
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed cyclization of alkyne-nitriles with organoboronic acids involving anti-carbometalation of alkynes†
Xingjie
Zhang
,
Xin
Xie
and
Yuanhong
Liu
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: yhliu@sioc.ac.cn
First published on 19th May 2016
Abstract
A nickel-catalyzed regioselective addition/cyclization of o-(cyano)phenyl propargyl ethers with arylboronic acids has been developed, which provides an efficient protocol for the synthesis of highly functionalized 1-naphthylamines with wide structural diversity. The reaction is characterized by a regioselective and anti-addition of the arylboronic acids to the alkyne and subsequent facile nucleophilic addition of the resulting alkenylmetal to the tethered cyano group. Mechanistic studies reveal that a Ni(I) species might be involved in the catalytic process.
Introduction
Transition-metal-catalyzed cascade reactions consisting of multiple carbometalation steps have attracted considerable attention in organic synthesis since these processes enable the rapid assembly of complex structures in an efficient, atom-economical and green manner.1 Among these reactions, organoboron compounds are one of the most widely used reagents, not only due to their chemical stability and ready availability, but also because they can undergo a series of addition reactions to unsaturated compounds such as alkynes, dienes, enones, aldehydes/ketones, nitriles and isocyanates etc. in the presence of a transition metal catalyst, especially Rh, Pd or Ni complexes.2 The development of cascade reactions by combining different types of these elemental reactions is undoubtedly important and attractive. In this regard, cascade reactions involving the addition of organoboron compounds to alkynes as the initial step have been realized mainly through Rh-![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1b,c,3 or Pd-catalysis,4 as reported by Murakami, Hayashi, Lu and other groups. These catalytic reactions generally proceed by syn-1,2-addition of the organometal species generated through transmetalation between the organoboron and the metal complex across the carbon–carbon triple bond, followed by nucleophilic attack of the resulting alkenylmetal on the remaining electrophile (Scheme 1, eqn (1)). So far most of the reported reactions proceed via formation of regioisomer A in which the R2 group of R2B(OH)2 locates on a carbon adjacent to the alkyne terminus R1, leading to an exo-alkene upon cyclization3,4 (Scheme 1, eqn (1)). Cyclizations involving the regioselective formation of the alkenylmetal with a metal α-to the R1 substituent such as syn-B are quite rare5 (Scheme 1, eqn (2)), possibly because the subsequent cyclization process will involve a highly strained transition state. Thus, the development of new cyclization systems with controlled regiochemistry towards B is highly challenging. During our studies on nickel-catalyzed reactions, we found that such a transformation could be achieved by the addition of organoboron compounds to benzene-tethered alkyne-nitriles utilizing nickel as the catalyst, possibly through the isomerization of syn-B to anti-B. Herein, we report the first example of a nickel-catalyzed carboarylative cyclization of alkyne-nitriles with organoboronic acids involving regioselective and anti-carbonickelation of alkynes, which provides an efficient protocol for the synthesis of highly functionalized 1-naphthylamines. In addition, mechanistic studies revealed that Ni(I) species6 rather than Ni(II) species were involved as the key intermediates, which has not been reported in Ni-catalyzed boron addition reactions.
