
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed C-3 direct arylation of pyridinium ions for the synthesis of 1-azafluorenes†
Jean-Nicolas
Desrosiers
*a,
Xudong
Wei
*a,
Osvaldo
Gutierrez
 b,
Jolaine
Savoie
a,
Bo
Qu
a,
Xingzhong
Zeng
a,
Heewon
Lee
a,
Nelu
Grinberg
a,
Nizar
Haddad
a,
Nathan K.
Yee
a,
Frank
Roschangar
a,
Jinhua J.
Song
a,
Marisa C.
Kozlowski
*b and
Chris H.
Senanayake
a
b,
Jolaine
Savoie
a,
Bo
Qu
a,
Xingzhong
Zeng
a,
Heewon
Lee
a,
Nelu
Grinberg
a,
Nizar
Haddad
a,
Nathan K.
Yee
a,
Frank
Roschangar
a,
Jinhua J.
Song
a,
Marisa C.
Kozlowski
*b and
Chris H.
Senanayake
a
aDepartment of Chemical Development US, Boehringer-Ingelheim Pharmaceuticals, Inc., 900 Ridgebury Road, Ridgefield, CT 06877, USA. E-mail: nick.desrosiers@boehringer-ingelheim.com; xudong.wei@boehringer-ingelheim.com
bDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA 19104-6323, USA. E-mail: marisa@sas.upenn.edu
First published on 19th May 2016
Abstract
The direct arylation of pyridine substrates using non-precious catalysts is underdeveloped but highly desirable due to its efficiency to access important motifs while being extremely cost-effective. The first nickel-catalyzed C-3 direct arylation of pyridine derivatives to provide a new approach to valuable 1-azafluorene pharmacophore frameworks was developed. This transformation is accomplished using air-stable nickel catalyst precursors combined with phenanthroline ligands and tolerates a variety of substituents. Computational studies suggest facile oxidative addition via the pyridinium form, deprotonation, and a subsequent carbo-nickelation cyclization. Nickel homolysis/recombination permits isomerization to the stereochemical array needed for the final elimination.
The direct arylation of pyridine derivatives is a highly efficient approach for the synthesis of various heteroaromatic building blocks.1 No pre-installment of a functional group on the pyridine core, such as a halogen or boron derivative, or stoichiometric metalating reagent is required to promote the arylation which translates into better step economy, cost-efficiency, as well as reduced waste.1,2 The possibility to achieve a pyridine direct arylation using a non-precious metal catalyst such as nickel is highly desirable since it is sustainable due to its natural abundance and its significant yearly production.3 Nickel is also inexpensive, potentially air-stable, and exhibits a distinctive reactivity.3
As a part of the Non-Precious Metal Consortium4 and the preparation of an active pharmaceutical ingredient, efforts were devoted toward the development of a sustainable synthesis of 1-azafluorenes. These heterocycles are valuable pharmacophores that exhibit a wide range of biological activities. For instance, indenopyridines have demonstrated efficacy as thrombin inhibitors, for arresting tumor cell growth, and for the treatment of inflammatory and central nervous system related diseases.5 These heterocycles also possess absorption and emission properties, which are widely utilized for the development of dyes6 and light-emitting materials.7 In addition, the reduced form, indeno-hexahydro-pyridine, can be found in the core of natural products such as haouamines that have potent anticancer activity.8 Given the versatility and numerous applications of 1-azafluorenes, it is surprising that little attention has been dedicated to the synthesis of these compounds.5,6,9 Herein, we describe the first nickel-catalyzed C-3 direct arylation of pyridine derivatives (Fig. 1) to provide a new approach to valuable 1-azafluorene pharmacophore frameworks. Computational studies and control experiments revealed key elements of the reaction pathway explaining the unprecedented reactivity of pyridinium ions.
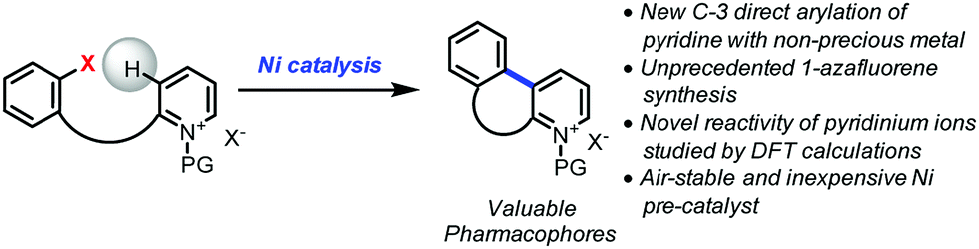 | ||
| Fig. 1 Nickel-catalyzed C-3 direct arylation of pyridine derivatives. | ||
Despite the numerous advantages of combining the effectiveness of a direct arylation with the cost-efficiency of nickel,10 there are relatively few examples of nickel-catalyzed direct arylation of pyridine derivatives. Notably, Chatani reported a C-2 selective C–H bond arylation of pyridines and quinolines via a nickel-catalyzed 1,2-addition with diorganozinc reagents.11 More recently, Doyle et al. described the C-2 arylation via Ni(II) π-allyl intermediates of N-acyl pyridinium ions with either boronic acids12 or aryl zinc halide reagents.13 In spite of these precedents, until now, the corresponding nickel-catalyzed C-3 direct arylations of pyridine derivatives have not been reported.14,15
Notwithstanding the absence of reported methodology for C-3 direct arylation of pyridines with nickel,16 we envisioned using this challenging approach as a general platform for the synthesis of 1-azafluorenes (Fig. 1). Even though substrate 1 seems poised for directed insertion to the C-3-position of pyridine after oxidative addition of the aryl-X group, initial experiments did not provide the desired 1-azafluorene product (Scheme 1). Reasoning that the nonaromatic dihydropyridine 2 could exploit an alternative carbo-metalation pathway, we were intrigued when it performed poorly under various reaction conditions during the optimization stage. Surprisingly, careful control studies revealed that the corresponding pyridinium ions 3 generated the 1-azafluorenes far more effectively. Mechanistic and computational studies revealed the source of the greater reactivity of the pyridinums providing useful design principles for future insertion–cyclization reactions.
