stock solutions (nominally 1.0 mg ml−1) were prepared in 0.1 M phosphate buffer (PB) pH 7.0 solution. The DNA solutions gave a UV absorbance ratio (A260/A280) of about 1.9, indicating that the DNA was free of protein.45 The concentration of base pairs (bp) in the DNA was determined by using a molar absorptivity of 6600 M−1 at 260 nm.46 Single stranded calf thymus DNA (CT-ssDNA) was obtained by boiling in water capped vials containing CT-dsDNA (1.0 mg ml−1) in 0.1 M PB pH 7.0 for 30 minutes followed by rapid cooling in an ice bath. The synthetic oligonucleotides (from Sigma-Aldrich Co.) used in this work were 25 or 100-mer sequences from the pathogen bacterium Helicobacter pylori (H. Pylori) or characteristic DNA sequences from the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Two different types of synthetic oligonucleotides were used – thiolated sequences (SH-HP1 and SH-WT11, SH-WT12) and non thiolated sequences (HP2C, HP2NC, HP2SNP) that have been used as analytes (see Table 2). Genomic DNA was isolated from peripheral blood leukocytes from cystic fibrosis patients by standardized procedures (commercial Kit Purogene from Qiagen GmbH, Hilden, Germany) as we described before.12 The polymerase chain reaction (PCR) samples consisted of wild type (WT11, WT12) and mutated (MUTF508del and SNPG542X) sequences from gene samples. Synthetic oligonucleotides and PCR amplified sequences of real DNA samples from healthy or cystic fibrosis patients are listed in Tables 2 and 3 respectively. All solutions were prepared just prior to use. Water was purified with a Millipore Milli-Q-System (18.2 MΩ cm).| Synthetic oligonucleotides (25bp) | ||
| Thiolated probe | 5′-HS(CH2)6-GCGTTCCAAAGGGCAGGATCATTGA | SH-HP1 |
| Complementary sequence | 5′-TCAATGATCCTGCCCTTTGGAACGC | HP2C |
| SNP sequence | 5′-TCAATGATCCTACCCTTTGGAAGCG | HP2SNP |
| Non complementary sequence | 5′-GACCGTCGAAGTAAAGGGTTCCATA | HP2NC |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Synthetic oligonucleotides (100bp) | ||
| Thiolated probe 11 | 5′-HS(CH2)6-CTCAGTTTTCCTGGATTATGCCTGGCACCATTAAAGAAAATATCATCTTTGGTGTTTCCTATGATGAATATAGATACAGAAGCGTCATCAAA GCATGCC | SH-WT11 |
| Thiolated probe 12 | 5′-HS(CH2)6-TTGGTAATAGGACATCTCCAAGTTTGCAGAGAAAGACAATATAGTTCTTGGAGAAGGTGGAATCACACTGAGTGGAGGTCAACGAGCAAGAATTTCTTTA | SH-WT12 |
| PCR samples | |||
|---|---|---|---|
| Complementary sequence exon 11 | 5′_AACCGATTGAATATGGAGCCAAATATATAATTTGGGTAGTGTGAAGGGTTCATATGCATAATCAAAAAGTTTTCACATAGTTTCTTACCTCTTCTAGTTGGCATGCTTTGATGACGCTTCTGTATCTATATTCATCATAGGAAACACCAAAGATGATATTTTCTTTAATGGTGCCAGGCATAATCCAGGAAAACTGAGAACAGAATGAAATTCTTCCACTGTGCTTAATTTTACCCTCTGAAGGCTCCAGTTCTCCCATAATCACCATTAGAAGTGAAGTCTGGAAATAAAACCCATCATTATTAGGTCATTATCAAATCACGCTCAGGATTCACTTGCCTCCAATTATCATCCTAAGCAGAAGTGTATATTC | WT11 | |
