
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium catalyzed regioselective B–C(sp) coupling via direct cage B–H activation: synthesis of B(4)-alkynylated o-carboranes†
Yangjian
Quan
,
Cen
Tang
and
Zuowei
Xie
 *
*
Department of Chemistry, State Key Laboratory of Synthetic Chemistry, The Chinese University of Hong Kong, Shatin N. T., Hong Kong, China. E-mail: zxie@cuhk.edu.hk
First published on 13th May 2016
Abstract
Pd-catalyzed carboxylic acid guided regioselective alkynylation of cage B(4)–H bonds in o-carboranes has been achieved for the first time using two different catalytic systems. In the presence of 5 mol% Pd(OAc)2 and 3 equiv. of AgOAc, the reaction of 1-COOH-2-R1-C2B10H10 with R3SiC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CBr in ClCH2CH2Cl gives 4-(R3SiCC)-2-R1-o-C2B10H10 in moderate to high yields. This reaction is compatible with alkynes possessing sterically bulky silyl groups such as iPr3Si or tBuMe2Si. Meanwhile, another catalytic system of Pd(OAc)2/AgOAc/K2HPO4 can catalyze the direct B(4)-alkynylation of 1-COOH-2-R1-C2B10H10 with terminal alkynes R2CCH in moderate to high yields. The latter has a broader substrate scope from bulky silyl to aromatic to carboranyl substituents. Desilylation of the resultant products affords carboranyl acetylene 4-(HCC)-2-R1-o-C2B10H10 which can undergo further transformations such as Sonogashira coupling, dimerization and click reactions. It is suggested that the above two catalytic systems may proceed via Pd(II)–Pd(IV)–Pd(II) and Pd(II)–Pd(0)–Pd(II) catalytic cycles, respectively. In addition, the silver salt is found to promote the decarboxylation reaction and thereby controls the mono-selectivity.
CBr in ClCH2CH2Cl gives 4-(R3SiCC)-2-R1-o-C2B10H10 in moderate to high yields. This reaction is compatible with alkynes possessing sterically bulky silyl groups such as iPr3Si or tBuMe2Si. Meanwhile, another catalytic system of Pd(OAc)2/AgOAc/K2HPO4 can catalyze the direct B(4)-alkynylation of 1-COOH-2-R1-C2B10H10 with terminal alkynes R2CCH in moderate to high yields. The latter has a broader substrate scope from bulky silyl to aromatic to carboranyl substituents. Desilylation of the resultant products affords carboranyl acetylene 4-(HCC)-2-R1-o-C2B10H10 which can undergo further transformations such as Sonogashira coupling, dimerization and click reactions. It is suggested that the above two catalytic systems may proceed via Pd(II)–Pd(IV)–Pd(II) and Pd(II)–Pd(0)–Pd(II) catalytic cycles, respectively. In addition, the silver salt is found to promote the decarboxylation reaction and thereby controls the mono-selectivity.
Introduction
The development of efficient synthetic methodologies to incorporate alkyne motifs has received broad interest, as they are not only important building blocks in natural products, pharmaceuticals and materials1 but also essential functional groups in cross-coupling, metathesis and cycloaddition reactions.2 Meanwhile, carboranyl acetylenes have proved to be useful basic units in molecular rods,3 nonlinear optical materials,4 supramolecular design,5 nanovehicles6 and metal–organic frameworks.7 As there is a lack of direct and efficient methodologies for the synthesis of B-alkynylated carboranes, the alkyne moieties in the aforementioned materials are generally connected to cage carbon atoms,3–8 which limits the application scope of the carborane derivatives.Though cage boron alkynylated carboranes can be prepared by two-step reactions, such as the selective iodination of an o-carborane, followed by Pd(0)-catalyzed cross-coupling with alkynyl Grignard reagents,9 the installation of iodo groups to specific positions on the carboranes is necessary (Scheme 1). However, the selective iodination of cage B(4,5,7,11)–H is rather challenging, if not impossible.8 Thus, we aim to develop new methodologies for the selective and direct alkynylation of carboranes via cage B–H activation.
 | ||
| Scheme 1 Selected examples of transition metal catalyzed formation of cage B–C(sp) and B–C(sp2) bonds in o-carboranes. | ||
Directing groups are essential in transition metal catalyzed C–H activation due to their ability to chelate the metal catalyst, position it for selective C–H cleavage, and reduce activation energy by stabilizing the metallacycle intermediates.10 Nevertheless, strategies using directing groups suffer from limitations when the directing groups are not present in the target molecules. To overcome this problem, the use of traceless directing groups is obviously an ideal method. Recently, the use of –COOH as a weak coordinating yet efficient directing group for transition metal catalyzed phenyl C–H activation has been documented, and has been found to be easily removed by decarboxylation after the reaction.10h Subsequently, carboxylic acid directed phenyl C–H olefination,11 arylation,12 alkylation,13 acylation,14 carboxylation,15 amination,16 hydroxylation17 and halogenation18 have been successfully developed. However, to the best of our knowledge, the direct alkynylation of C–H bonds guided by –COOH is still elusive, although nitrogen-based directing-group-guided transition-metal catalyzed phenyl C–H alkynylation has been recently documented using alkynyl halides,19 hypervalent iodine-alkyne reagents20 and terminal alkynes21 as the alkynylating reagents. Meanwhile, oxidative coupling of two C–H bonds for the formation of a C–C bond has received growing interest due to its benefits which include atom-economy, step-economy and less waste.22 Compared with the achievements of phenyl C–H bond oxidative coupling, the regioselective and direct oxidative coupling of an organic C–H bond with a cage B–H bond in o-carboranes is very rare.23
Very recently, our group has developed a transition metal catalyzed –COOH guided cage B–H alkenylation23,24 and arylation25 of o-carboranes, in which the carboxyl group is removed in a one-pot fashion. Inspired by these results and other cage B–H activation reactions,26–29 we have extended our research to investigate direct cage B–H alkynylation by alkynyl halides through a Pd(II)–Pd(IV)–Pd(II) catalytic cycle and by terminal alkynes via a Pd(II)–Pd(0)–Pd(II) catalytic cycle. These new findings are reported in this article (Scheme 1).