1b,c,3 or Pd-catalysis,4 as reported by Murakami, Hayashi, Lu and other groups. These catalytic reactions generally proceed by syn-1,2-addition of the organometal species generated through transmetalation between the organoboron and the metal complex across the carbon–carbon triple bond, followed by nucleophilic attack of the resulting alkenylmetal on the remaining electrophile (Scheme 1, eqn (1)). So far most of the reported reactions proceed via formation of regioisomer A in which the R2 group of R2B(OH)2 locates on a carbon adjacent to the alkyne terminus R1, leading to an exo-alkene upon cyclization3,4 (Scheme 1, eqn (1)). Cyclizations involving the regioselective formation of the alkenylmetal with a metal α-to the R1 substituent such as syn-B are quite rare5 (Scheme 1, eqn (2)), possibly because the subsequent cyclization process will involve a highly strained transition state. Thus, the development of new cyclization systems with controlled regiochemistry towards B is highly challenging. During our studies on nickel-catalyzed reactions, we found that such a transformation could be achieved by the addition of organoboron compounds to benzene-tethered alkyne-nitriles utilizing nickel as the catalyst, possibly through the isomerization of syn-B to anti-B. Herein, we report the first example of a nickel-catalyzed carboarylative cyclization of alkyne-nitriles with organoboronic acids involving regioselective and anti-carbonickelation of alkynes, which provides an efficient protocol for the synthesis of highly functionalized 1-naphthylamines. In addition, mechanistic studies revealed that Ni(I) species6 rather than Ni(II) species were involved as the key intermediates, which has not been reported in Ni-catalyzed boron addition reactions.
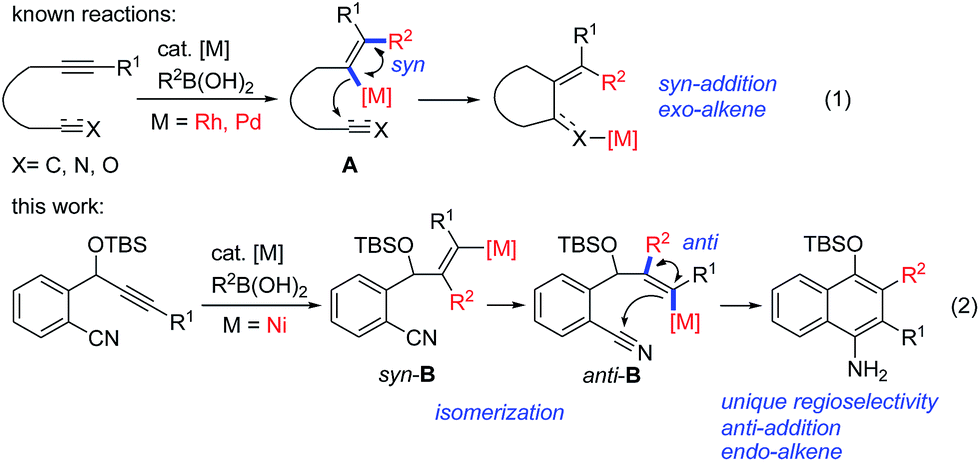 | ||
| Scheme 1 Metal-catalyzed cascade addition/cyclization reactions. | ||
Results and discussion
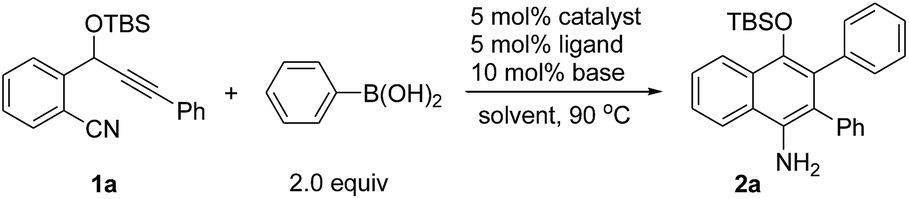
We chose the nickel-catalyzed reaction of o-(cyano)phenyl propargyl ether71a and phenylboronic acid as a model reaction for the optimization of the reaction conditions. Initially, we examined the reactions in the presence of Ni(COD)2 and various phosphine ligands such as PPh3 in 1,4-dioxane at 90 °C. However, only a trace of the desired cyclization product was observed, along with some byproducts (Table 1, entry 1). Replacing Ni(COD)2 with a Ni(II) complex, Ni(acac)2·2H2O (acac = acetylacetonate), afforded the cyclized product 4-OTBS-substituted 1-naphthylamine 2a, albeit in only 17–18% yields (entries 2–3). To our delight, the addition of 10 mol% of tBuOK as a base improved the yield of 2a dramatically to 66% within a short reaction time (entry 4). The results suggest that a base is necessary for this reaction, possibly for promoting the transmetalation step by formation of a borate8 with the organoboronic acid. The structure of 2a also revealed that arylation in the initial step occurred regioselectively on the alkyne carbon that is closer to the OTBS group. Subsequently, the effects of bases, phosphine ligands and solvents were evaluated. Of the various bases, Cs2CO3 gave the best result (73%, entry 5). Increasing the catalytic loading of Cs2CO3 to 20 mol% had little effect on the yield of 2a (entry 8). However, when a stoichiometric amount of Cs2CO3 was used, the yield was reduced rapidly (entry 9). It was remarkable