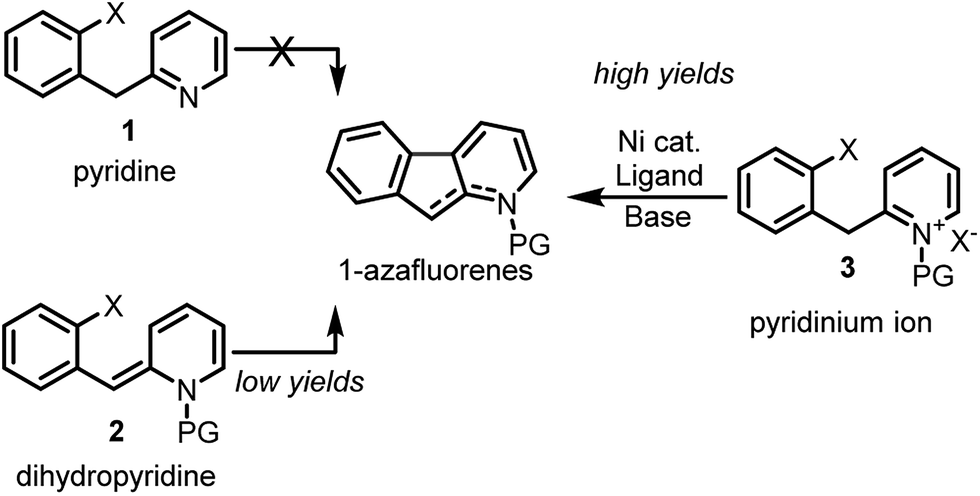 | ||
| Scheme 1 Nickel-catalyzed C-3 direct arylation to generate 1-azafluorenes. | ||
Further reaction optimization of the pyridinium ion showed that the N-benzyl derived 4a was optimal due to the reactivity it induces, ease of substrate preparation, and ease of cleavage of the protecting group. Screening (Table 1) of air-stable nickel(II) sources, including Ni(acac)2, (TMEDA)NiTolCl,17 and NiX2 (X = Br and Cl), revealed that NiCl2(dme) afforded superior yields with the use of PPh3 as ligand (entry 1–4). Further, bisphosphine and tridentate nitrogen ligands improved yields to 45 and 69%, respectively (entry 5–7). Inorganic bases including acetates and carbonates were not advantageous (entries 9–11).
|
|
||||
|---|---|---|---|---|
| Entry | Ni source | Base | Ligand (mol%) | Yieldb (%) |
| a Reaction conditions: Ni catalyst (5 mol%), ligand (6–12 mol%), base (5.0 equiv.) in DMF at 120 °C. b HPLC assay % yield determined by using a calibration curve. c binap = 2,2′-bis(diphenyl-phosphino)-1,1′-binapthalene. d iPr-pybox = 2,6-bis[isopropyl-2-oxazolin-2-yl]pyridine. | ||||
| 1 | Ni(acac)2 | DBU | PPh3 (12) | 3 |
| 2 | (TMEDA)NiTolCl | DBU | PPh3 (12) | 7 |
| 3 | NiBr2 | DBU | PPh3 (12) | 9 |
| 4 | NiCl2(DME) | DBU | PPh3 (12) | 21 |
| 5 | NiCl2(DME) | DBU | rac-binapc (6) | 45 |
| 6 | NiCl2(DME) | DBU | iPr-pyboxd (6) | 69 |
| 7 | NiCl2(DME) | DBU | Terpyridine (6) | 13 |
| 8 | NiCl2(DME) | DBU | 2,2′-Bipyridine (6) | 70 |
| 9 | NiCl2(DME) | NaOAc | L1 (6) | 0 |
| 10 | NiCl2(DME) | KOAc | L1 (6) | 38 |
| 11 | NiCl2(DME) | K2CO3 | L1 (6) | 75 |
| 12 | NiCl2(DME) | DBU | L1 (6) | 98 |
| 13 | NiCl2(DME) | DBU | L2 (6) | 92 |
| 14 | NiCl2(DME) | DBU | L3 (6) | 32 |
| 15 | NiCl2(DME) | DBU | L4 (6) | 57 |
| 16 | NiCl2(DME) | DBU | L5 (6) | 87 |
| 17 | NiCl2(DME) | DBU | L6 (6) | 22 |
| 18 | NiCl2(DME) | DBU | L7 (6) | 12 |
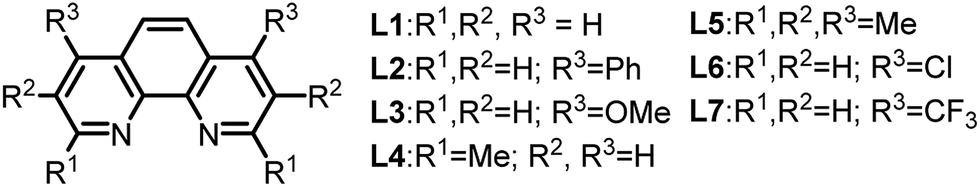
|
||||
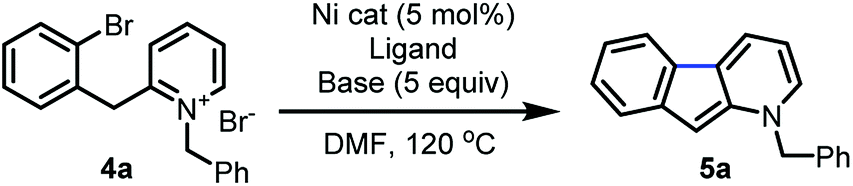
Among all ligands tested in presence of DBU, phenanthroline derivatives were optimal (entry 12–18) with nickel(II) catalysts, and 1,10-phenanthroline gave rise to 98% yield (entry 12). Introduction of electron-donating, withdrawing or aromatic substituents on 1,10-phenanthroline did not enhance the rate or yield of the reaction.