Results and discussion
Alkynylation using alkynyl halides
The initial reaction of 1-COOH-2-CH3-o-C2B10H10 (1a) with iPr3SiCCBr in the presence of 10 mol% Pd(OAc)2 and 1 equiv. of AgOAc in toluene at 90 °C for 6 h did not give any of the desired product (entry 1, Table 1). Replacement of toluene with 1,2-dichloroethane (DCE) afforded the desired coupling product 4-(iPr3SiCC)-2-CH3-o-C2B10H10 in 40% GC yield (entry 2, Table 1). Increasing the amount of AgOAc to 3 equiv. resulted in 90% GC yield of 3a (entry 4, Table 1). Higher or lower reaction temperatures led to decreased yields of 3a (entries 5 and 6, Table 1). Lowering the catalyst loading to 5 mol% did not change the reaction efficiency (entry 7, Table 1). In view of the yields of 3a, entry 7 in Table 1 was chosen as the optimal reaction conditions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat (mol%) | Additive (equiv.) | Solvent | Temp (°C) | Yieldb (%) |
| a Reactions were conducted on a 0.05 mmol scale in 0.5 mL of solvent in a closed flask for 6 h; DCE = 1,2-dichloroethane; TFA = trifluoroacetate. b GC yields. | |||||
| 1 | Pd(OAc)2 (10) | AgOAc (1) | Toluene | 90 | N.R. |
| 2 | Pd(OAc)2 (10) | AgOAc (1) | DCE | 90 | 40 |
| 3 | Pd(OAc)2 (10) | AgOAc (2) | DCE | 90 | 67 |
| 4 | Pd(OAc)2 (10) | AgOAc (3) | DCE | 90 | 90 |
| 5 | Pd(OAc)2 (10) | AgOAc (3) | DCE | 80 | 70 |
| 6 | Pd(OAc)2 (10) | AgOAc (3) | DCE | 100 | 75 |
| 7 | Pd(OAc)2 (5) | AgOAc (3) | DCE | 90 | 89 |
| 8 | Pd(OAc)2 (2.5) | AgOAc (3) | DCE | 90 | 78 |
| 9 | Pd(TFA)2 (5) | AgOAc (3) | DCE | 90 | 82 |
| 10 | Pd(OAc)2 (5) | Ag2CO3 (2) | DCE | 90 | 75 |
| 11 | Pd(OAc)2 (5) | Ag2O (2) | DCE | 90 | 63 |
| 12 | Pd(OAc)2 (5) | AgNO3 (3) | DCE | 90 | 30 |
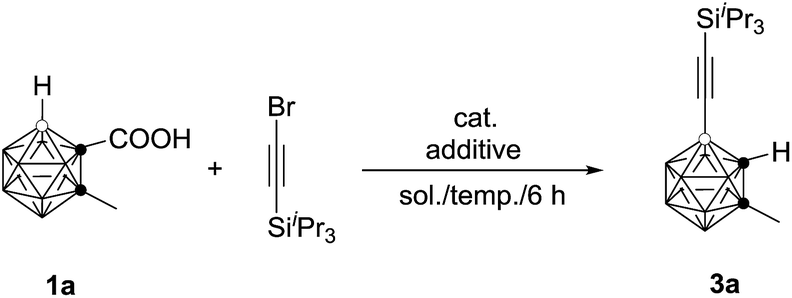
A variety of carborane monocarboxylic acids (1) were examined under the chosen optimal reaction conditions, and the results are compiled in Table 2. All alkyl, alkenyl and aryl substituents on cage C(2), regardless of electronic properties, afforded the coupling products 3 in high isolated yields (entries 1–10 and 13, Table 2). For the heteroatom containing substrate 1j, the product 3j was isolated in 78% yield (entry 10, Table 2), whereas that bearing a thiophenyl group (1l) afforded the product 3l in 54% yield (entry 12, Table 2) probably due to the interaction of Pd with the S atom. Meanwhile, substrate 1k with a naphthyl substituent on cage C(2) gave 3k in only 40% isolated yield (entry 11, Table 2). For R1 = H, an inseparable mixture was produced (entry 14, Table 2). When R1 = Me3Si, the desilylation species 3n was isolated in 41% yield after work up (entry 15, Table 2).
