
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective synthesis of Iboga alkaloids and vinblastine via rearrangements of quaternary ammoniums†
Yun
Zhang
b,
Yibin
Xue
a,
Gang
Li
a,
Haosen
Yuan
a and
Tuoping
Luo
*a
aKey Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, Beijing National Laboratory for Molecular Science (BNLMS), College of Chemistry and Molecular Engineering, Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China. E-mail: tuopingluo@pku.edu.cn
bLaboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China
First published on 16th May 2016
Abstract
An efficient and novel strategy for the enantioselective syntheses of various iboga alkaloids has been developed. The salient features include a gold-catalyzed oxidation of a terminal alkyne followed by cyclization, a Stevens rearrangement and a tandem sequence that combines the gold-catalyzed oxidation, cyclization and [1,2]-shift. The catharanthine analogs provided by our approach were further converted to the vinca alkaloid vinblastine and its analogs, which confirmed the remarkable sensitivity of the cytotoxicity to the C20′ substituent of vinblastine.
Introduction
The total synthesis of complex natural product small molecules invites the examination of various methodologies in a complicated system, which at times reveals current limitations and inspires new advances.1 Importantly, total synthesis could also provide valuable analogs to explore the structure–activity relationships of targeted chemotypes.2 We have been interested in using rearrangement reactions that lead to dramatic changes in molecular skeletons to develop novel and efficient synthetic routes towards various biologically active natural products.3 Herein, we describe a concise and collective synthesis4 of iboga alkaloids and vinblastine which further substantiates these concepts.The iboga alkaloid family of natural products comprises over 60 members of monoterpene indoles that share a common pentacyclic skeleton of ibogamine (Fig. 1).5 Among the various neurological activities of ibogamine (1) and ibogaine (2), the most exciting one is their capability to attenuate the addiction to a number of drugs, although the molecular mechanism of action remains largely elusive.6 Ibogaine, as the most abundant alkaloid in the root bark of the shrub Tabernanthe iboga, has even been studied in a clinical setting.6 Interestingly, both enantiomers of ibogamine are not only active in reducing the self-administration of cocaine and morphine in rats but are also devoid of tremorigenic activity—a side effect exhibited by ibogaine, which hence deserves further investigation.7 Catharanthine (3) and its derivative dihydrocatharanthine (4) have recently been identified as among the most potent TRPM8 antagonists and modulate cold-induced pain signals as well as mammalian thermoregulation.8 More importantly, the conversion of catharanthine to the potent anti-cancer drug vinblastine (5) via a one-pot procedure has boosted the value of this iboga alkaloid, and its derivatives have led to vinblastine analogs revealing insightful structure–activity relationships.9
 | ||
| Fig. 1 Representative iboga alkaloids and vinblastine. | ||
Despite the variety of synthetic approaches towards different iboga alkaloids that have been reported, the enantioselective total syntheses remain relatively sparse.10–13 Since Trost's group published the elegant synthesis of enantioenriched ibogamine in 1978,11 the preparation of chiral isoquinuclidine fragments followed by the construction of a C2–C16 bond in the late stages (catharanthine numbering, throughout) has become the focus of asymmetric synthesis studies.12 The only two exceptions are the efficient syntheses of (−)-ibogamine and (−)-catharanthine by White's group and Oguri's group respectively, both employing the asymmetric Diels–Alder reaction.13 An alternative approach to prepare such a privileged skeleton, especially in an enantioselective manner, would be a valuable addition to current synthetic endeavours and more importantly, would enable flexible structural changes of this chemotype.