With the optimized reaction conditions in hands, the substrate scope was surveyed (Scheme 2). Substitution with electron-withdrawing groups at C-6 such as esters (5b and 5c), amides (5d), nitriles (5e) and sulfonamides (5f) was well tolerated (77–95%). Notably, the aryl iodides underwent full conversion more rapidly (1–3 h) than the corresponding aryl bromides (8–12 h). 6,7-Difluoro and 7-methoxy substrates 5g and 5h provided the desired 1-azafluorenes with moderate yields. Substitution of the pyridine ring (5i) at C-4 also was well tolerated. This method was found to be specific to azafluorenes since a longer tether did not afford the fused 6-membered ring 5j with high efficiency.
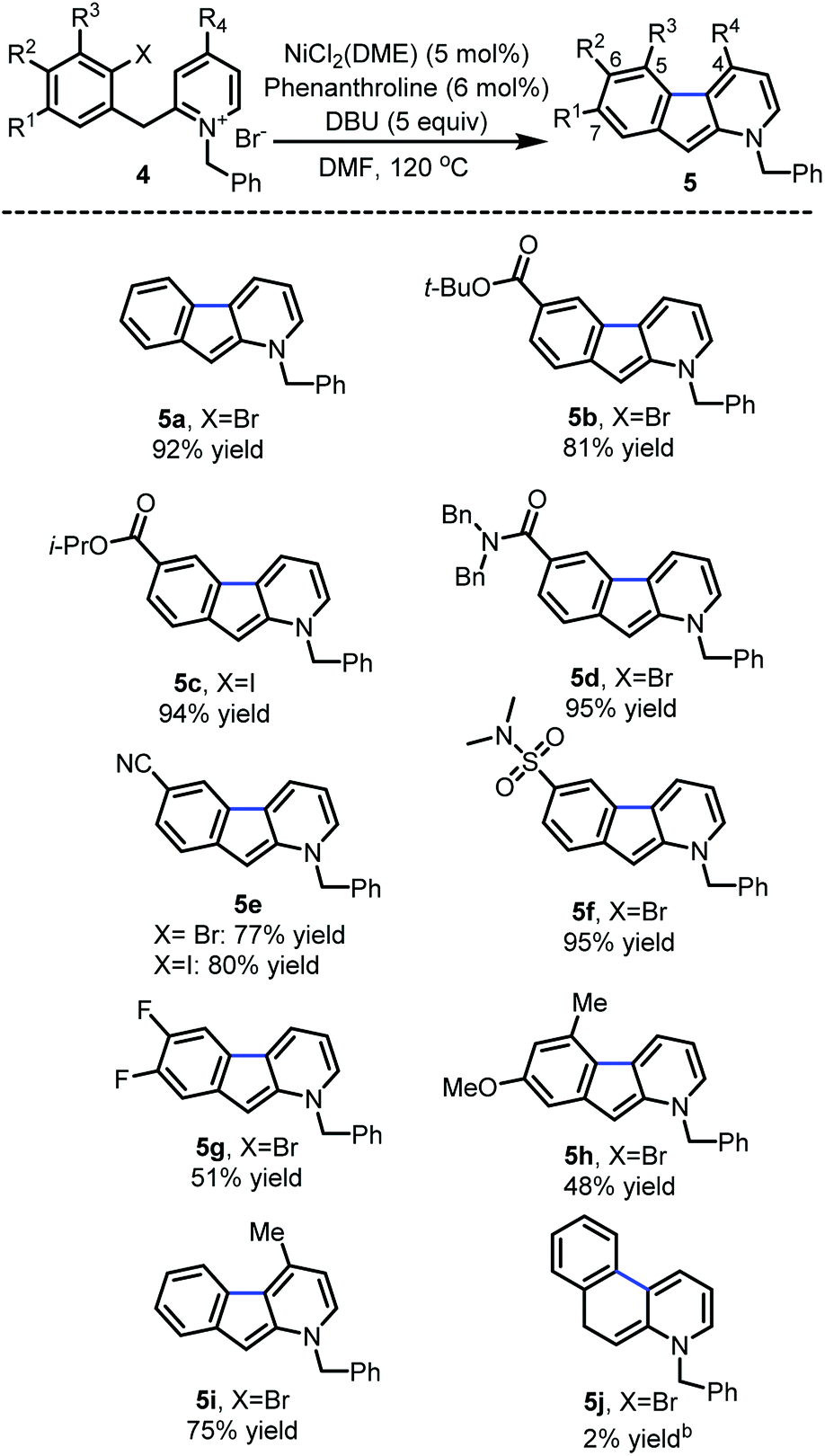 | ||
| Scheme 2 Reaction scope of C-3 arylation of pyridinium ionsa. aReaction conditions: Ni catalyst (5 mol%), 1,10-phenanthroline (6 mol%), DBU (5.0 equiv.) in DMF at 120 °C for 1–12 h; isolated yield determined after flash chromatography. bDetermined by 1H NMR assay yield using dimethyl fumarate as an internal standard. | ||
The usefulness of cyclized N-benzyl derivatives 5 was established by performing a straightforward deprotection to afford the corresponding 1-azafluorene 6 or the indeno-hexahydro-pyridine 7 (Scheme 3). Specifically, hydrogenolysis of the benzyl protecting group under a low pressure of hydrogen (100 psi) at room temperature with Pd/C provided 6 in 91% isolated yield. At higher hydrogen pressure and temperature, reduction of the pyridine ring can also be accomplished to furnish cis-fused piperidine 7 in 82% yield.