|
|
|||
|---|---|---|---|
| Entry | R1 | R2 (2) | Isolated yield (%) |
| a Reactions were conducted on a 0.2 mmol scale of 1 in a closed flask. b Me3Si was removed after work up. c N.R. = no reaction. | |||
| 1 | Me (1a) | iPr3Si | 81 (3a) |
| 2 | Et (1b) | iPr3Si | 76 (3b) |
| 3 | iPr (1c) | iPr3Si | 75 (3c) |
| 4 | Bn (1d) | iPr3Si | 73 (3d) |
| 5 | Ph (1e) | iPr3Si | 77 (3e) |
| 6 | 4-MeC6H4 (1f) | iPr3Si | 82 (3f) |
| 7 | 3,5-(CH3)2C6H3 (1g) | iPr3Si | 70 (3g) |
| 8 | 4-CF3C6H4 (1h) | iPr3Si | 81 (3h) |
| 9 | 4-ClC6H4 (1i) | iPr3Si | 72 (3i) |
| 10 | 4-MeOC6H4 (1j) | iPr3Si | 78 (3j) |
| 11 | 1-Naphenyl (1k) | iPr3Si | 40 (3k) |
| 12 | 2-Thiophenyl (1l) | iPr3Si | 54 (3l) |
| 13 | EtCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Et) (1m) C(Et) (1m) |
iPr3Si | 80 (3m) |
| 14 | H (1n) | iPr3Si | Messy |
| 15 | Me3Si (1o) | iPr3Si | 41b (3n) |
| 16 | Me (1a) | t BuMe2Si | 70 (3p) |
| 17 | Me (1a) | Me3Si | N.R.c |

In contrast to R1 at cage C(2), the scope of R2 is highly limited in such a coupling reaction. tBuMe2SiCCBr worked well to give 3p in 70% isolated yield (entry 16, Table 2). However, less hindered Me3SiCCBr was not reactive, probably due to its propensity to coordinate with a Pd center via the π bond (entry 17, Table 2). Such a phenomenon was also observed in phenyl C–H alkynylations using R3SiCCBr as reagents.30 It was noted that other alkynyl bromides such as PhCCBr and tBuCCBr were not compatible with this reaction.
Alkynylation using terminal alkynes
As the previous method has a limited substrate scope, we wanted to develop a more atom- and step-economic method for cage B–H alkynylation using terminal alkynes as reagents. We commenced our studies by screening for a suitable base for the oxidative coupling of cage B–H in 1-COOH-2-CH3-o-C2B10H10 (1a) with iPr3SiCCH under the aforementioned optimal reaction conditions. No reaction was observed in the absence of a base (entry 1, Table 3). The addition of 2 equiv. of K2HPO4 afforded the target product 3a in 30% GC yield with iPr3SiCC–CCiPr3Si as the side product (entry 2, Table 3). To inhibit the formation of a homocoupling side product, iPr3SiCCH was added slowly via a syringe pump, leading to a significantly increased yield of 3a to 56% GC yield (entry 3, Table 3). The yield was further improved to 75% if 2 equiv. of the terminal alkyne was used (entry 4, Table 3). Replacement of 1,2-dichloroethane (DCE) with toluene resulted in a slightly higher yield of 3a (entry 5, Table 3). Decreasing the reaction temperature to 80 °C afforded 3a in 86% GC yield (entry 6, Table 3). In view of the yields of 3a, entry 6 in Table 3 was chosen as the optimal reaction conditions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat (mol%) | Additive (equiv.) | Solvent | Temp (°C) | Yieldb (%) |
| a Reactions were conducted on a 0.05 mmol scale of 1a in 0.5 mL of solvent in the presence of 2 equiv. of K2HPO4 in a closed flask for 10 h; DCE = 1,2-dichloroethane; TFA = trifluoroacetate; dba = dibenzylideneacetone. b GC yields. c Without K2HPO4. d Terminal alkyne was added dropwise by a syringe pump over a period of 10 h. e Two equiv. of terminal alkyne was added. | |||||
| 1c | Pd(OAc)2 (5) | AgOAc (3) | DCE | 90 | N.R. |
| 2 | Pd(OAc)2 (5) | AgOAc (3) | DCE | 90 | 30 |
| 3 | Pd(OAc)2 (5) | AgOAc (3) | DCE | 90 | 56d |
| 4 | Pd(OAc)2 (5) | AgOAc (3) | DCE | 90 | 75d,e |
| 5 | Pd(OAc)2 (5) | AgOAc (3) | Toluene | 90 | 78d,e |
| 6 | Pd(OAc)2 (5) | AgOAc (3) | Toluene | 80 | 86d,e |
| 7 | Pd(OAc)2 (5) | AgOAc (3) | Toluene | 70 | Trace |
| 8 | Pd(OAc)2 (3) | AgOAc (3) | DCE | 90 | 18 |
| 9 | Pd(TFA)2 (5) | AgOAc (3) | DCE | 90 | 26 |
| 10 | Pd2(dba)3 (5) | AgOAc (3) | DCE | 90 | 21 |
| 11 | Pd(OAc)2 (5) | Ag2CO3 (2) | DCE | 90 | 15 |
| 12 | Pd(OAc)2 (5) | Ag2O (2) | DCE | 90 | 12 |
| 13 | Pd(OAc)2 (5) | AgNO3 (3) | DCE | 90 | Trace |

This reaction has a much broader substrate scope (R2 = silyl, phenyl and carboranyl). The results are compiled in Table 4. For R1 = alkyl groups, the isolated yields of 3 are comparable to those observed in Table 2. However, if R1 = aryl unit such as 1g, the isolated yield of 3g is 30% (entry 4, Table 4), which is significantly lower than that of 70% shown in entry 7, Table 2. On the other hand, compounds 1n (R1 = H) and 1o (R1 = Me3Si) give 3n in 35% and 74% yields, respectively (entries 5 and 6, Table 4). These yields are much higher than those found in the previous reaction (entries 14 and 15, Table 2). The reasons for this phenomenon are not clear at this stage.