Results and discussion
While seeking a unified strategy to access iboga alkaloids with and without the methoxycarbonyl group at C16, we envisioned two late-stage intermediates 6a and 6b (Fig. 2). The C20 carbonyl group of 6 could be a versatile handle for the preparation of bioactive natural products and small-molecule probes. Inspired by the transannular cyclization accomplished by Kutney and co-workers,10b as well as recent advances in the fragmentation of the C16–C21 bond,14 we decided to explore the [1,2]-Stevens rearrangement of ammonium ylide 7 to construct the C16–C21 bond and give the structurally compact product 6.15 Given that zwitterion 7 could be generated from the quaternary ammonium cation 8 upon treatment with base due to the enhanced acidity at C21, 6 would therefore be accessible from 8 in one step. This key precursor 8 could be prepared by intramolecular alkylation of the tertiary amine 9, for which we hypothesized that the recently developed gold-catalyzed conversion of terminal alkynes to α-chloromethyl ketones could find application.16 Thus, the tertiary amine 10 became the precursor for 9, which could be traced back to the known amide 11via propargylation and reduction.17,18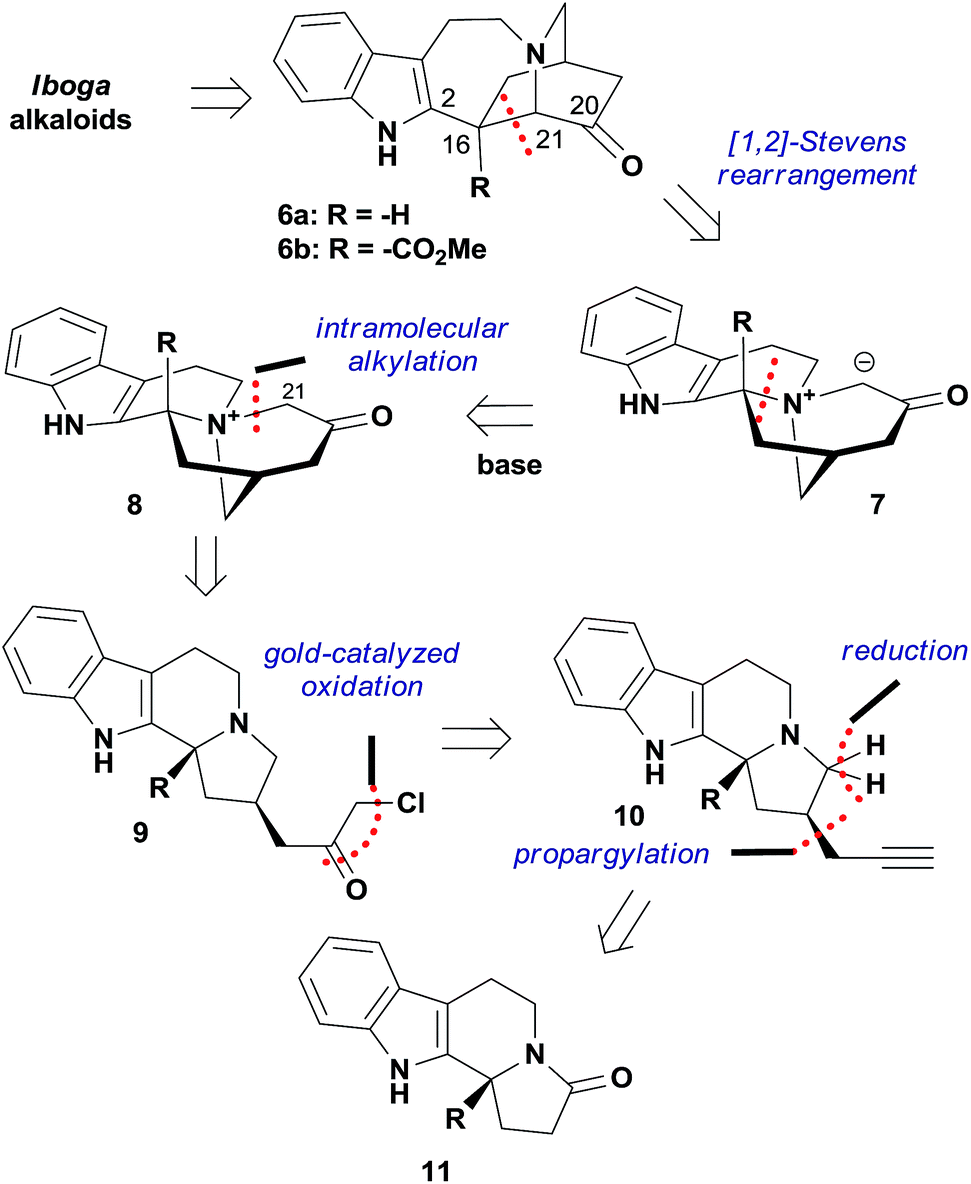 | ||
| Fig. 2 Retrosynthetic analysis of iboga alkaloids based on the [1,2]-Stevens rearrangement reaction. | ||
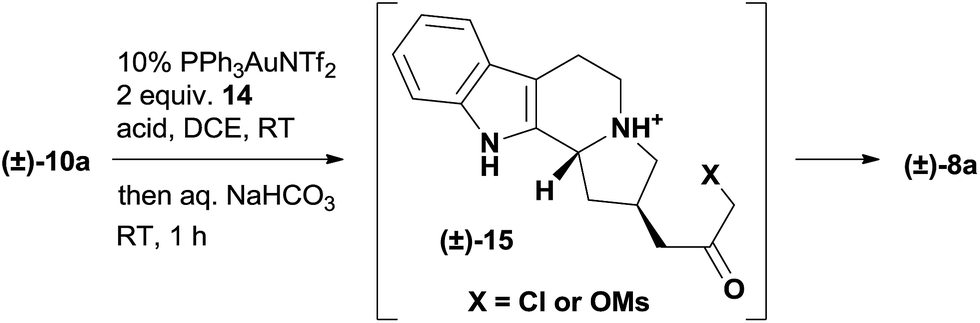
We commenced our studies with the chiral amide 11a, which was prepared from tryptamine in 3 steps via the organocatalytic Pictet–Spengler reaction reported by Jacobsen's group (Scheme 1).17 The introduction of the propargyl group was achieved with the protection of the nitrogen atom, and the following deprotection afforded a pair of readily separable diastereomers, where the desired stereoisomer 13a was isolated as the major product in 52% yield over 3 steps. The subsequent reduction of 13a by LiAlH4 produced the tertiary amine 10a smoothly in 85% yield. We ultimately developed a one-pot procedure that converted 10a to the quaternary ammonium compound 8a in good yield (see the ESI† for the determination of the counteranion).