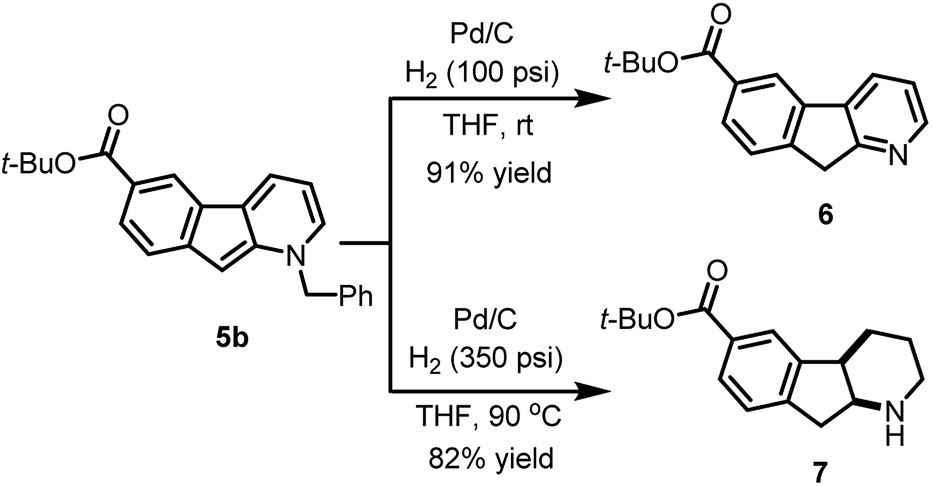 | ||
| Scheme 3 Deprotection and hydrogenation of protected 1-azafluorene. | ||
In terms of the mechanism, various pathways are reported to achieve a direct arylation and several possibilities were considered including concerted deprotonative metalation, Ni(I) or Ni(II) single electron abstraction of the halide to initiate a radical sequence, and carbo-metalation process. As acid sources are present, the protonation state (pyridinium ion or dihydropyridine) was also evaluated.
DFT studies were initiated using bipyridine as ligand and the neutral version of N-methyl 2-(2-bromobenzyl)pyridine as a model substrate.18,19Fig. 2 illustrates several of the pathways identified.20 Ligand dissociation (COD) followed by coordination of the Ni(0) to the aryl ring of the dihydropyridine generates adduct A, which is subject to two distinct fates. An energetically accessible oxidative addition (barrier = 20.3 kcal mol−1 from uncoordinated substrates) viaA-TS (blue pathway) leads to the much more thermodynamically favorable Ni(II) adduct B.21 Deprotonative metalation pathways have been reported in related systems,22 and transition states were located for the bromide-(B′-TS-Br, shown) or base-mediated (not shown, see ESI†) deprotonation from B. However, all deprotonative metalation pathways were substantially higher in energy (>12.2 kcal mol−1) relative to migratory insertion viaB-TS (5.4 kcal mol−1) which gives rise to the C-anti adduct (see ESI† for energetics of the deprotonative metalation pathway). However, the lack of a hydrogen syn to the Ni center in C-anti disfavors β-hydride elimination to form the product from this species.
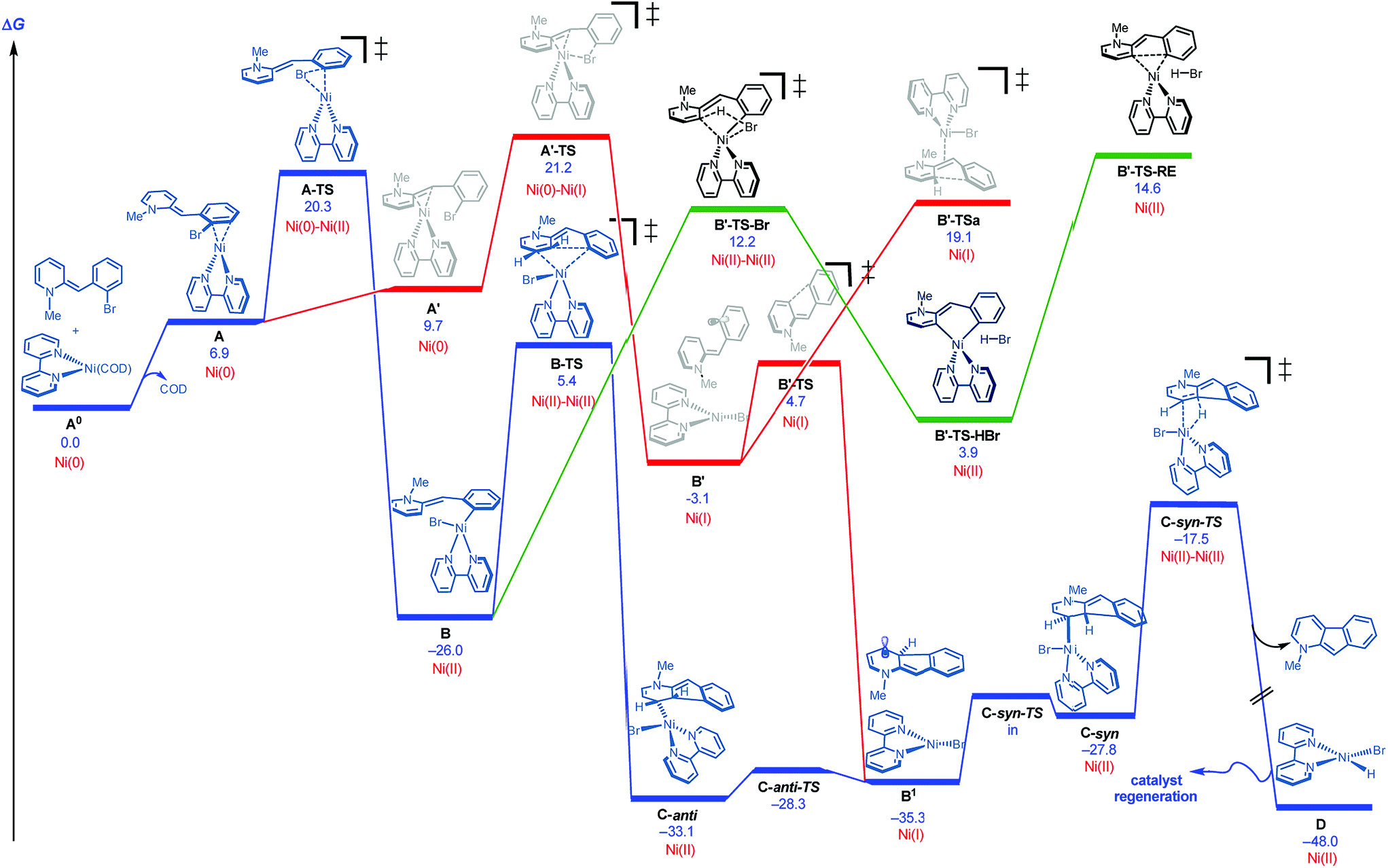 | ||
| Fig. 2 Cyclization pathways commencing from neutral dihydripyridine (free energies are given in kcal mol−1 for calculations with UM06/6-311+G(d,p)-SMD-nitromethane//UB3LYP/6-31G(d)-gas). | ||