|
|
|||
|---|---|---|---|
| Entry | R1 | R2 (2) | Isolated yield (%) |
| a Reactions were conducted on a 0.2 mmol scale of 1 in a closed flask. b Me3Si was removed after work up. c 3 equiv. of terminal alkyne was used. | |||
| 1 | Me (1a) | iPr3Si | 79 (3a) |
| 2 | iPr (1c) | iPr3Si | 86 (3c) |
| 3 | Bn (1d) | iPr3Si | 70 (3d) |
| 4 | 3,5-(CH3)2C6H3 (1g) | iPr3Si | 30 (3g) |
| 5 | H (1n) | iPr3Si | 35 (3n) |
| 6 | Me3Si (1o) | iPr3Si | 74b (3n) |
| 7 | Me (1a) | t BuMe2Si | 72 (3p) |
| 8 | Me (1a) | Ph | 52c (3r) |
| 9 | Me (1a) | 2-CH3C6H4 | 65c (3s) |
| 10 | Me (1a) | 2,6-(CH3)2C6H3 | 73c (3t) |
| 11 | Me (1a) | 2-iPrC6H4 | 80c (3u) |
| 12 | Me (1a) |

|
82c (3v) |
| 13 | Me (1a) | 4-CH3C6H4 | 48c (3w) |
| 14 | Me (1a) | 4-CF3C6H4 | 44c (3x) |

More importantly, this catalytic system is compatible with phenyl acetylene, producing the corresponding product 3r in 52% isolated yield (entry 8, Table 4). The coupling efficiency was largely enhanced from R2 = Ph, 2-MeC6H4, 2,6-(Me)2C6H3, 2-iPrC6H4 to 1-nC6H13-o-C2B10H10, affording the corresponding products, 3s, 3t, 3u and 3v, in 65%, 73%, 80% and 82% isolated yields, respectively (entries 9–12, Table 4). It should be noted that o-carboranyl is a strong electron-withdrawing unit.4c In view of the isolated yields of 3w and 3x (entries 13 and 14, Table 4), the electronic effects on the reactions are not obvious as –CH3 and –CF3 have significantly different electronic properties. The above data (entries 8–14, Table 4) indicate strongly that bulkier substituents favor the formation of coupling products.
Transformation of 3a
To demonstrate the applications of the resultant compounds 3 as building blocks, further transformation of 3a was carried out. The iPr3Si group in 3a was readily removed by treatment with TBAF (TBAF = tetra-n-butylammonium fluoride) to afford quantitatively the terminal alkyne 4a (Scheme 2). Like other terminal alkynes, compound 4a can undergo various transformations to give different kinds of carborane-incorporated functional molecules. Sonogashira coupling of 4a with iodobenzene or 2-bromothiophene generated 3r or 5a in 92% and 90% isolated yields, respectively. Glaser–Hay homocoupling of 4a gave 1,4-dicarboranyldiacetylene (6a) in 84% isolated yield. A click reaction of 4a with phenyl azide afforded carborane-functionalized 1,2,3-triazole (7a) in 95% isolated yield. | ||
| Scheme 2 Transformations of 3a. | ||
All new compounds 3 and 4a–7a were fully characterized by 1H, 13C, and 11B NMR spectroscopy as well as high-resolution mass spectrometry (HRMS).31 Molecular structures of 4a and 6a were further confirmed by single-crystal X-ray analyses and are shown in Fig. 1. Experimental details are included in the ESI.†
 | ||
| Fig. 1 Molecular structures of 4a (top) and 6a (bottom) (only the terminal alkyne H atom is shown for clarity). | ||
Reaction mechanism
To gain some insight into the reaction mechanism, the following control experiments were carried out. No reaction was observed if 1a was treated with 1 equiv. of iPr3SiCCBr in the presence of 20 mol% Pd(dba)2 (dba = dibenzylideneacetone) in DCE at 90 °C for 6 h in the absence of AgOAc. On the other hand, under the same reaction conditions, replacement of Pd(dba)2 with Pd(OAc)2 gave the alkynylation product 3a in 30% GC yield (Scheme 3a). Similarly, in the presence of 20 mol% Pd(OAc)2, the reaction of 1a with 2 equiv. of iPr3SiCCH afforded 3a in 16% GC yield without AgOAc as the oxidant. While, no 3a was observed when 20 mol% Pd(dba)2 was used instead of Pd(OAc)2 (Scheme 3b). These results suggest that both cross-coupling reactions are initiated by Pd(II) not Pd(0).