| Mutated sequence: mutation F508del | 5′_AACCGATTGAATATGGAGCCAAATATAATTTGGGTAGTGTGAAGGGTTCATATGCATAATCAAAAAGTTTTCACATAGTTTCTTACCTCTTCTAGTTGGCATGCTTTGATGACGCTTCTGTATCTATATTCATCATAGGAAACACCA___ATGATATTTTCTTTAATGGTGCCAGGCATAATCCAGGAAAACTGAGAACAGAATGAAATTCTTCCACTGTGCTTAATTTTACCCTCTGAAGGCTCCAGTTCTCCCATAATCACCATTAGAAGTGAAGTCTGGAAATAAAACCCATCATTATTAGGTCATTATCAAATCACGCTCAGGATTCACTTGCCTCCAATTATCATCCTAAGCAGAAGTGTATATTC | MUTF508del | |
| Complementary sequence exon 12 | 5′_ACTAGCCATAAAACCCCAGGATTTTTTCAATTCCAGAAACAGAATATAAAGCAATAGAGAAATGTCTGTAATTTTTTTACATGAATGACATTTACAGCAAATGCTTGCTAGACCAATAATTAGTTATTCACCTTGCTAAATTCTTGCTCGTTGACCTCCACTCAGTGTGATTCCACCTTCTCCAAGAACTATATTGTCTTTCTCTGCAAACTTGGAGATGTCCTATTACCAAAAATAGAAAATTAGAGAGTCACTTTTAGTATGCTCAATCTGAATTTGAAAGGCACATCTTCCTTCTAATGT | WT12 | |
| Mutated sequence: mutation G542X | 5′_ACTAGCCATAAAACCCCAGGATTTTTTCAATTCCAGAAACAGAATATAAAGCAATAGAGAAATGTCTGTAATTTTTTTACATGAATGACATTTACAGCAAATGCTTGCTAGACCAATAATTAGTTATTCACCTTGCTAAAGAATTCTTGCTCGTTGACCTCCACTCAGTGTGATTCCACCTTCTC![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) AAGAACTATATTGTCTTTCTCTGCAAACTTGGAGATGTCCTATTACCAAAAATAGAAAATTAGAGAGTCACTTTTAGTATGCTCAATCTGAATTTGAAAGGCACATCTTCCTTCTAATGT AAGAACTATATTGTCTTTCTCTGCAAACTTGGAGATGTCCTATTACCAAAAATAGAAAATTAGAGAGTCACTTTTAGTATGCTCAATCTGAATTTGAAAGGCACATCTTCCTTCTAATGT |
SNPG542X | |
[N(CH3)4][3,3′-Fe(1,2-closo-C2B9H11)2], [N(CH3)4][FESANE], was synthesized as reported in the literature.47
Apparatus
UV-visible spectra were recorded using a double beam PharmaSpec UV-1700 series spectrophotometer from Shimadzu Corporation, operating from 200 nm to 800 nm in 1.0 cm quartz cells. A thermostatic bath in conjunction with the spectrophotometer was used to control the cuvette temperature in the measurements. The sequencing of the DNA samples was carried out using a Sanger Sequencher 3730xL (Array 36 cm; POP 7; Applied Biosystems). The final concentration of the DNA sample solutions was determined by UV-vis molecular absorption spectrometry using a Thermo Scientific NanoDrop™ 1000 Spectrophotometer (NanoDrop Technologies). PCR fragments were generated in a BIO-RAD thermal cycler (DNA Engine Tetrad2; Peltier Thermal Cycler; BIO-RAD Laboratories Inc.). Circular dichroism measurements were carried out using a spectropolarimeter J715 from JASCO. Electrochemical measurements were carried out at room temperature in a homemade one-compartment electrochemical cell coupled to an Autolab PGSTAT 30 potentiostat from Eco-Chemie (KM Utrecht, the Netherlands) using the software package GPES 4.9 (General Purpose Elec. Experiments). Integrated screen-printed gold electrodes (4 mm diameter, AuSPEs) from DropSens S.L (Oviedo, Spain) were used as working electrodes. The format of these screen-printed electrodes includes a gold working electrode, a