These results together with the finding that 1-chloro-2,4-dinitrobenzene, a single electron transfer inhibitor,23 was deleterious to the reaction,24 motivated us to examine single electron transfer pathways. From a different coordination complex (A′), halide abstraction by the Ni(0) (viaA′-TS) was found to be higher in energy (1–5 kcal mol−1) relative to oxidative addition (A-TS) depending on the method used (see ESI†). On the other hand, halide abstraction commencing with Ni(I)25 was found to be much higher in energy and hence not viable (see ESI†). At this juncture, distinction of the Ni(0) oxidative addition vs. halide abstraction was not obvious from the calculations alone. In addition, neither of these pathways satisfactorily explained the requirement of a proton source: either the pyridinium substrate needs to be employed or DBU·HBr needs to be introduced to the neutral dihydropyridine.26
On this basis, further modeling was undertaken starting with the cationic (pyridinium) version of the substrate (Fig. 3). Notably, oxidative addition (A2-TS, 13.5 kcal mol−1) was found to be far more facile than halide abstraction (A″-TS, >38 kcal mol−1, not shown, see ESI†) when the charged species is considered. Furthermore, oxidative addition of the charged substrate is more facile than oxidative addition of the neutral analog (13.5 vs. 20.3 kcal mol−1). It appears that several effects give rise to this behavior. The neutral dihydropyridine is planar as a result of conjugation, but this conjugation is disrupted in A-TS due to steric interactions with the catalyst during oxidation addition. In contrast, the pyridinium is not conjugated and is not planar in its starting form; as a result, much less strain builds up in A2-TS (see ESI† for structures). Thus, the oxidative addition step is favored via the protonated pyridinium form, and then a facile deprotonation occurs, so that intramolecular migratory insertion can occur via the lower energy pathway B-TS (Fig. 2). Migratory insertion on cationic oxidative addition adduct B′ in Fig. 3 causes a loss of aromaticity in the pyridinium ring and is precluded energetically (transition state barrier from B′ is 49.6 kcal mol−1; not shown, see ESI†) vs. migratory insertion of neutral oxidative addition adduct B (31.1 kcal mol−1viaB-TS in Fig. 2).