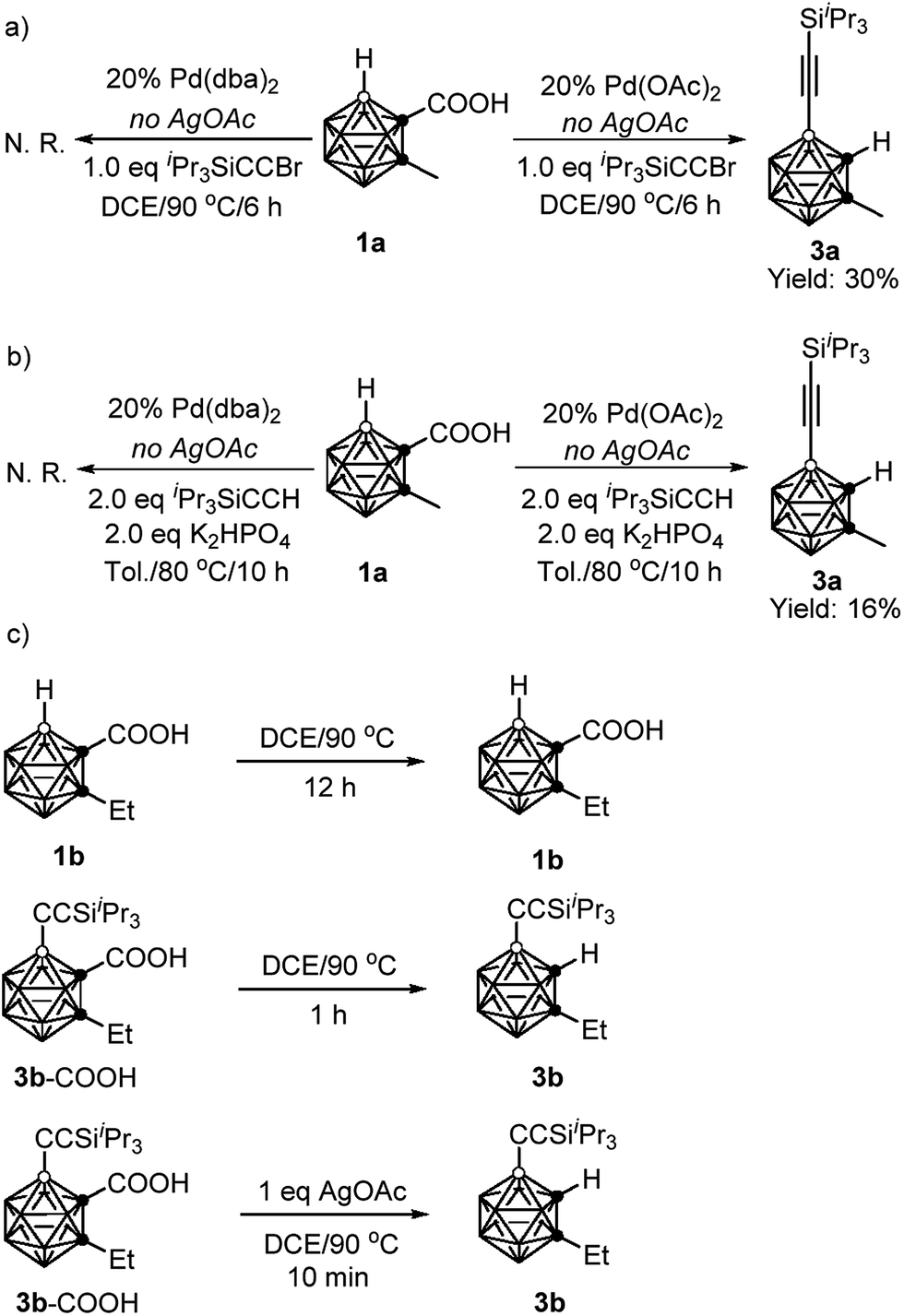 | ||
| Scheme 3 Control experiments. | ||
Decarboxylation of carboranyl carboxylic acids (1b and 3b–COOH) was also examined (Scheme 3c). Compound 1b was stable after heating at 90 °C for 12 h in DCE, whereas 3b–COOH underwent complete decarboxylation within one hour under the same reaction conditions. Notably, it only took ten minutes to convert 3b–COOH to 3b in the presence of 1 equiv. of AgOAc. These results clearly indicate that the introduction of an alkynyl group at the cage B(4) site can induce the decarboxylation, and the addition of a silver salt can accelerate such decarboxylation, which is crucial for controlling the mono-selectivity.
On the basis of the aforementioned experimental data, two plausible reaction mechanisms are proposed in Scheme 4. For the Pd(II)–Pd(IV)–Pd(II) catalytic cycle: an exchange reaction of 1 with Pd(OAc)2, followed by regioselective electrophilic attack at the more electron-rich cage B(4) site yields the intermediate A as the charge distribution on the cage follows the trend B(9,12) > B(8,10) > B(4,5,7,11) > B(3,6).32 Oxidative addition of R2CCBr affords a Pd(IV) intermediate B.25,33 Reductive elimination produces the intermediate C, which undergoes a salt metathesis reaction, protonation and decarboxylation to give the final product 3 and regenerates the catalyst Pd(OAc)2. Meanwhile, another catalytic system involves a Pd(II)–Pd(0)–Pd(II) cycle. An acid–base reaction between K2HPO4 and carboranyl carboxylic acid 1 gives the potassium salt 1′.34 Coordination of the oxygen atom of 1′ to the Pd(II) center, followed by subsequent regioselective electrophilic attack at the more electron-rich cage B(4) site generates the intermediate D. Ligand exchange by acetylide gives a carboranyl-palladium acetylide intermediate E.21b,35 Reductive elimination affords the cage B(4)-alkynylated intermediate F and Pd(0). Decarboxylation of F results in the formation of the final product 3, meanwhile Pd(0) is oxidized by AgOAc to regenerate Pd(OAc)2. It is noted that AgOAc acts as a bromide captor in the Pd(II)–Pd(IV)–Pd(II) catalytic cycle, but as an oxidant to regenerate Pd(II) from Pd(0) in the Pd(II)–Pd(0)–Pd(II) catalytic cycle. However, in both cross-coupling reactions, AgOAc plays a crucial role in promoting decarboxylation and thereby controlling the mono-selectivity.