 | ||
| Scheme 1 Preparation of the quaternary ammonium compound 8a. Reagents and conditions: (a) Boc2O (3.0 equiv.), TEA (1.1 equiv.), DMAP (0.2 equiv.), DCM, RT, 14 h, 89%; (b) LDA (1.2 equiv.), propargyl bromide 12 (2.5 equiv.), −78 °C to RT, 2 h, THF; (c) TFA (5.0 equiv.), DCM, RT, 16 h; 58% over two steps; (d) LiAlH4 (3.0 equiv.), THF, 80 °C, 1 h, 85%; (e) PPh3AuNTf2 (5 mol%), 14 (2.0 equiv.), MsOH (1.3 equiv.), TFA (1.1 equiv.), AgOTf (2 mol%), DCE, RT, 6 h; then NaHCO3 (sat.), AgOTf (1.0 equiv.), RT, 73%. | ||
The extensive optimization of this gold-catalyzed reaction followed by intramolecular alkylation was carried out using racemic 10a (Table 1 and S1†). The basicity of the tertiary amine 10a is detrimental to the cationic gold catalysis and needs to be neutralized with the addition of another equivalent of acid.16b,19 Using a 10 mol% (Ph3P)AuNTf2 catalyst and 2 equiv. of MsOH additive, we examined a variety of oxidants and identified 2-bromopyridine N-oxide 14 as the optimal one (Table S1†). The formation of intermediate 15 was supported by LCMS analysis, and the intermediate then underwent facile cyclization upon the treatment of the reaction mixture with a saturated aqueous solution of sodium bicarbonate. The end product 8a was obtained in 47% yield with the recovery of 39% starting material 10a (Table 1, entry 1). Inspired by the screening of the acid additives to improve the conversion in similar transformations,20 we discovered that complete consumption of 10a was achieved in 20 h at room temperature with 1.3 equiv. MsOH and 1.1 equiv. TFA as the acid additives to afford 8a in 63% isolated yield (entry 2). The addition of 3 mol% AgOTf significantly accelerated the reaction,21 which gave 8a in 69% yield on an even larger scale (200 mg scale, entry 3). We intentionally lowered the gold catalyst loading to 5 mol% for the gram scale reaction and found that the first step went to completion in 8 h at room temperature but 8a was isolated in only 51% yield after workup (entry 4). The conditions for the gram scale reaction were further improved by discovering that the addition of 1 equiv. AgOTf effectively promoted the cyclization step, which eventually afforded 8a in 74% isolated yield (entry 5).
|
|
|||
|---|---|---|---|
| Entry | Acid | Time | Yieldb |
| a [10a] = 0.1 M (0.12 mmol). b Isolated yield after flash chromatography. c 39% starting material 10a was recovered. d 200 mg scale reaction, 3% AgOTf was added as an additive. e 1 g scale reaction, 5% PPh3AuNTf2, 2% AgOTf as an additive. f 3 g scale reaction, 5% PPh3AuNTf2, 2% AgOTf as an additive, 1 equiv. AgOTf was added with NaHCO3 (s, aq.) to facilitate the cyclization. | |||
| 1 | 2.1 equiv. MsOH | 24 h | 47%c |
| 2 | 1.3 equiv. MsOH, 1.1 equiv. TFA | 20 h | 63% |
| 3 | 1.3 equiv. MsOH, 1.1 equiv. TFA | 6 h | 69%d |
| 4 | 1.3 equiv. MsOH, 1.1 equiv. TFA | 8 h | 51%e |
| 5 | 1.3 equiv. MsOH, 1.1 equiv. TFA | 8 h | 74%f |
With abundant 8a in hand, we proceeded to test the Stevens rearrangement. Initial efforts in base, solvent and temperature screening proved unfruitful, leading to either starting material recovery or decomposition (Table S2†). Inspired by the development of organocatalytic sigmatropic reactions,22 we shifted our focus to exploiting a novel Stevens rearrangement through the intermediacy of an enamine.23 By examining a variety of amines (Table S3†), we identified that 5 equiv. of piperidine 16 could promote the desired transformation in methanol even if the isolated yield of 6a was only 11% after heating at 170 °C for 8 h in a sealed tube (Table 2, entry 1).24 We therefore turned to microwave technology and found that it was more effective than conventional thermal conditions (entry 2).25 Through a series of optimization procedures including the amount of 16 (entry 3), solvent (entry 4), concentration and heating sequence (entry 5), the iboga alkaloid framework 6a was eventually obtained from 8a in 50–60% isolated yield (over 90% yield based on starting material recovery). Furthermore, when N-methylmorpholine was employed in place of piperidine 16 under the optimized reaction conditions, we did not observe the formation of 6a and the starting material 8a was recovered in 88% yield, thus suggesting that the formal Stevens rearrangement was not base mediated. To the best of our knowledge, this transformation represents the first example of Stevens rearrangement through secondary amine catalysis.