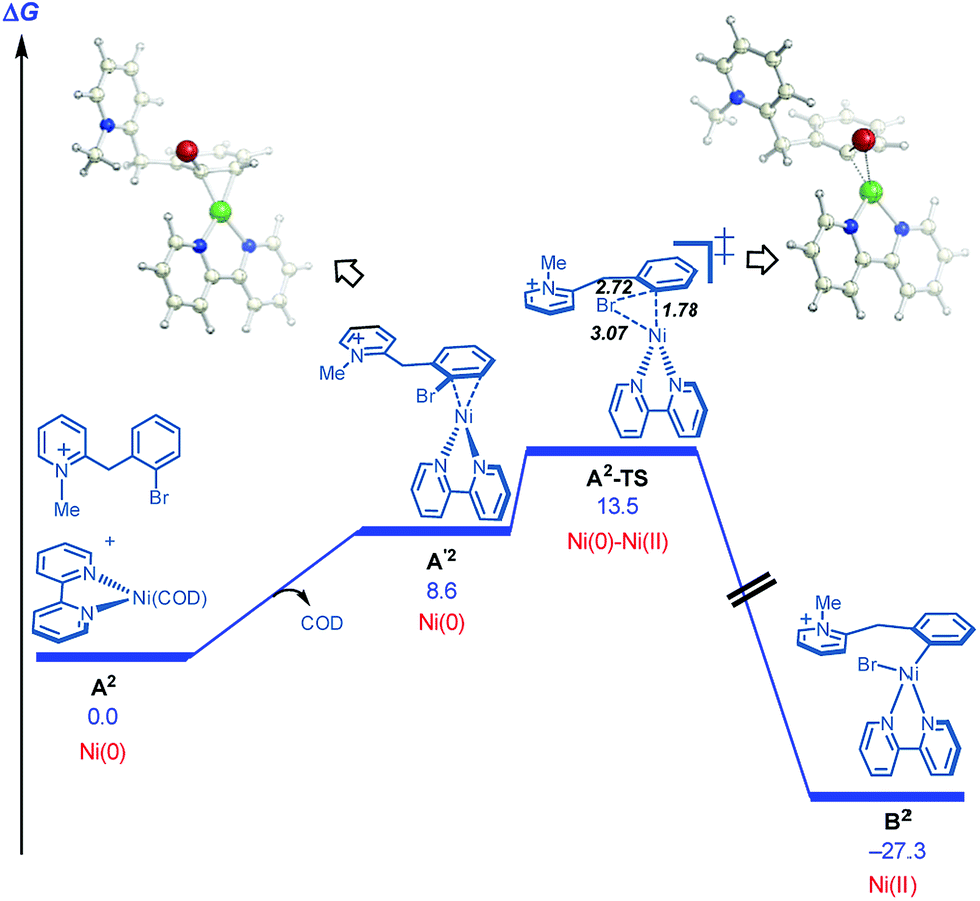 | ||
| Fig. 3 Cyclization pathways commencing from the pyridinium iona. aFree energies are given in kcal mol−1 for calculations with UM06/6-311+G(d,p)-SMD-nitromethane//UB3LYP/6-31G(d)-gas phase. | ||
The remaining issue to resolve is how C-anti can undergo β-hydride elimination to the product. Due to the highly conjugated nature of the radical produced, calculations show that homolysis of the Ni–C bond of C-anti is facile leading to B1 (Fig. 2). Isomerization and recombination leads to C-syn, which can, in turn, undergo low barrier β-hydride elimination (viaC-syn-TS) to the product. This result also accounts for why an electron transfer inhibitor is deleterious.23
Overall, a carbo-nickelation accounts for the observed direct arylation products (Scheme 4). A notable feature is that substrate protonation distorts conjugation of the substrate positioning the requisite C–Br in a less sterically encumbered environment which facilitates oxidative addition. Subsequent deprotonation is required for migratory insertion. The ability of nickel to undergo facile homolysis provides a low energy pathway for a cis to trans isomerization, which is needed for β-hydride elimination to product. Reduction of the Ni(II) pre-catalyst and of the Ni(II) hydride species could occur at 120 °C via either oxidation of DBU27 or oxidative decomposition of DMF.28 An alternative pathway for nickel reduction could be a redox process between Ni(II) and phenanthroline to generate Ni(I)19a which can then react with the aryl bromide to afford B2 after a bimetallic oxidative addition.29
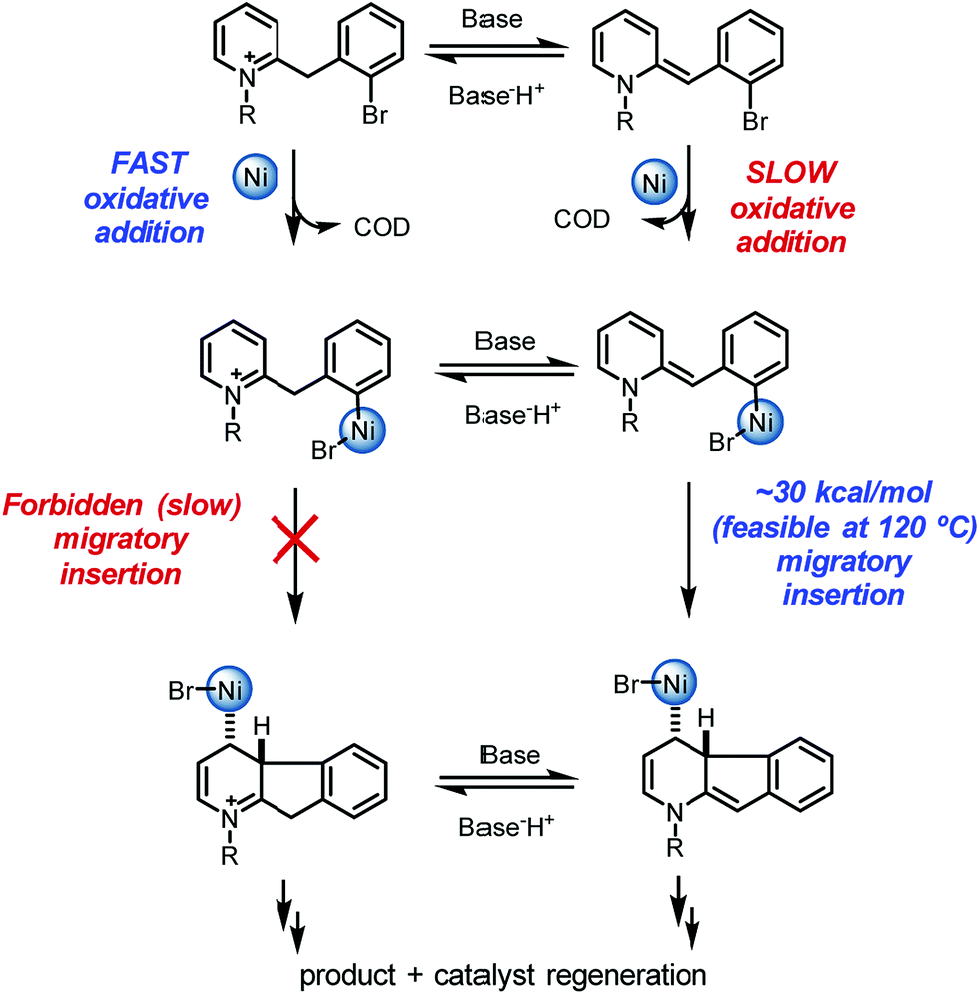 | ||
| Scheme 4 Overall mechanism of the pyridinium direct arylation. | ||
In conclusion, an unprecedented C-3 direct arylation of pyridinium ions was developed using air-stable nickel catalyst precursors to achieve a practical synthesis of 1-azafluorenes. This strategy is compatible with a variety of substituents either on the aryl halide or pyridine core. The N-benzyl protected 1-azafluorenes readily undergo hydrogenolysis and reduction to afford valuable pharmacophores. This method represents a significant advance in non-precious metal catalysis considering that prior methods only permit C-2 direct arylation. Computational studies combined with control experiments revealed that the oxidation addition step is sensitive to steric hindrance which can be mitigated by use of the pyridinium ion. The subsequent cyclization takes place only after isomerization under basic conditions. Finally, the homolysis/recombination provides the required configuration for β-hydride elimination. These mechanistic findings provided principles for the design of new nickel catalyzed reactions. Future reports will include results on multi-kilo scale using this methodology and efforts towards achieving the intermolecular version of this transformation.
Acknowledgements