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusion
We have developed two catalytic systems for regioselective and efficient alkynylation of cage B(4)–H bonds in o-carboranes using alkynyl bromides or terminal alkynes as alkynylating agents, where –COOH acts as a traceless directing group. A series of new cage B(4)-alkynylated o-carborane derivatives has been prepared for the first time, which could find many applications in the synthesis of carborane-based materials.3–7 This opens up a new window for the functionalization of carboranes by direct oxidative coupling of the cage B–H and organic C–H bonds. This work also offers a useful reference for selective C–H alkynylation using carboxylic acid as a traceless directing group in other aromatic systems.On the basis of control experiments and literature work, two catalytic cycles are proposed for the above two reactions: a Pd(II)–Pd(IV)–Pd(II) cycle for using alkynyl bromides as coupling agents and a Pd(II)–Pd(0)–Pd(II) cycle for employing terminal alkynes as coupling partners. The latter has a broader substrate scope than the former. This work also gives some hints for the development of new catalytic systems for the functionalization of carboranes.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (973 Program, Grant No. 2012CB821600) and the Research Grants Council of The Hong Kong Special Administration Region (Project No. CUHK7/CRF/12G and 14304115).Notes and references
- Acetylene Chemistry: Chemistry, Biology and Material Science, ed F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed.
- (a) A. Fürstner and P. W. Davies, Chem. Commun., 2005, 2307–2320 RSC; (b) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (c) M. G. Finn and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1231–1232 RSC; (d) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC; (e) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed.
- (a) J. Müller, K. Base, T. F. Magnera and J. Michl, J. Am. Chem. Soc., 1992, 114, 9721–9722 CrossRef; (b) P. F. H. Schwab, M. D. Levin and J. Michl, Chem. Rev., 1999, 99, 1863–1933 CrossRef CAS PubMed.
- (a) M. F. Hawthorne and A. Moderna, Chem. Rev., 1999, 99, 3421–3434 CrossRef CAS PubMed; (b) J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein and K. A. Stephenson, Coord. Chem. Rev., 2002, 232, 173–230 CrossRef CAS; (c) J. Guo, D. Liu, J. Zhang, Q. Maio and Z. Xie, Chem. Commun., 2015, 51, 12004–12007 RSC.
- (a) H. Jude, H. Disteldorf, S. Fischer, T. Wedge, A. M. Hawkridge, A. M. Arif, M. F. Hawthorne, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2005, 127, 12131–12139 CrossRef CAS PubMed; (b) B. H. Northrop, H.-B. Yang and P. J. Stang, Chem. Commun., 2008, 5896–5908 RSC; (c) J. M. Ludlow III, M. Tominaga, Y. Chujo, A. Schultz, X. Lu, T. Xie, K. Guo, C. N. Moorefield, C. Wesdemiotis and G. R. Newkome, Dalton Trans., 2014, 43, 9604–9611 RSC.
- (a) C. J. Villagómez, T. Sasaki, J. M. Tour and L. Grill, J. Am. Chem. Soc., 2010, 132, 16848–16854 CrossRef PubMed; (b) S. Khatua, J. M. Guerrero, K. Claytor, G. Vives, A. B. Kolomeisky, J. M. Tour and S. Link, ACS Nano, 2009, 3, 351–356 CrossRef CAS PubMed; (c) P.-T. Chiang, J. Mielke, J. Godoy, J. M. Guerrero, I. Alemany, C. J. Villagomez, A. Saywell, L. Grill and J. M. Tour, ACS Nano, 2012, 6, 592–597 CrossRef CAS PubMed; (d) M. Koshino, T. Tanaka, N. Solin, K. Suenaga, H. Isobe and E. Nakamura, Science, 2007, 316, 853 CrossRef CAS PubMed.
- (a) A. K. Singh, A. Sadrzadeh and B. I. Yakobson, J. Am. Chem. Soc., 2010, 132, 14126–14129 CrossRef CAS PubMed; (b) O. K. Farha, A. M. Spokoyny, K. L. Mulfort, M. F. Hawthorne, C. A. Mirkin and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 12680–12681 CrossRef CAS PubMed.
- For selected reviews, see: (a) R. N. Grimes, Carboranes, 2nd edn, Elsevier, Oxford, 2011 Search PubMed; (b) N. S. Hosmane, Boron Science: New Technologies and Applications, Taylor & Francis Books/CRC, Boca Raton, FL, 2011 Search PubMed; (c) D. Olid, R. Núñez, C. Viñas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318–3336 RSC; (d) Z. Qiu, Tetrahedron Lett., 2015, 56, 963–971 CrossRef CAS; (e) Z. Xie, Sci. China: Chem., 2014, 57, 1061–1063 CrossRef CAS.
- (a) A. Himmelspach and M. Finze, Eur. J. Inorg. Chem., 2010, 2012–2024 CrossRef CAS and references therein; (b) M. Tominaga, Y. Morisaki and Y. Chujo, Macromol. Rapid Commun., 2013, 34, 1357–1362 CrossRef CAS PubMed; (c) W. Jiang, D. E. Harwell, M. D. Mortimer, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1996, 35, 4355–4359 CrossRef CAS PubMed; (d) W. Jiang, C. B. Knobler, C. E. Curtis, M. D. Mortimer and M. F. Hawthorne, Inorg. Chem., 1995, 34, 3491–3498 CrossRef CAS.