that, unlike the use of more than one equivalent of base in most of the transition metal-catalyzed reactions involving organoborons, here only catalytic amounts of base were needed. Increasing the catalyst loading did not improve the yield of the product (entry 10). Triarylphosphine ligands and the N-heterocyclic carbene ligand IPr (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene) were also effective, while ligands such as PPh2Me and PCy3 were less efficient (entries 11–17). Changing the solvent to THF or toluene afforded 2a in satisfactory yields of 64–68% (entries 18–19). Addition of one equivalent of H2O as a promoter or proton source did not afford a better result (entry 20). Ni(acac)2 also catalyzed the reaction efficiently (entry 21). When Ni(COD)2 was used as the catalyst, only trace amounts of 2a were obtained (entry 22). Without the phosphine ligand, the reaction also proceeded to afford 2a in 65% yield, albeit with a longer reaction time (entry 23). Without a nickel catalyst, no reaction occurred (entry 24). On the basis of the above optimization studies, the reaction conditions shown in Table 1, entry 5 were chosen as the best conditions.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Solvent | Time [h] | Yielda [%] |
| a Isolated yields. The yields of the recovered 1a were shown in parentheses. b 10 mol% of the ligand was used. c 20 mol% of Cs2CO3 was used. d 1.0 equiv. of Cs2CO3 was used. e 10 mol% Ni(acac)2·2H2O, 10 mol% P(p-CF3C6H4)3 and 20 mol% Cs2CO3 were used. f One equiv. of H2O was added. | ||||||
| 1 | Ni(COD)2 | PPh3b | — | 1,4-Dioxane | 3 | Trace |
| 2 | Ni(acac)2·2H2O | PPh3 | — | 1,4-Dioxane | 24 | 18 |
| 3 | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | — | 1,4-Dioxane | 24 | 17 |
| 4 | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | t BuOK | 1,4-Dioxane | 4 | 66 |
| 5 | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | Cs2CO3 | 1,4-Dioxane | 3 | 73 |
| 6 | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | CsF | 1,4-Dioxane | 6 | 67 |
| 7 | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | K2CO3 | 1,4-Dioxane | 10 | 31 |
| 8 | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | Cs2CO3c | 1,4-Dioxane | 3 | 68 |
| 9 | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | Cs2CO3d | 1,4-Dioxane | 10 | 8 (59) |
| 10e | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | Cs2CO3 | 1,4-Dioxane | 3 | 74 |
| 11 | Ni(acac)2·2H2O | PPh3 | Cs2CO3 | 1,4-Dioxane | 5 | 69 |
| 12 | Ni(acac)2·2H2O | P(p-MeC6H4)3 | Cs2CO3 | 1,4-Dioxane | 4 | 68 |
| 13 | Ni(acac)2·2H2O | P(C6F5)3 | Cs2CO3 | 1,4-Dioxane | 5 | 63 |
| 14 | Ni(acac)2·2H2O | PPh2Me | Cs2CO3 | 1,4-Dioxane | 7 | 45 |
| 15 | Ni(acac)2·2H2O | PCy3 | Cs2CO3 | 1,4-Dioxane | 17 | 55 |
| 16 | Ni(acac)2·2H2O | IPr | Cs2CO3 | 1,4-Dioxane | 6 | 64 |
| 17 | Ni(acac)2·2H2O | IPr | t BuOK | 1,4-Dioxane | 7 | 62 |
| 18 | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | Cs2CO3 | THF | 8 | 64 |
| 19 | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | Cs2CO3 | Toluene | 3 | 68 |
| 20f | Ni(acac)2·2H2O | P(p-CF3C6H4)3 | Cs2CO3 | 1,4-Dioxane | 6 | 57 |
| 21 | Ni(acac)2 | P(p-CF3C6H4)3 | Cs2CO3 | 1,4-Dioxane | 3 | 72 |
| 22 | Ni(COD)2 | P(p-CF3C6H4)3b | Cs2CO3 | 1,4-Dioxane | 9 | Trace |
| 23 | Ni(acac)2·2H2O | — | Cs2CO3 | 1,4-Dioxane | 5 | 65 |
| 24 | — | P(p-CF3C6H4)3 | Cs2CO3 | 1,4-Dioxane | 10 | (99) |