silver pseudo reference electrode and a gold counter electrode. All experiments were carried out by substituting the Ag ink pseudo reference with an external Ag/AgCl reference electrode. The electrodes were connected using a SPE Connector (DropSens S. L.) as the interface. IR spectra were obtained on a PerkinElmer® Universal ATR Accessory spectrophotometer. The 1H- and 1H{11B}-NMR (300.13 MHz), 13C{1H}-NMR (75.47 MHz) and 11B- and 11B{1H}-NMR (96.29 MHz) spectra were recorded on a Bruker ARX 300 instrument equipped with the appropriate decoupling accessories. All NMR spectra were performed in deuterated acetone solvent at 22 °C. The 11B- and 11B{1H}-NMR shifts were referenced to external BF3·OEt2, while the 1H, 1H{11B}, and 13C{1H}-NMR shifts were referenced to SiMe4. Chemical shifts are reported in units of parts per million downfield from the reference, and all coupling constants are in Hz. The mass spectra were recorded in the negative ion mode using a Bruker Biflex MALDI-TOF-MS [N2 laser; λexc 337 nm (0.5 ns pulses); voltage ion source 20.00 kV (Uis1) and 17.50 kV (Uis2)].Procedures
General procedure of cationic exchange resin. To get the sodium counterbalancing species, approximately 100 mg of the starting compound, [N(CH3)4][FESANE] and [N(CH3)4][Cl6-FESANE], which are water-insoluble, were dissolved in a minimum volume of acetonitrile/water (50
:50). Then each solution was passed repeatedly through a cation exchange resin, previously loaded with sodium. The solvent was finally evaporated. The partial or complete solubilization of the new salts in distilled water and the disappearance of the [NMe4]+ peaks in the IR and 1H NMR spectra was a clear indication that the complete exchange to sodium was successful.
[Na·2.5H2O][FESANE]. IR (KBr): ν [cm−1] = 3590, 3562, 3521 (vs, νs(H2O)), 3037, 3021 (vs, νs(Cc–H)), 2566, 2521 (vs, νs(B–H)), 1636, 1605 (vs, ν(H2O)). 1H{11B} NMR: δ [ppm] = 69.30 (br s), 45.65 (s, B(10)–H), 39.69 (br s), 1.32 (br s, B–H), −7.8 (br s, B–H). 11B NMR: δ [ppm] = 103.7 (s, 2B), 21.5 (s, 4B), 0.6 (s, 4B), −31.1 (s, 2B), −403.5 (s, 4B), −453.5 (s, 2B). 13C{1H} NMR: δ [ppm] = −438.19 (br. s, Cc). MALDI-TOF MS: m/z (%) = 321.16 [M, 100%]−. UV-vis in H2O at 2.5 × 10−5 M: 271 (2.066) nm.
[Na·2.5H2O][Cl6-FESANE]. IR (KBr): ν [cm−1] = 3576 (vs, νs(H2O)), 3039 (vs, νs(Cc–H)), 2567 (vs, νs(B–H)), 1612 (vs, ν(H2O)). 1H NMR: δ [ppm] = 39.20 (br s, 4H, Cc–H), −6.59, −11.8, −15.54, −24.10 (br s, 12H, B–Ht). 11B NMR: δ [ppm] = 126.7, 104.4, 45.8, 38.4, 29.4, 7.6, 3.3, −11.6, −28.8, −70.3, −334.0, −351.6, −412.3, −423.4, −498.1, −510.5, −542.8. MALDI-TOF MS: m/z (%) = 493.16 [M − Cl, 20.8%]−, 527.13 [M, 100%]−, 562.10 [M + Cl, 14.0%]−. UV-vis in H2O at 10−4 M: 247 (0.212), 295 (0.404), 335 (0.384), 607 (0.003) nm.