We thank the Boehringer-Ingelheim-Pfizer-Glaxo-SmithKline-Abbvie Non-Precious Metal Catalysis Consortium for general discussions in this area. 3D structures of Fig. 2 were generated using CYLview.30 We thank the National Institutes of Health (GM087605 to M. C. K.) for financial support. Computational support was provided by XSEDE on SDSC Gordon (TG-CHE120052).Notes and references
- For reviews on direct arylation of (hetero)arenes, see: (a) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (b) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed; (c) J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, 20–40 CrossRef CAS; (d) R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17–117 CrossRef CAS.
- (a) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed; (b) L.-C. Campeau and K. Fagnou, Chem. Soc. Rev., 2007, 36, 1058–1068 RSC.
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 299–309 CrossRef CAS PubMed; (b) Modern Organonickel Chemistry, ed. Y. Tamaru, Wiley-VCH, Weinheim, 2005 Search PubMed; (c) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed.
- Non-Precious Metal Consortium established in 2012 (see Acknowledgements section).
- (a) K. J. Stauffer, P. D. Williams, H. G. Selnick, P. G. Nantermet, C. L. Newton, C. F. Homnick, M. M. Zrada, S. D. Lewis, B. J. Lucas, J. A. Krueger, B. L. Pietrak, E. A. Lyle, R. Singh, C. Miller-Stein, R. B. White, B. Wong, A. A. Wallace, G. R. Sitko, J. J. Cook, M. A. Holahan, M. Stranieri-Michener, Y. M. Leonard, J. J. Lynch Jr, D. R. McMasters and Y. Yan, J. Med. Chem., 2005, 48, 2282–2293 CrossRef CAS PubMed; (b) B. E. Evans, K. F. Gilbert, J. M. Hoffman and K. E. Rittle, GB2355457, 2000; (c) A. L. Salzman, WO2007/117289, 2007; (d) C. Hamblett, S. Kattar, D. Mampreian, J. Method, T. Miller and P. Tempest, WO2007/136605, 2007; (e) N. S. Prostakov, A. T. Soldatenkov, V. O. Fedorov, V. M. Bagdadi and M. M. Borisov, Pharm. Chem. J., 1987, 21, 408–411 CrossRef.
- (a) N. S. Prostakov, A. T. Soldatenkov, V. O. Fedorov, S. Mobio and M. A. Galiullin Chem, Heterocycl. Compd., 1980, 16, 1149–1153 CrossRef; (b) D. G. Krotko, K. V. Fedotov and A. I. Tolmachev, Dyes Pigm., 2005, 65, 183–189 CrossRef CAS.
- (a) Y. G. Kim, Y. J. Cho, H. J. Kwon, B. O. Kim, S. M. Kim and S. S. Yoon, WO2010/114266, 2010; (b) T. Takasu, R. Nomura, S. Shitagaki and S. Seo, WO2009/119869, 2009.
- L. Garrido, E. Zubia, M. J. Ortega and J. Salva, J. Org. Chem., 2003, 68, 293 CrossRef CAS PubMed.
- (a) N. S. Prostakov, A. A. Obynochnyi, N. M. Kolyadina and A. T. Soldatenkov, Russ. Chem. Rev., 1997, 66, 121–138 CrossRef; (b) N. S. Prostakov, Russ. Chem. Rev., 1969, 38, 774–782 CrossRef; (c) G. A. Urbina, Synth. Commun., 1979, 9, 245–250 CrossRef; (d) C. Mayor and C. Wentrup, J. Am. Chem. Soc., 1975, 97, 7467–7480 CrossRef CAS.
- For a review on nickel-catalyzed C–H bond functionalization reactions, see: X.-H. Cai and B. Xie, ARKIVOC, 2015, 184–211 CAS.