- For selected reviews, see: (a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (b) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed; (c) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed; (d) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486–1495 CrossRef CAS PubMed; (e) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 43, 936–946 CrossRef PubMed; (f) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (g) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (h) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (i) J.-Q. Yu and Z. Shi, C–H Activation, Springer, Berlin, 2010 Search PubMed.
- (a) M. Miura, T. Tsuda, T. Satoh, S. Pivsa-Art and M. Nomura, J. Org. Chem., 1998, 63, 5211–5215 CrossRef CAS; (b) L. Achermann and J. Pospech, Org. Lett., 2011, 13, 4153–4155 CrossRef PubMed; (c) S. Mochida, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 5776–5779 CrossRef CAS PubMed; (d) K. Ueura, T. Satoh and M. Miura, Org. Lett., 2007, 9, 1407–1409 CrossRef CAS PubMed; (e) R. K. Chinnagolla and M. Jeganmohan, Chem. Commun., 2012, 48, 2030–2032 RSC.
- (a) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS PubMed; (b) D.-H. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676–17677 CrossRef CAS PubMed; (c) H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879–9884 CrossRef CAS PubMed; (d) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429–9432 CrossRef CAS PubMed; (e) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS PubMed; (f) Y. Zhang, H. Zhao, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 127, 3888–3892 CrossRef.
- (a) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS PubMed; (b) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 6097–6100 CrossRef CAS PubMed.
- (a) P. Mamone, G. Danoun and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 6704–6708 CrossRef CAS PubMed; (b) J. Miao and H. Ge, Org. Lett., 2013, 15, 2930–2933 CrossRef CAS PubMed.
- R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082–14083 CrossRef CAS PubMed.
- (a) Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654–14655 CrossRef CAS PubMed; (b) S. Bhadra, W. I. Dzik and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 2959–2962 CrossRef CAS PubMed.
- (a) K.-H. Ng, F.-N. Ng and W.-Y. Yu, Chem. Commun., 2012, 48, 11680–11682 RSC; (b) F.-N. Ng, Z. Zhou and W.-Y. Yu, Chem.– Eur. J., 2014, 20, 4474–4480 CrossRef CAS PubMed.
- T.-S. Mei, R. Giri, N. Maugel and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 5215–5297 CrossRef CAS PubMed.
- (a) Y. Ano, M. Tobisu and N. Chatani, Org. Lett., 2012, 14, 354–357 CrossRef CAS PubMed; (b) Y. Ano, M. Tobisu and N. Chatani, Synlett, 2012, 23, 2763–2767 CrossRef CAS.
- (a) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780–4787 CrossRef CAS PubMed; (b) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 2722–2726 CrossRef CAS PubMed.
- (a) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590–11593 CrossRef CAS PubMed; (b) S. H. Kim, S. H. Park and S. Chang, Tetrahedron, 2012, 68, 5162–5166 CrossRef CAS; (c) J. Dong, F. Wang and J. You, Org. Lett., 2014, 16, 2884–2887 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (b) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- For a recently reported example of Heck-type reaction of o-carborane with styrene, see: H. Lyu, Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2015, 54, 10623–10626 CrossRef CAS PubMed.
- Y. Quan and Z. Xie, J. Am. Chem. Soc., 2014, 136, 15513–15516 CrossRef CAS PubMed.
- Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 1295–1298 CrossRef CAS PubMed.
- Selected examples for catalytic B–H activation of C2B10H12: (a) E. L. Hoel, M. Talebinasab-Savari and M. F. Hawthorne, J. Am. Chem. Soc., 1977, 99, 4356–4367 CrossRef CAS; (b) M. G. L. Mirabelli and L. G. Sneddon, J. Am. Chem. Soc., 1988, 110, 449–453 CrossRef CAS; (c) K. Cao, Y. Huang, J. Yang and J. Wu, Chem. Commun., 2015, 7257–7260 RSC; (d) Z. Zheng, W. Jiang, A. A. Zinn, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1995, 34, 2095–2100 CrossRef CAS; (e) Z. Zheng, C. B. Knobler, M. D. Mortimer, G. Kong and M. F. Hawthorne, Inorg. Chem., 1996, 35, 1235–1243 CrossRef CAS PubMed; (f) Z. Qiu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2013, 135, 12192–12195 CrossRef CAS PubMed; (g) Y. Quan and Z. Xie, J. Am. Chem. Soc., 2015, 137, 3502–3505 CrossRef CAS PubMed; (h) D. Liu, L. Dang, Y. Sun, H.-S. Chan, Z. Lin and Z. Xie, J. Am. Chem. Soc., 2008, 130, 16103–16110 CrossRef CAS PubMed.