Interaction with DNA
The ratio of the equilibrium surface-binding constants (Kox/Kred) was estimated from the E0′ shifts (ΔE0′) of the DNA/Au electrodes with respect to E0′ of the unmodified Au electrodes and using eqn (1) according to the model of Bard and Carter:40
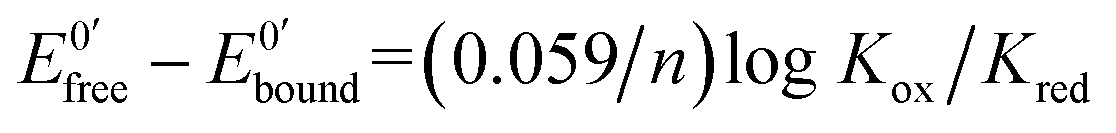 | (1) |
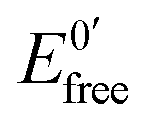 and
and 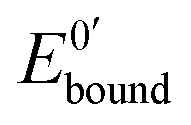 represent the formal potentials of the metallacarborane in solution and bound to DNA and Kox and Kred are the respective equilibrium binding constants to DNA for the oxidized and reduced forms of the metallacarborane.
represent the formal potentials of the metallacarborane in solution and bound to DNA and Kox and Kred are the respective equilibrium binding constants to DNA for the oxidized and reduced forms of the metallacarborane.
Electrochemical experiments in the presence of DNA were performed at a fixed concentration of metallacarboranes (1.0 mM), varying the concentration of CT-dsDNA (from 0 to 500 μM).
Biosensor development
In the case of DNA oligonucleotides, 10 μl of a 40 μM thiolated synthetic sequence (SH-HP1, SH-WT11 or SH-WT12) were transferred onto a clean AuSPE. Afterwards, the electrode was kept for 24 hours at room temperature. Then it was soaked in sterile water for at least 30 min.
Conclusions
The synthesis and characterization of two different metallacarboranes, [Na·2.5H2O] [3,3-Fe(1,2-closo-C2B9H11)2] and [Na·2.5H2O][3,3-Fe-(8,9,12-Cl3-1,2-closo-C2B9H8)2], and their interactions with CT-dsDNA and CT-ssDNA were thoroughly investigated. From these studies, it is clear that there is a strong interaction between these anionic metallacarboranes and DNA, and that they can be accumulated at a surface-immobilized DNA layer of screen-printed electrodes. Using these findings and the fact that both compounds are electrochemical active as a starting point, they have been employed as redox indicators of DNA hybridization in the development of electrochemical DNA biosensors as screening methods for the detection of gene mutations. A wide range of applications of this type of biosensor is foreseen because their fabrication and measuring protocols are simple, economical, and non-time consuming. In addition, these biosensors can be easily adapted for the detection of mutations, as a method having immense potential for greatly simplifying the development of point-of-care molecular diagnostic analysis systems.Acknowledgements
This work has been supported by the Comunidad Autónoma de Madrid (project No. S2009/PPQ- 363 1642), NANOAVANSENS (project No. S2013/MIT-3029), the Spanish Ministerio de Economía y Competitividad (projects No. CTQ2015- 71955-REDT (ELECTROBIONET), CTQ2014-53334-C2-1 R and CTQ2013-44670 R), and the Generalitat de Catalunya (2014/SGR/149). I. Fuentes is enrolled in the PhD program of the UAB. E. Lorenzo thanks the Fulbright scholarship-Salvador de Madariaga program from Spanish Ministerio de Economía y Competitividad.References
- P. Palaska, E. Aritzoglou and S. Girousi, Talanta, 2007, 72, 1119–1206 CrossRef PubMed.
- A. Erdem, Talanta, 2007, 74, 318–325 CrossRef CAS PubMed.
- J. Wang, Trends Anal. Chem., 2002, 21, 226–232 CrossRef CAS.
- A. Shabani, A. W. H. Mak, I. Gerges, L. A. Cuccia and M. F. Lawrence, Talanta, 2006, 70, 615–623 CrossRef CAS PubMed.