|
|
|||
|---|---|---|---|
| Entry | Conditionsa | Conversionb | Yieldc |
| a [8a] = 0.1 M (0.075 mmol). b Conversion was calculated based on the recovery of 8a. c Isolated yield after column chromatography. d The reaction was carried out in a sealed tube. e The reaction vial was pretreated by N,O-bis(trimethylsilyl)acetamide. f [8a] = 0.5 M (1 mmol). g The heating sequence was composed of 12 cycles with each cycle including irradiation at 150 °C for 15 min and at 50 °C for 15 min. | |||
| 1 | 5 equiv. 16, MeOH, 170 °C, 8 hd | 46% | 13% |
| 2 | 5 equiv. 16, MeOH, 120 °C (mW), 12 h | N.D. | 21% |
| 3 | 0.4 equiv. 16, MeOH, 120 °C (mW), 8 h | N.D. | 32%e |
| 4 | 0.4 equiv. 16, HFIP, 150 °C (mW), 3 h | 45% | 47%e,f |
| 5 | 0.4 equiv. 16, HFIP, 150 °C (mW), 3 h | 58% | 56%e,f,g |

The stage was set for the late-stage functional group manipulations of 6a (Scheme 2). First, the addition of an organocerium reagent, prepared from ethylmagnesium bromide, to the ketone afforded 17 as a single diastereomer, effectively completing the formal synthesis of (+)-catharanthine (3).10d,12c The Wittig reaction was then employed to convert 6a to olefin 18, where the Z configuration of the trisubstituted olefin was assigned by a NOESY experiment (see ESI† for details). Hydrogenation of alkene 18 using activated Pd/C as the catalyst afforded epiibogamine 19 in 97% yield.26 The high stereoselectivity could be attributed to the preferential addition of H2 to the less hindered side of the molecule. Therefore we turned to the radical-based hydrogenation of electron-neutral alkenes initiated by hydrogen atom transfer.27 While manganese and cobalt-based catalyst precursors also produced 19 as the predominant product (Table S4†), we were delighted to find that Fe(acac)3, the precatalyst reported by Baran and co-workers for reductive alkene coupling,28 afforded separable 19 and (+)-ibogamine 1 in 34% and 26% yields, respectively. The key intermediate 6a could be expediently decorated to other interesting derivatives with an iboga alkaloid skeleton. For instance, the reductive cyanation of ketone 6a with tosylmethylisocyanide produced a pair of separable diastereomers 20 and 21 in 25% and 54% yields, respectively.29 The structures of racemic 19 and 20 were determined unambiguously by X-ray crystallography,30 while the analytical data of 1 and 17 corresponded well with that in the literature.12a,c
 | ||
| Scheme 2 Formal synthesis of catharanthine (3) and the total synthesis of ibogamine (1). Reagents and conditions: (a) CeCl3 (2.5 equiv.), EtMgBr (2.0 equiv.), THF, 0.5 h, 72%; (b) tBuOK (3.0 equiv.), Ph3PEtBr (3.0 equiv.), THF, 2 h, 92%; (c) H2, Pd/C (2.0 equiv.), MeOH, 2 h, 97%; (d) PhSiH3 (2.5 equiv.), Fe(acac)3 (0.8 equiv.), TBHP (1.5 equiv.), EtOH, 60 °C, 6 h; 19, 34%; 1, 26%; (e) tBuOK (2.5 equiv.), TosMIC (1.3 equiv.), EtOH (1.7 equiv.), DME, 12 h; 20, 25%; 21, 54%. | ||
Encouraged by the completion of (+)-ibogamine (1), we moved towards the synthetic study of the iboga alkaloids with the methoxycarbonyl group at C16 (Scheme 3). The amide 13b was prepared from the known compound 11b with excellent enantiopurity18 following the same procedures depicted in Scheme 1, while the undesired diastereomeric amide could also be converted to 13b readily (see ESI† for details). The selective reduction of the amide carbonyl group in 13b subsequently afforded the tertiary amine 10b.31 We fortunately isolated a trace amount of the rearranged product 6b after the work-up of the gold-catalyzed oxidation reaction of 10b, indicating that the [1,2]-shift was quite facile in the presence of the C16 methoxycarbonyl group. Therefore, the gold-catalyzed oxidation was followed by the addition of a saturated aqueous solution of sodium bicarbonate and excess triethylamine to promote the cyclization and rearrangement. Gratifyingly, this one-pot procedure afforded ketone 6b in 51% yield from 10b under mild reaction conditions. The Wittig reaction of 6b gave rise to 22—a catharanthine isomer with an exocyclic versus endocyclic double bond. Hydrogenation of 22 afforded dihydrocatharanthine (4) in 79% yield. Interestingly, 22 differs from a known compound derived from catharanthine in the olefin geometry.32 Eventually, employing the conditions reported by Boger and coworkers,32a we successfully made vinblastine (5) in 50% yield by coupling 22 with commercially available vindoline 23.