Next, we proceeded to investigate the scope of this new cascade addition/cyclization reaction catalyzed by Ni(acac)2·2H2O. The reactivity of various organoboronic acids was first examined using 1a as a reaction partner (Table 2). During this process, we found that the 5 mol% catalyst loading was not effective in some cases and thus 10 mol% Ni(acac)2·2H2O, 10 mol% P(p-CF3C6H4)3 and 20 mol% Cs2CO3 were used in most of the cases to achieve better product yields. As shown in Table 2, a wide range of diversely substituted aryl- or heteroaryl-boronic acids were suitable for this reaction, leading to the desired 1-naphthylamines 2a–2s in generally good to high yields. Arylboronic acids bearing electron-donating groups such as p-Me, p-tBu and p-MeO or electron-withdrawing groups such as p-F, p-Cl, p-CF3, p-CN, p-CO2Et and p-Ac on the aryl ring underwent the cyclization smoothly to provide the corresponding 1-naphthylamines 2b–2j in 64–77% yields, and these functional groups were well tolerated during the reaction. Of note is that the CN and Ac groups remained intact under the reaction conditions, and no nickel-catalyzed boron additions to these groups were observed. The results indicated that electron-poor or -rich aryl substituents on the arylboronic acid had little influence on the yields of products 2. The sterically demanding o-MeO substituted arylboronic acid afforded 2k with a longer reaction time and a lower yield of 47%, indicating that the reaction is markedly influenced by steric effects. Arylboronic acids with –MeO or –Cl substituents at the 3-, 3,4- or 3,5-positions of the phenyl ring, or with a biphenyl or 2-naphthyl ring transformed into products 2l–2m and 2o–2r efficiently in good yields. The use of 2-fluorophenylboronic acid gave 2n in 41% yield. 2-Thienylboronic acid also participated in this cascade reaction, albeit with a lower yield of 2s. However, when alkylboronic acids such as n-butylboronic acid were employed, no desired product was obtained.

The scope of o-(cyano)phenyl propargyl ethers was then examined (Table 3). A variety of electron-donating and -withdrawing groups on the aryl rings at the alkyne terminus were found to be compatible, such as p-Me, p-OMe, p-F, p-Cl, p-CF3 and p-CO2Me substituents, and the corresponding products 2t–2y were formed in 61–76% yields. Interestingly, in contrast to the results of the reaction of 1a with 2-thienylboronic acid, the presence of a 2-thienyl or 3-benzothienyl ring as the alkyne terminal did not have much influence on the reaction, and the corresponding products 2z and 2za were obtained in high yields of 80% and 74%, respectively. A 9H-fluorene substituent, which is a very useful unit in organofunctional materials, was also successfully incorporated into the product 2zb in 70% yield. Alkenyl or alkyl-substituted alkynes, such as cyclohexenyl, cyclopropyl and propyl-substituted ones, however, afforded the desired products 2zc–2ze in low yields of 23–35%. The structure of the 1-naphthylamine product was unambiguously confirmed by X-ray crystallographic analysis of 2o.9 1-Naphthylamines are important structural motifs found in a variety of biologically active substances. They also act as useful building blocks in synthetic chemistry and the dyestuffs industry. However, efficient methods for their synthesis are quite limited.10 Our reaction provides a convenient route to 1-naphthylamines.