- X. H. Lin, P. Wu, W. Chen, Y. F. Zhang and X. H. Xia, Talanta, 2007, 72, 468–471 CrossRef CAS PubMed.
- W. M. McIntosh, X. Zhang and H. Ju, Talanta, 2007, 74, 387–392 CrossRef PubMed.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS PubMed.
- M. U. Ahmed, M. M. Hossain and E. Tamiya, Electroanalysis, 2007, 20, 616–626 CrossRef.
- L. Bouffier, B. S. Wang, A. Roget, T. Livache, M. Demeunynck and P. Mailley, Anal. Bioanal. Chem., 2014, 406, 1163–1172 CrossRef CAS PubMed.
- V. Vyskocil, J. Labuda and J. Barek, Anal. Bioanal. Chem., 2010, 397, 233–241 CrossRef CAS PubMed.
- Y. Wahyuni Hartati, S. N. Topkaya, I. P. Maksum and M. Ozsoz, Adv. Anal. Chem., 2013, 3, 20–27 Search PubMed.
- T. García-Mendiola, T. Barreiro Martinez, F. Pariente, J. Molano and E. Lorenzo, Electroanalysis, 2014, 26, 1–11 CrossRef.
- T. García, M. Revenga-Parra, H. D. Abruña, F. Pariente and E. Lorenzo, Anal. Chem., 2008, 80, 77–84 CrossRef PubMed.
- R. N. Grimes, Carboranes, Academic Press, London, United Kingdom, 2011 Search PubMed.
- A. Pepiol, F. Teixidor, R. Sillanpää, M. Lupu and C. Viñas, Angew. Chem., 2011, 50, 12491–12495 CrossRef CAS PubMed.
- P. Bauduin, S. Prevost, P. Farras, F. Teixidor, O. Diat and T. Zemb, Angew. Chem., Int. Ed., 2011, 50, 5298–5300 CrossRef CAS PubMed.
- D. Brusselle, P. Bauduin, L. Girard, A. Zaulet, C. Viñas, F. Teixidor, I. Ly and O. Diat, Angew. Chem., Int. Ed., 2013, 52, 12114–12118 CrossRef CAS PubMed.
- M. J. Hardie and C. L. Raston, Chem. Commun., 2001, 905–906 RSC.
- R. Ziólkowski, A. B. Olejniczak, L. Górski, J. Janusik, Z. J. Leśnikowski and E. Malinowska, Bioelectrochemistry, 2012, 87, 78–83 CrossRef PubMed.
- F. Jelen, A. B. Olejniczak, A. Kourilova, Z. J. Lesnikowski and E. Palecek, Anal. Chem., 2009, 81, 840–844 CrossRef CAS PubMed.
- I. Grabowska, A. Stachyra, A. Góra-Sochacka, A. Sirko, A. B. Olejniczak, Z. J. Leśnikowski, J. Radecki and H. Radecka, Biosens. Bioelectron., 2014, 51, 170–176 CrossRef CAS PubMed.
- P. Stepnickam, Ferrocenes: Ligands, Materials and Biomolecules, John Wiley & Sons Ltd, United Kingdom, 2008 Search PubMed.
- I. B. Sivaev and V. I. Bregadze, Collect. Czech. Chem. Commun., 1999, 64, 783–805 CrossRef CAS.
- I. Rojo, F. Teixidor, C. Viñas, R. Kivekäs and R. Sillanpää, Chem.–Eur. J., 2003, 9, 4311–4323 CrossRef CAS PubMed.
- P. González-Cardoso, A. I. Stoica, P. Farràs, A. Pepiol, C. Viñas and F. Teixidor, Chem.–Eur. J., 2010, 16, 6660–6665 CrossRef PubMed.
- M. Lupu, A. Zaulet, F. Teixidor, E. Ruiz and C. Viñas, Chem.–Eur. J., 2015, 21, 6888–6897 CrossRef CAS PubMed.
- M. Corsini, F. Fabrizi de Biani and P. Zanello, Coord. Chem. Rev., 2006, 250, 1351–1372 CrossRef CAS.