 | ||
| Scheme 3 Syntheses of dihydrocatharanthine (4) and vinblastine (5). Reagents and conditions: (a) Boc2O (3.0 equiv.), TEA (1.1 equiv.), DMAP (0.2 equiv.), DCM, RT, 14 h, 87%; (b) LDA (1.2 equiv.), 12 (2.5 equiv.), THF, 12 h; (c) TFA (5.0 equiv.), DCM, 16 h, 33% over two steps; (d) trimethyloxonium tetrafluoroborate (2.5 equiv.), 2,6-di-tert-butylpyridine (3.5 equiv.), DCM, 12 h; then NaBH4 (0.5 equiv.), MeOH, 0.5 h, 87%; (e) PPh3AuNTf2 (5 mol%), 14 (2.0 equiv.), MsOH (2.1 equiv.), AgOTf (2 mol%), DCE, RT, 5 h; then NaHCO3 (sat.), TEA (3.0 equiv.), RT, 3 h, 51%; (f) tBuOK (3.0 equiv.), Ph3PEtBr (3.0 equiv.), THF, 2 h, 83%; (g) H2, PtO2 (0.3 equiv.), MeOH, 15 h, 79%; (h) vindoline 23 (1.2 equiv.), HCl-CF3CH2OH, FeCl3 (5.0 equiv.), 2 h; Fe2(ox)3 (30 equiv.), O2; then NaBH4 (20 equiv.), 0 °C, 0.5 h, 50%. | ||
It is noteworthy that the chiral compound 6b, which was prepared from L-tryptophan in 8 steps, would be a valuable synthetic intermediate towards vinblastine analogs. To illustrate this point, compounds 24 and 25, vinblastine analogs differing only in the C20′ substituent, were readily prepared by employing the Wittig reaction of 6b followed by biomimetic coupling (Fig. 3). We also prepared fluoroalkene 27 using reagent 26,33 where the E configuration of the olefin was assigned by a NOESY experiment (see ESI† for details). Interestingly, the coupling of 27 with vindoline (23) afforded aldehyde 28 in 68% yield (see Fig. S1† for a proposed mechanism). The cytotoxicities of 24 and 25 were measured in the HCT116 cell line using vinblastine (5) as a positive control. Our data indicated that 24 was over 100-fold less active than vinblastine, and that 25 was even less active than 24. Based on a 40-step total synthesis, Fukuyama's group has reported inactive vinblastine analogs with C20′ acetylene functionalities that differ significantly in size and shape with the ethyl group of the natural product.34 Herein we showed that even a subtle change—with the C20′ alkyl substituent length extended for one (24) or two more carbons (25)—was enough to dramatically decrease the potency. This could be rationalized by the X-ray crystallographic analysis of the vinblastine–tubulin interactions, in which the C20′ ethyl substituent of vinblastine is embedded in a hydrophobic binding site.35 Interestingly, the aldehyde analog of vinblastine, compound 28, almost lost the ability to inhibit the growth of HCT116 cells. However, compound 29, obtained by the condensation of 28 with hydrazine, showed decent cytotoxicity (IC50 = 959 nM). This observation implies the necessity of a hydrogen bond donor around the C20′ position,36 although further investigation is needed to provide more insight into the hydrazone analog.
 | ||
| Fig. 3 Synthesis of vinblastine analogs and their cell growth inhibitory activity. | ||
Conclusions
In summary, we have accomplished a unique and general route for the enantioselective synthesis of iboga alkaloids by developing a Stevens rearrangement through secondary amine catalysis and an oxidation/cyclization/rearrangement tandem sequence. The precise mechanism of the rearrangement remains to be investigated to identify whether a radical or an ionic intermediate is involved. Nonetheless, both reactions have the potential to be applied in the synthesis of a myriad of complex alkaloids. This study nicely exemplifies the total synthesis of complex natural products serving as not only a driving force for advancing the synthetic methodology but also as an important source for providing analogs. Furthermore, this practical approach to modify iboga alkaloids and vinblastine paves the way for studies into their pronounced pharmacological properties using state-of-the-art chemical biology technologies, which are underway in our group and will be reported in due course.Acknowledgements
This work was supported by generous start-up funds from the College of Chemistry and Molecular Engineering, Peking University and Peking-Tsinghua Center for Life Sciences, and the National Science Foundation of China (Grant No. 21472003 and 31521004). We thank Dr Nengdong Wang and Prof. Wenxiong Zhang (Peking University) for their help in analyzing the X-ray crystallography data, and Prof. Jian Wang (Tsinghua University) for his help in chiral HPLC analysis.Notes and references
- R. W. Hoffmann, Angew. Chem., Int. Ed., 2013, 52, 123–130 CrossRef CAS PubMed.