| a The yields given are for the isolated products. |
|---|

|
To understand the reaction mechanism, we tried to isolate the possible reaction intermediates. First, a stoichiometric reaction of Ni(acac)2 (1 equiv.), IPr (1 equiv.), PhB(OH)2 (2 equiv.) and tBuOK (2 equiv.) was carried out (Scheme 2, eqn (1)). It was found that in addition to a biphenyl product, a red crystalline compound IPrNi(acac) 3 was isolated in 62% yield. Complex 3 is paramagnetic as indicated by the appearance of broad signals in the 1H NMR spectrum. The X-ray crystal analysis of 3 (ref. 9) clearly shows a rare, three-coordinate distorted T-shaped Ni(I) structure.6e,f When Cs2CO3 was used instead of tBuOK, the same Ni(I) complex was also observed, albeit in a low yield of 14%. In this case, a large amount of precipitate could be observed. The precipitate was assumed to be the PhB(OH)2–Cs2CO3 adduct. Thus, the formation of this low-soluble adduct might prevent further reaction with the nickel complex. In fact, stirring a 1:1 mixture of PhB(OH)2 and Cs2CO3 in 1,4-dioxane at 90 °C resulted in the formation of a large amount of white precipitate. This might also explain why the use of 1 equivalent of Cs2CO3 provided the cyclized product 2a in a low yield (Table 1, entry 9). Complex 3 might be formed by the comproportionation of Ni(0) and Ni(II) species.6c,d To confirm this point, the stoichiometric reaction of Ni(COD)2 with Ni(acac)2 in the presence of IPr was carried out. To our delight, the same complex 3 was formed in moderate yield (Scheme 2, eqn (2)). This Ni(I) complex 3 was found to catalyze the cyclization of 1a with PhB(OH)2 to afford 2a (Scheme 2, eqn (3)), implying that the Ni(I) species was likely involved in the catalytic process. Recently, a Ni(I) species was proposed to be relevant in Ni-catalyzed cross-coupling reactions.6 Our results demonstrate that Ni(I) may also be involved in the Ni-catalyzed addition reactions of organoboron reagents to alkynes. In addition, the reaction of allene 4 with PhB(OH)2 under Ni-catalyzed conditions afforded indeno[1,2-b]quinoline 5via base-promoted cyano-Schmittel cyclization,7 while the desired 2a was not observed (Scheme 2, eqn (4)), indicating that the reaction does not involve the allene intermediate.
 | ||
| Scheme 2 Mechanistic studies. | ||
The origin of the regioselectivity in this reaction is not clear yet; it is possibly controlled by electronic factors in π–complex 12 (see Scheme 3).11 To understand the role of the OTBS group, alkyne-nitrile 6 without the OTBS group was synthesized. The Ni-catalyzed reaction of 6 with PhB(OH)2 afforded only 10% of the desired naphthylamine 7, along with small amounts of an unidentified byproduct (Scheme 2, eqn (5)). The results indicate that the presence of the OTBS group is crucial for this reaction.
In addition, it was found that in the presence of a radical scavenger such as TEMPO, the desired 2a and an N-arylated product 8 were formed in 74% combined yield (Scheme 2, eqn (6)). 8 was possibly formed by the Ni-catalyzed oxidative amination of the arylboronic acid with amine 2a.12,13 The results indicate that the reaction was not inhibited by TEMPO, and this suggests that a radical species is not involved in this system.