- (a) M. Tobisu, I. Hyodo and N. Chatani, J. Am. Chem. Soc., 2009, 131, 12070–12071 CrossRef CAS PubMed; (b) I. Hyodo, M. Tobisu and N. Chatani, Chem.–Asian J., 2012, 7, 1357–1365 CrossRef CAS PubMed.
- (a) T. J. A. Graham, J. D. Shields and A. G. Doyle, Chem. Sci., 2011, 2, 980–984 RSC; (b) J. D. Shields, D. T. Ahneman, T. J. A. Graham and A. G. Doyle, Org. Lett., 2014, 16, 142–145 CrossRef CAS PubMed; (c) K. T. Sylvester, K. Wu and A. G. Doyle, J. Am. Chem. Soc., 2012, 134, 16967–16970 CrossRef CAS PubMed.
- S. T. Chau, J. P. Lutz, K. Wu and A. G. Doyle, Angew. Chem., Int. Ed., 2013, 52, 9153–9156 CrossRef CAS PubMed.
- For palladium-catalyzed C-3 inter-molecular arylations of pyridine derivatives, see: (a) P. Guo, J. M. Joo, S. Rakshit and D. Sames, J. Am. Chem. Soc., 2011, 133, 16338–16341 CrossRef CAS PubMed; (b) M. Wasa, B. T. Worrell and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 1275–1277 CrossRef CAS PubMed; (c) M. Ye, G.-L. Gao, A. J. F. Edmunds, P. A. Worthington, J. A. Morris and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19090–19093 CrossRef CAS PubMed; (d) G. L. Gao, W. Xia, P. Jain and J.-Q. Yu, Org. Lett., 2016, 18, 744–747 CrossRef CAS PubMed.
- For examples of palladium-catalyzed C-3 intra-molecular direct arylations of pyridine derivatives, see: (a) S. K. Singh, A. L. Ruchelman, T.-K. Li, A. Liu, L. F. Liu and E. J. LaVoie, J. Med. Chem., 2003, 46, 2254–2257 CrossRef CAS PubMed; (b) C. Meyers, G. Rombouts, K. T. J. Loones, A. Coelho and B. U. W. Maes, Adv. Synth. Catal., 2008, 350, 465–470 CrossRef CAS.
- For nickel-catalyzed alkyne insertion into pyridines, see (a) C.-C. Tsai, W.-C. Shih, C.-H. Fang, C.-Y. Li, T.-G. Ong and G. P. A. Yap, J. Am. Chem. Soc., 2010, 132, 11887–11889 CrossRef CAS PubMed; (b) Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666–13668 CrossRef CAS PubMed; (c) Y. Nakao, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 2448–2449 CrossRef CAS PubMed.
- (a) J. D. Shields, E. E. Gray and A. G. Doyle, Org. Lett., 2015, 17, 2166–2169 CrossRef CAS PubMed; (b) J. Magano and S. Monfette, ACS Catal., 2015, 5, 3120–3123 CrossRef CAS.
- All calculations were carried out using Gaussian09, see: M. J. Frisch, et al., Gaussian09, Revision A. 02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed, see ESI† for full reference and computational details.
- For examples of mechanistic/DFT studies on nickel-catalyzed systems, see: (a) G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175–13183 CrossRef CAS PubMed; (b) X. Lin and D. L. Phillips, J. Org. Chem., 2008, 73, 3680–3688 CrossRef CAS PubMed; (c) B.-L. Lin, L. Liu, Y. Fu, S.-W. Luo, Q. Chen and Q.-X. Guo, Organometallics, 2004, 23, 2114–2123 CrossRef CAS.
- For a recent computational report on nickel catalysis from our group, see: O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed.
- Similar energetics were observed when iodine-substrate (see ESI†).
- (a) Y. Aihara and N. Chantani, J. Am. Chem. Soc., 2014, 136, 898–901 CrossRef CAS PubMed; (b) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2014, 136, 1789–1792 CrossRef CAS PubMed; (c) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2015, 137, 4924–4927 CrossRef CAS PubMed.
- (a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442–7445 CrossRef CAS PubMed; (b) M. A. Prasad and M. V. Sangaranarayanan, Electrochim. Acta, 2005, 51, 242–246 CrossRef CAS; (c) G. A. Russell, E. G. Janzen and E. T. Strom, J. Am. Chem. Soc., 1963, 86, 1807–1814 CrossRef.
- The use of 1-chloro-2,4-dinitrobenzene (0.5 equiv.) under optimal conditions led to a complete inhibition of product formation (0% yield).
- (a) S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192–16197 CrossRef CAS PubMed; (b) C. Liu, S. Tang, D. Liu, J. Yuan, L. Zheng, L. Meng and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 3638–3641 CrossRef CAS PubMed.
- When dihydropyridine 2 (Scheme 1, PG = N-benzyl) was treated under optimal conditions, 15% yield was obtained. When 1 equivalent of DBU·HBr was used as an additive, the desired product was obtained with 77% yield. When 1 equivalent of TBAB was used, 34% was obtained.
- M. C. Henningsen, S. Jeropoulos and E. H. Smith, J. Org. Chem., 1989, 54, 3015–3018 CrossRef CAS.
- (a) G. H. Hugar and S. T. Nandibewoor, Indian J. Chem., Sect. A, 1993, 32, 1056–1059 Search PubMed; (b) I. Pastoriza-Santos and L. M. Lis-Marzan, Langmuir, 1999, 15, 948–951 CrossRef CAS.
- J. Breitenfeld, J. Ruiz, M. D. Wodrich and X. Hu, J. Am. Chem. Soc., 2013, 135, 12004–12012 CrossRef CAS PubMed.
- C. Y. Legault, CYLview, version 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc01457g |
| This journal is © The Royal Society of Chemistry 2016 |