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS PubMed.
- (a) W. Ren, Y. Bian, Z. Zhang, H. Shang, P. Zhang, Y. Chen, Z. Yang, T. Luo and Y. Tang, Angew. Chem., Int. Ed., 2012, 51, 6984–6988 CrossRef CAS PubMed; (b) X. Yu, F. Su, C. Liu, H. Yuan, S. Zhao, Z. Zhou, T. Quan and T. Luo, J. Am. Chem. Soc., 2016, 138, 6261–6270 CrossRef CAS PubMed; rearrangement reactions have been used extensively in the organic synthesis of natural products. For some recent examples, see: (c) C. L. Martin, L. E. Overman and J. M. Rohde, J. Am. Chem. Soc., 2010, 132, 4894–4906 CrossRef CAS PubMed; (d) L. Jørgensen, S. J. McKerrall, C. A. Kuttruff, F. Ungeheuer, J. Felding and P. S. Baran, Science, 2013, 341, 878–882 CrossRef PubMed; (e) C. W. Plummer, C. S. Wei, C. E. Yozwiak, A. Soheili, S. O. Smithback and J. L. Leighton, J. Am. Chem. Soc., 2014, 136, 9878–9881 CrossRef CAS PubMed; (f) C. P. Ting and T. J. Maimone, J. Am. Chem. Soc., 2015, 137, 10516–10519 CAS.
- S. B. Jones, B. Simmons, A. Mastracchio and D. W. C. MacMillan, Nature, 2011, 475, 183–188 CrossRef CAS PubMed.
- K. R. Alper, Alkaloids: Chem. Biol. Perspect., 2001, 56, 1–38 CAS.
- (a) P. Popik, R. T. Layer and P. Skolnick, Pharmacol. Rev., 1995, 47, 235–253 CAS; (b) C. Jenks, Nat. Prod. Lett., 2002, 16, 71–76 CrossRef CAS PubMed.
- S. D. Glick, M. E. Kuehne, J. Raucci, T. E. Wilson, D. Larson, R. W. Keller Jr and J. N. Carlson, Brain Res., 1994, 657, 14–22 CrossRef CAS PubMed.
- Y. Terada, M. Kitajima, F. Taguchi, H. Takayama, S. Horie and T. Watanabe, J. Nat. Prod., 2014, 77, 1831–1838 CrossRef CAS PubMed.
- (a) H. Ishikawa, D. A. Colby and D. L. Boger, J. Am. Chem. Soc., 2008, 130, 420–421 CrossRef CAS PubMed; (b) For a recent review, see: J. E. Sears and D. L. Boger, Acc. Chem. Res., 2015, 48, 653–662 CrossRef CAS PubMed.
- For a recent review, see: (a) G. K. Jana, S. Paul and S. Sinha, Org. Prep. Proced. Int., 2011, 43, 541–573 CrossRef CAS; (b) J. P. Kutney, R. T. Brown and E. Piers, J. Am. Chem. Soc., 1964, 86, 2287–2288 CrossRef CAS; (c) G. Buchi, D. L. Coffen, K. Kocsis, P. E. Sonnet and F. E. Ziegler, J. Am. Chem. Soc., 1965, 87, 2073–2075 CrossRef; (d) G. Büchi, P. Kulsa, K. Ogasawara and R. L. Rosati, J. Am. Chem. Soc., 1970, 92, 999–1005 CrossRef; (e) J. W. Huffman, G. Shanmugasundaram, R. Sawdaye, P. C. Raveendranath and R. C. Desai, J. Org. Chem., 1985, 50, 1460–1464 CrossRef CAS; (f) M. E. Kuehne and P. J. Reider, J. Org. Chem., 1985, 50, 1464–1467 CrossRef CAS; (g) W. Nagata, H. Hirai, T. Okumura and K. Kawata, J. Am. Chem. Soc., 1968, 90, 1650–1651 CrossRef CAS PubMed; (h) S. Hiari, K. Kawata and W. Nagata, Chem. Commun., 1968, 1016–1017 RSC; (i) T. Imanishi, N. Yagi, H. Shin and M. Hanaoka, Tetrahedron Lett., 1981, 22, 4001–4004 CrossRef CAS; (j) T. Imanishi, N. Yagi and M. Hanaoka, Chem. Pharm. Bull., 1985, 33, 4202–4211 CrossRef CAS; (k) G. R. Krow, D. A. Shaw, B. Lynch, W. Lester, S. W. Szczepanski, K. Raghavachari and A. E. Derome, J. Org. Chem., 1988, 53, 2258–2262 CrossRef CAS; (l) C. Herdeis and C. Hartke-Karger, Liebigs Ann. Chem., 1991, 99–104 CrossRef CAS; (m) P. Rosenmund, W. H. Haase, J. Bauer and R. Frische, Chem. Ber., 1975, 108, 1871–1895 CrossRef CAS; (n) S. I. Sallay, J. Am. Chem. Soc., 1967, 89, 6762–6763 CrossRef CAS PubMed; (o) M. Ikezaki, T. Wakamastu and Y. Ban, J. Chem. Soc. D, 1969, 88–89 RSC; (p) K. J. Henrey, P. A. Grieco Jr and W. J. DuBay, Tetrahedron Lett., 1996, 37, 8289–8292 CrossRef; (q) W. G. Bornmann and M. E. Kuhene, J. Org. Chem., 1992, 57, 1752–1760 CrossRef CAS; (r) A. C. Kruegel, S. Rakshit, X. Li and D. Sames, J. Org. Chem., 2015, 80, 2062–2071 CrossRef CAS PubMed.