- C. Masalles, S. Borrós, C. Viñas and F. Teixidor, Adv. Mater., 2000, 12, 1199–1202 CrossRef CAS.
- J. L. Mergny, G. Duval-Valentin, C. H. Nguyen, L. Perrouault, B. Faucon and M. Rougée, et al. , Science, 1992, 256, 1681–1684 CrossRef CAS PubMed.
- J. L. Mergny, G. Duval-Valentin, C. H. Nguyen, L. Perrouault, B. Faucon and M. Rougée, et al. , Science, 1992, 256, 1681–1684 CrossRef CAS PubMed.
- C. Kumar, R. Turner and E. Asuncion, J. Photochem. Photobiol., A, 1993, 74, 231–238 CrossRef CAS.
- M.-J. Li, T.-Y. Lan, Z.-S. Lin, C. Yi and G.-N. Chen, J. Biol. Inorg. Chem., 2013, 18, 993–1003 CrossRef CAS PubMed.
- N. I. Kirillova, A. S. Zhdanov, A. I. Gusev, V. N. Kirin, S. P. Knyazev and T. V. Sokolova, Metalloorg. Khim., 1989, 2, 859–861 CAS.
- V. G. Vaidyanathan, R. Vijayalakshmi, V. Subramanian and B. U. Nair, Bull. Chem. Soc. Jpn., 2002, 75, 1143–1149 CrossRef CAS.
- M. F. Hawthorne, D. C. Young, T. D. Andrews, D. V. Howe, R. L. Pilling, A. D. Pitts, M. Reintjes, L. F. Warren Jr and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 879–896 CrossRef CAS.
- R. Hajian and M. Tavakol, E-J. Chem., 2012, 9, 471–480 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods. Fundamentals and Applications, John Wiley and Sons, 1980 Search PubMed.
- M. V. Del Pozo, C. Alonso, F. Pariente and E. Lorenzo, Biosens. Bioelectron., 2005, 20(8), 1549–1558 CrossRef CAS PubMed.
- D. W. Pang and H. Abruña, Anal. Chem., 1998, 70, 3162–3169 CrossRef CAS PubMed.
- M. T. Carter, M. Rodriguez and A. J. Bard, J. Am. Chem. Soc., 1989, 111(24), 8901–8911 CrossRef CAS.
- B. Kerem, J. M. Rommens, J. A. Buchanan, D. Markiewicz, T. K. Cox, A. Chakravarti, M. Buchwald and L. C. Tsui, Science, 1989, 245(4922), 1073–1080 CrossRef CAS PubMed.
- J. R. Riordan, J. M. Rommens, B. Kerem, N. Alon, R. Rozmahel, Z. Grzelczak, J. Zielenski, S. Lok, N. Playsic and J. L. Chou, Science, 1989, 245(4922), 1066–1072 CrossRef CAS PubMed.
- T. García-Mendiola, M. Ramos Cerro, J. M. López-Moreno, F. Pariente and E. Lorenzo, Bioelectrochemistry, 2016, 111, 115–122 CrossRef PubMed.
- R. E. Wilentz, C. H. Chung, P. D. J. Sturm, A. Musler, T. A. Sohn, G. J. A. Offerhaus, C. J. Yeo, R. H. Hruban and R. J. C. Slebos, Cancer, 1998, 82, 96–103 CrossRef CAS PubMed.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- P. Doty and S. A. Rice, Biochim. Biophys. Acta, 1955, 16(3), 446–448 CrossRef CAS.
- M. F. Hawthorne, D. C. Young and P. A. Wegner, J. Am. Chem. Soc., 1965, 87, 1818–1819 CrossRef CAS.
- D. W. Pang, Y. P. Qi, Z. L. Wang, J. K. Cheng and J. W. Wang, Electroanalysis, 1995, 7(8), 774–777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc01567k |
| This journal is © The Royal Society of Chemistry 2016 |