Based on the above results, we propose the following reaction mechanism (Scheme 3). Initially, transmetalation of the arylboronic acid with the Ni(II) complex, promoted by a base, provides diarylnickel(II) species 9, together with HOBO, HCsCO3 and Cs(acac).149 undergoes reductive elimination to form a Ni(0) species. The observation of biphenyl15 in the catalytic reaction of 1a with PhB(OH)2 also indicates that Ni(II) was reduced in the reaction process. This Ni(0) species comproportionates with Ni(II) to afford Ni(I) complex 10, which undergoes transmetalation with the arylboronic acid to give arylnickel(I) species 11. Regioselective 1,2-addition of arylnickel(I) species 11 to the alkyne moiety in a syn-fashion takes place to give an alkenylnickel(I) intermediate syn-13. cis-to-trans isomerization of 13,16 possibly through a carbene-like zwitterionic resonance species17 yields alkenylnickel(I) intermediate anti-13 with a metal trans-to the Ar substituent. It was noted that most of the metal-catalyzed reactions of organoborons to alkynes gave the syn-addition product while few reactions produced the anti-addition product.2r,17 The regio- and stereochemistry for the addition process here are consistent with those observed for the cobalt(II)-catalyzed hydroarylation of propargyl-alcohols or -carbamates with arylboronic acids.18 The cyano group may play a role in facilitating the cis–trans isomerization by stabilizing the metal species and directing the subsequent addition reaction. Nucleophilic attack of the alkenylmetal in anti-13 to the cyano group forms a cyclized intermediate 14. Subsequent protonation of 14 produces the N–H imine 15 and a nickel(I) species 16. Tautomerization of 15 affords the observed product 2. 16 undergoes transmetalation with ArB(OH)2 to regenerate the arylnickel(I) catalyst 11.
 | ||
| Scheme 3 Possible reaction mechanism. | ||
Conclusions
In summary, we have developed a nickel-catalyzed regioselective addition/cyclization of o-(cyano)phenyl propargyl ethers with arylboronic acids, which provides an efficient protocol for the synthesis of highly functionalized 1-naphthylamines with wide structural diversity. The reaction is characterized by a regioselective and anti-addition of the arylboronic acids to the alkyne and subsequent facile nucleophilic addition of the resulting alkenylmetal to the tethered cyano group. Mechanistic studies reveal that a Ni(I) species might be involved in the catalytic process. Further mechanistic studies and the extension to alkynes tethered with a wide variety of electrophiles are currently ongoing in our laboratory.Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 21125210, 21421091) for financial support.Notes and references
- For reviews, see: (a) E. Negishi, C. Copéret, S. Ma, S.-Y. Liou and F. Liu, Chem. Rev., 1996, 96, 365–393 CrossRef CAS; (b) T. Miura and M. Murakami, Chem. Commun., 2007, 217–224 RSC; (c) S. W. Youn, Eur. J. Org. Chem., 2009, 2597–2605 CrossRef CAS.