- B. M. Trost, S. A. Godleski and J. P. Genêt, J. Am. Chem. Soc., 1978, 100, 3930–3931 CrossRef CAS.
- (a) D. M. Hodgson and J.-M. Galano, Org. Lett., 2005, 7, 2221–2224 CrossRef CAS PubMed; (b) S. Höck and H.-J. Borschberg, Helv. Chim. Acta, 2006, 89, 542–557 CrossRef; (c) L. Moisan, P. Thuéry, M. Nicolas, E. Doris and B. Rousseau, Angew. Chem., Int. Ed., 2006, 45, 5334–5336 CrossRef CAS PubMed; (d) M. Harada, K. N. Asaba, M. Iwai, N. Kogure, M. Kitajima and H. Takayama, Org. Lett., 2012, 14, 5800–5803 CrossRef CAS PubMed; (e) K. Ishihara, H. Yamada and M. Akakura, Chem. Commun., 2014, 50, 6357–6360 RSC; (f) M. Hatano, Y. Goto, A. Izumiseki, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2015, 137, 13472–13475 CrossRef CAS PubMed.
- (a) J. D. White and Y. Choi, Org. Lett., 2000, 2, 2373–2376 CrossRef CAS PubMed; (b) H. Mizoguchi, H. Oikawa and H. Oguri, Nat. Chem., 2014, 6, 57–64 CrossRef CAS PubMed.
- J. W. Beatty and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 10270–10273 CrossRef CAS PubMed and references cited therein.
- (a) T. S. Stevens, E. M. Creighton, A. B. Gordon and M. MacNicol, J. Chem. Soc., 1928, 3193–3197 RSC; (b) I. E. Markó in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 3, pp. 913–974 Search PubMed; (c) M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395–398 CrossRef CAS PubMed; (d) J. A. Vanecko, H. Wan and F. G. West, Tetrahedron, 2006, 62, 1043–1062 CrossRef CAS; (e) P. Tuzina and P. Somfai, Org. Lett., 2009, 11, 919–921 CrossRef CAS PubMed; (f) T. M. Bott, J. A. Vanecko and F. G. West, J. Org. Chem., 2009, 74, 2832–2836 CrossRef CAS PubMed; (g) M. Valpuesta, M. Ariza, A. Díaz and R. Suau, Eur. J. Org. Chem., 2010, 4393–4401 CAS; (h) L. Palombi, Catal. Commun., 2011, 12, 485–488 CrossRef CAS; (i) J. Clayden, M. Donnard, J. Lefranc and D. J. Tetlow, Chem. Commun., 2011, 47, 4624–4639 RSC; (j) S. Harada, M. Kono, T. Nozaki, Y. Menjo, T. Nemoto and Y. Hamada, J. Org. Chem., 2015, 80, 10317–10333 CrossRef CAS PubMed.
- (a) W. He, L. Xie, Y. Xu, J. Xiang and L. Zhang, Org. Biomol. Chem., 2012, 10, 3168–3171 RSC; (b) E. L. Noey, Y. Luo, L. Zhang and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 1078–1084 CrossRef CAS PubMed.
- I. T. Raheem, P. S. Thiara, E. A. Peterson and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 13404–13405 CrossRef CAS PubMed.
- (a) P. Magnus, M. Ladlow, C. S. Kim and P. Boniface, Heterocycles, 1989, 28, 951–956 CrossRef CAS; (b) P. Magnus, J. S. Mendoza, A. Stamford, M. Ladlow and P. Willis, J. Am. Chem. Soc., 1992, 114, 10232–10245 CrossRef CAS.