- Selected examples for stoichiometric B–H activation of C2B10H12: (a) E. L. Hoel and M. F. Hawthorne, J. Am. Chem. Soc., 1973, 95, 2712–2713 CrossRef CAS; (b) M. R. Churchill, J. J. Hackbarth, A. Davison, D. D. Traficante and S. S. Wreford, J. Am. Chem. Soc., 1974, 96, 4041–4042 CrossRef CAS; (c) G.-X. Jin, J.-Q. Wang, C. Zhang, L.-H. Weng and M. Herberhold, Angew. Chem., Int. Ed., 2005, 44, 259–262 CrossRef CAS PubMed; (d) M. Herberhold, H. Yan, W. Milius and B. Wrackmeyer, Angew. Chem., Int. Ed., 1999, 38, 3689–3691 CrossRef; (e) R. Zhang, L. Zhu, G. Liu, H. Dai, Z. Lu, J. Zhao and H. Yan, J. Am. Chem. Soc., 2012, 134, 10341–10344 CrossRef CAS PubMed; (f) Z. Wang, H. Ye, Y. Li, Y. Li and H. Yan, J. Am. Chem. Soc., 2013, 135, 11289–11298 CrossRef CAS PubMed; (g) J.-Y. Bae, Y.-J. Lee, S.-J. Kim, J. Ko, S. Cho and S. O. Kang, Organometallics, 2000, 19, 1514–1521 CrossRef CAS; (h) Z. J. Yao, W. B. Yu, Y. J. Lin, S. L. Huang, Z. H. Li and G. X. Jin, J. Am. Chem. Soc., 2014, 136, 2825–2832 CrossRef CAS PubMed; (i) Y. Quan, Z. Qiu and Z. Xie, J. Am. Chem. Soc., 2014, 136, 7599–7602 CrossRef CAS PubMed; (j) A. V. Puga, F. Teixidor, R. Sillanpää, R. Kivekäs and C. Viñas, Chem. Commun., 2011, 47, 2252–2254 RSC; (k) G. Barberà, C. Viñas, F. Teixidor, R. Sillanpää and R. Kivekäs, Inorg. Chem., 2006, 45, 3496–3498 CrossRef PubMed; (l) A. M. Spokoyny, M. G. Reuter, C. L. Stern, M. A. Ratner, T. Seideman and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 9482–9483 CrossRef CAS PubMed; (m) M. Y. Tsang, C. Viñas, F. Teixidor, J. G. Planas, N. Conde, R. SanMartin, M. T. Herrero, E. Domínguez, A. Lledós, P. Vidossich and D. Choquesillo-Lazarte, Inorg. Chem., 2014, 53, 9284–9285 CrossRef CAS PubMed; (n) N. Fey, M. F. Haddow, R. Mistry, N. C. Norman, A. G. Orpen, T. J. Reynolds and P. G. Pringle, Organometallics, 2012, 31, 2907–2913 CrossRef CAS.
- Selected examples for catalytic B–H activation of CB11H12− and C2B9H112−: (a) J. D. Hewes, C. W. Kreimendahl, T. B. Marder and M. F. Hawthorne, J. Am. Chem. Soc., 1984, 106, 5757–5759 CrossRef CAS; (b) E. Molinos, G. Kociok-Köhn and A. S. Weller, Chem. Commun., 2005, 3609–3611 RSC; (c) E. Molinos, S. K. Brayshaw, G. Kociok-Köhn and A. S. Weller, Organometallics, 2007, 26, 2370–2382 CrossRef CAS; (d) I. Rojo, F. Teixidor, R. Kivekäs, R. Sillanpää and C. Viñas, J. Am. Chem. Soc., 2003, 125, 14720–14721 CrossRef CAS PubMed. Selected examples for stoichiometric B–H activation of CB11H12− and C2B9H112−: (e) R. E. King, S. B. Miller, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1983, 22, 3549–3554 Search PubMed; (f) F. Teixidor, J. Casabó, A. M. Romerosa, C. Viñas, J. Rius and C. Miravitlles, J. Am. Chem. Soc., 1991, 113, 9895–9896 CrossRef CAS; (g) M. G. Davidson, M. A. Fox, T. G. Hibbert, J. K. Howard, A. Mackinnon, I. S. Neretin and K. Wade, Chem. Commun., 1999, 1649–1650 RSC; (h) V. I. Bregadze, I. D. Kosenko, I. A. Lobanova, Z. A. Starikova, I. A. Godovikov and I. B. Sivaev, Organometallics, 2010, 29, 5366–5372 CrossRef CAS; (i) J. N. Francis, C. J. Jones and M. F. Hawthorne, J. Am. Chem. Soc., 1972, 94, 4878–4881 CrossRef CAS.
- Selected examples for catalytic B–H activation of boranes: (a) H. C. Johnson, C. L. McMullin, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2013, 52, 9776–9780 CrossRef CAS PubMed; (b) E. W. Corcoran and L. G. Sneddon, J. Am. Chem. Soc., 1984, 106, 7793–7800 CrossRef CAS; (c) E. W. Corcoran and L. G. Sneddon, J. Am. Chem. Soc., 1985, 107, 7446–7450 CrossRef CAS; (d) M. J. Pender, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2001, 123, 12222–12231 CrossRef CAS PubMed; (e) T. Peymann, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1999, 121, 5601–5602 CrossRef CAS; (f) R. Wilczynski and L. G. Sneddon, Inorg. Chem., 1981, 20, 3955–3962 CrossRef CAS.
- J. He, M. Wasa, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387–3390 CrossRef CAS PubMed.
- For detailed experimental procedures and complete characterization data, see ESI.†.
- F. Teixidor, G. Barberà, A. Vaca, R. Kivekäsc, R. Sillanpää, O. Oliva and C. Viñas, J. Am. Chem. Soc., 2005, 127, 10158–10159 CrossRef CAS PubMed.
- A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2096–2098 CrossRef CAS PubMed.
- R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS PubMed.
- X. Jie, Y. Shang, P. Hu and W. Su, Angew. Chem., Int. Ed., 2013, 52, 3630–3633 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1455897 and 1455898. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00901h |
| This journal is © The Royal Society of Chemistry 2016 |