- For selected papers, see: for Rh: (a) M. Sakai, M. Ueda and N. Miyaura, Angew. Chem., Int. Ed., 1998, 37, 3279–3281 CrossRef CAS; (b) T. Hayashi, K. Inoue, N. Taniguchi and M. Ogasawara, J. Am. Chem. Soc., 2001, 123, 9918–9919 CrossRef CAS PubMed; (c) C. G. Frost and K. J. Wadsworth, Chem. Commun., 2001, 2316–2317 RSC; (d) M. Murakami and H. Igawa, Chem. Commun., 2002, 390–391 RSC; (e) T. Matsuda, M. Makino and M. Murakami, Org. Lett., 2004, 6, 1257–1259 CrossRef CAS PubMed; (f) H. Shimizu and M. Murakami, Chem. Commun., 2007, 2855–2857 RSC; (g) T. Miura, Y. Takahashi and M. Murakami, Chem. Commun., 2007, 3577–3579 RSC; (h) T. Matsuda, Y. Suda and A. Takahashi, Chem. Commun., 2012, 48, 2988–2990 RSC. For Pd: (i) T. Nishikata, Y. Yamamoto and N. Miyaura, Angew. Chem., Int. Ed., 2003, 42, 2768–2770 CrossRef CAS PubMed; (j) C. H. Oh, T. W. Ahn and R. Reddy, Chem. Commun., 2003, 2622–2623 RSC; (k) Y. Bai, J. Yin, W. Kong, M. Mao and G. Zhu, Chem. Commun., 2013, 49, 7650–7652 RSC. For Ni: (l) E. Shirakawa, G. Takahashi, T. Tsuchimoto and Y. Kawakami, Chem. Commun., 2001, 2688–2689 RSC; (m) E. Shirakawa, G. Takahashi, T. Tsuchimoto and Y. Kawakami, Chem. Commun., 2002, 2210–2211 RSC; (n) K. Hirano, H. Yorimitsu and K. Oshima, Adv. Synth. Catal., 2006, 348, 1543–1546 CrossRef CAS; (o) J. D. Sieber, S. Liu and J. P. Morken, J. Am. Chem. Soc., 2007, 129, 2214–2215 CrossRef CAS PubMed; (p) K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9, 5031–5033 CrossRef CAS PubMed; (q) Y.-C. Wong, K. Parthasarathy and C.-H. Cheng, Org. Lett., 2010, 12, 1736–1739 CrossRef CAS PubMed; (r) D. W. Robbins and J. F. Hartwig, Science, 2011, 333, 1423–1427 CrossRef CAS PubMed . So far only two reports involving Ni-catalyzed hydroarylation of alkynes with arylboronic acids have been published, see ref. 2l and r.
- (a) R. Shintani, K. Okamoto, Y. Otomaru, K. Ueyama and T. Hayashi, J. Am. Chem. Soc., 2005, 127, 54–55 CrossRef CAS PubMed; (b) T. Miura, M. Shimada and M. Murakami, J. Am. Chem. Soc., 2005, 127, 1094–1095 CrossRef CAS PubMed; (c) T. Matsuda, M. Makino and M. Murakami, Angew. Chem., Int. Ed., 2005, 44, 4608–4611 CrossRef CAS PubMed; (d) T. Miura, M. Shimada and M. Murakami, Synlett, 2005, 667–669 CAS; (e) T. Miura, H. Nakazawa and M. Murakami, Chem. Commun., 2005, 2855–2856 RSC; (f) T. Miura, T. Sasaki, T. Harumashi and M. Murakami, J. Am. Chem. Soc., 2006, 128, 2516–2517 CrossRef CAS PubMed; (g) T. Miura, Y. Takahashi and M. Murakami, Org. Lett., 2007, 9, 5075–5077 CrossRef CAS PubMed ; for Rh-catalyzed reaction of arylboronic acids with alkyne-nitriles to exo-alkenes, see ref. 3e.
- (a) J. Song, Q. Shen, F. Xu and X. Lu, Org. Lett., 2007, 9, 2947–2950 CrossRef CAS PubMed; (b) T. Miura, T. Toyoshima, Y. Takahashi and M. Murakami, Org. Lett., 2008, 10, 4887–4889 CrossRef CAS PubMed; (c) H. Tsukamoto, T. Suzuki, T. Uchiyama and Y. Kondo, Tetrahedron Lett., 2008, 49, 4174–4177 CrossRef CAS; (d) X. Han and X. Lu, Org. Lett., 2010, 12, 108–111 CrossRef CAS PubMed; (e) K. Shen, X. Han and X. Lu, Org. Lett., 2012, 14, 1756–1759 CrossRef CAS PubMed.
- For a Pd(0)-catalyzed cyclization of alkynals or alkynones involving formal anti-addition of organoboronic reagents via a different reaction mechanism, see: H. Tsukamoto, T. Ueno and Y. Kondo, J. Am. Chem. Soc., 2006, 128, 1406–1407 CrossRef CAS PubMed.