- (a) P. de Frémont, N. Marion and S. P. Nolan, J. Organomet. Chem., 2009, 694, 551–560 CrossRef; (b) R. L. Lalonde, W. E. Brenzovich, D. Benitez, E. Tkatchouk, K. Kelley, W. A. Goddard and F. D. Toste, Chem. Sci., 2010, 1, 226–233 RSC.
- (a) L. Ye, L. Cui, G. Zhang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 3258–3259 CrossRef CAS PubMed; (b) E. P. A. Talbot, M. Richardson, J. M. McKenna and F. D. Toste, Adv. Synth. Catal., 2014, 356, 687–691 CrossRef CAS PubMed.
- J. Han, N. Shimizu, Z. Lu, H. Amii, G. B. Hammond and B. Xu, Org. Lett., 2014, 16, 3500–3503 CrossRef CAS PubMed.
- (a) G. Valero and A. Moyano, in Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, ed. P. I. Dalko, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, pp. 1191–1224 Search PubMed; (b) A. McNally, B. Evans and M. J. Gaunt, Angew. Chem., Int. Ed., 2006, 45, 2116–2119 CrossRef CAS PubMed.
- (a) H. G. Lindwall and J. S. Maclennan, J. Am. Chem. Soc., 1932, 54, 4739–4744 CrossRef CAS; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed and references cited therein; (c) D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed and references cited therein.
- A proposed mechanism for 16-catalyzed formation of 6a:
when 8a and 16 were heated at 80 °C in HFIP, the formation of enamine intermediate A was supported by LCMS analysis. - For a recent review, see: G. B. Dudley, R. Richert and A. E. Stiegman, Chem. Sci., 2015, 6, 2144–2152 RSC.
- The synthesis of racemic epiibogamine has been reported. See: (a) G. Büchi, D. L. Coffen, K. Kocsis, P. E. Sonnet and F. E. Ziegler, J. Am. Chem. Soc., 1966, 88, 3099–3109 CrossRef; (b) G. K. Jana and S. Sinha, Tetrahedron Lett., 2012, 53, 1671–1674 CrossRef CAS.
- (a) K. Iwasaki, K. K. Wan, A. Oppedisano, S. W. M. Crossley and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 1300–1303 CrossRef CAS PubMed; (b) S. M. King, X. Ma and S. B. Herzon, J. Am. Chem. Soc., 2014, 136, 6884–6887 CrossRef CAS PubMed.
- (a) J. C. Lo, Y. Yabe and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 1304–1307 CrossRef CAS PubMed; (b) J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS PubMed.
- O. H. Oldenziel, D. Van Leusen and A. M. Van Leusen, J. Org. Chem., 1977, 42, 3114–3118 CrossRef CAS.
- CCDC-1433182 (19), CCDC-1433188 (20) and CCDC-1433180 (S4) contain the supplementary crystallographic data for this paper.†.
- M. Ito, C. W. Clark, M. Mortimore, J. B. Goh and S. F. Martin, J. Am. Chem. Soc., 2001, 123, 8003–8010 CrossRef CAS PubMed.
- (a) H. Ishikawa, D. A. Colby, S. Seto, P. Va, A. Tam, H. Kakei, T. J. Rayl, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 4904–4916 CrossRef CAS PubMed; (b) E. Giovanelli, S. Leroux, L. Moisan, H. Carreyre, P. Thuéry, D.-A. Buisson, A. Meddour, J.-M. Coustard, S. Thibaudeau, B. Rousseau, M. Nicolas, P. Hellier and E. Doris, Org. Lett., 2011, 13, 4116–4119 CrossRef CAS PubMed.
- L. Zhu, C. Ni, Y. Zhao and J. Hu, Tetrahedron, 2010, 66, 5089–5100 CrossRef CAS.
- T. Miyazaki, S. Yokoshima, S. Simizu, H. Osada, H. Tokuyama and T. Fukuyama, Org. Lett., 2007, 9, 4737–4740 CrossRef CAS PubMed.
- (a) B. Gigant, C. Wang, R. B. G. Ravelli, F. Roussi, M. O. Steinmetz, P. A. Curmi, A. Sobel and M. Knossow, Nature, 2005, 435, 519–522 CrossRef CAS PubMed; (b) Y. Wang, F. W. Benz, Y. Wu, Q. Wang, Y. Chen, X. Chen, H. Li, Y. Zhang, R. Zhang and J. Yang, Mol. Pharmacol., 2016, 89, 233–242 CrossRef CAS PubMed.
- E. K. Leggans, K. K. Duncan, T. J. Barker, K. D. Schleicher and D. L. Boger, J. Med. Chem., 2013, 56, 628–639 CrossRef CAS PubMed.

Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all compounds are provided. CCDC 1433180, 1433182 and 1433188. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00932h |
| This journal is © The Royal Society of Chemistry 2016 |