- (a) T. J. Anderson, G. D. Jones and D. A. Vicic, J. Am. Chem. Soc., 2004, 126, 8100–8101 CrossRef CAS PubMed; (b) G. D. Jones, C. McFarland, T. J. Anderson and D. A. Vicic, Chem. Commun., 2005, 4211–4213 RSC; (c) G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164–4172 CrossRef CAS PubMed; (d) L. M. Guard, M. Mohadjer Beromi, G. W. Brudvig, N. Hazari and D. J. Vinyard, Angew. Chem., Int. Ed., 2015, 54, 13352–13356 CrossRef CAS PubMed. For Ni(I) complexes containing NHC ligands, see: Ni(IPr)2Cl: (e) S. Miyazaki, Y. Koga, T. Matsumoto and K. Matsubara, Chem. Commun., 2010, 46, 1932–1934 RSC; Ni(IMes)2X: (f) K. Zhang, M. Conda-Sheridan, S. R. Cooke and J. Louie, Organometallics, 2011, 30, 2546–2552 CrossRef CAS PubMed.
- For our recent results on cyano-Schmittel cyclization of (o-cyano)phenylpropargyl ethers, see: X. You, X. Xie, H. Chen, Y. Li and Y. Liu, Chem.–Eur. J., 2015, 21, 18699–18705 CrossRef CAS PubMed.
- Y. Makida, E. Marelli, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2014, 50, 8010–8013 RSC.
- CCDC 1440646 (2o) and 1440647(3) contain the supplementary crystallographic data for this paper.†.
- (a) P. Sagar, R. Fröhlich and E.-U. Würthwein, Angew. Chem., Int. Ed., 2004, 43, 5694–5697 CrossRef CAS PubMed; (b) Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 10028–10029 CrossRef CAS PubMed; (c) S.-C. Zhao, X.-Z. Shu, K.-G. Ji, A.-X. Zhou, T. He, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2011, 76, 1941–1944 CrossRef CAS PubMed; (d) R. S. Reddy, P. K Prasad, B. B. Ahuja and A. Sudalai, J. Org. Chem., 2013, 78, 5045–5050 CrossRef CAS PubMed; (e) P. Gao, J. Liu and Y. Wei, Org. Lett., 2013, 15, 2872–2875 CrossRef CAS PubMed; (f) W. P. Hong, A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 13664–13667 CrossRef CAS PubMed.
- A. Arcadi, M. Aschi, M. Chiarini, G. Ferrara and F. Marinelli, Adv. Synth. Catal., 2010, 352, 493–498 CrossRef CAS.
- D. S. Raghuvanshi, A. K. Gupta and K. N. Singh, Org. Lett., 2012, 14, 4326–4329 CrossRef CAS PubMed.
- The reaction of amine 2a with phenylboronic acid under the reaction conditions shown in Scheme 2, eqn (6) gave 8 in 43% yield.
- P.-S. Lin, M. Jeganmohan and C.-H. Cheng, Chem.–Asian J., 2007, 2, 1409–1416 CrossRef CAS PubMed.
- Without ligand, only biphenyl was observed. With 10 mol% of P(p-CF3C6H4)3 as the ligand, in addition to biphenyl, 4,4′-bis(trifluoromethyl)biphenyl and 4-(trifluoromethyl)biphenyl were also observed, possibly due to aryl transfer from the phosphine ligand. See: I. Colon and D. R. Kelsey, J. Org. Chem., 1986, 51, 2627–2637 CrossRef CAS.
- A. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2005, 127, 15706–15707 CrossRef CAS PubMed.
- M. Murakami, T. Yoshida, S. Kawanami and Y. Ito, J. Am. Chem. Soc., 1995, 117, 6408–6409 CrossRef CAS.
- P.-S. Lin, M. Jeganmohan and C.-H. Cheng, Chem.–Eur. J., 2008, 14, 11296–11299 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1440646 and 1440647. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01191h |
| This journal is © The Royal Society of Chemistry 2016 |