
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Au–Pd alloy nanoparticles supported on layered double hydroxide for heterogeneously catalyzed aerobic oxidative dehydrogenation of cyclohexanols and cyclohexanones to phenols†
Xiongjie
Jin
,
Kento
Taniguchi
,
Kazuya
Yamaguchi
and
Noritaka
Mizuno
*
Department of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: tmizuno@mail.ecc.u-tokyo.ac.jp; Fax: +81-3-5841-7220
First published on 6th May 2016
Abstract
Phenol, an important industrial chemical, is widely produced using the well-developed cumene process. However, demand for the development of a novel alternative method for synthesizing phenol from benzene has been increasing. Herein, we report a novel system for the synthesis of phenols through aerobic oxidative dehydrogenation of cyclohexanols and cyclohexanones, including ketone–alcohol (KA) oil, catalyzed by Mg–Al-layered double hydroxide (LDH)-supported Au–Pd alloy nanoparticles (Au–Pd/LDH). Alloying of Au and Pd and basicity of LDH are key factors in achieving the present transformation. Although monometallic Au/LDH, Pd/LDH, and their physical mixture showed almost no catalytic activity, Au–Pd/LDH exhibited markedly high catalytic activity for the dehydrogenative phenol production. Mechanistic studies showed that β-H elimination from Pd-enolate species is accelerated by Au species, likely via electronic ligand effects. Moreover, the effect of supports was critical; despite the high catalytic performance of Au–Pd/LDH, Au–Pd bimetallic nanoparticles supported on Al2O3, TiO2, MgO, and CeO2 were ineffective. Thus, the basicity of LDH plays a deterministic role in the present dehydrogenation possibly through its assistance in the deprotonation steps. The synthetic scope of the Au–Pd/LDH-catalyzed system was very broad; various substituted cyclohexanols and cyclohexanones were efficiently converted into the corresponding phenols, and N-substituted anilines were synthesized from cyclohexanones and amines. In addition, the observed catalysis was truly heterogeneous, and Au–Pd/LDH could be reused without substantial loss of its high performance. The present transformation is scalable, utilizes O2 in air as the terminal oxidant, and generates water as the only by-product, highlighting the potential practical utility and environmentally benign nature of the present transformation. Dehydrogenative aromatization of cyclohexanols proceeds through (1) oxidation of cyclohexanols to cyclohexanones; (2) dehydrogenation of cyclohexanones to cyclohexenones; and (3) disproportionation of cyclohexenones to afford the desired phenols. In the present Au–Pd/LDH-catalyzed transformation, the oxidation of the Pd–H species is included in the rate-determining step.
Introduction
Phenol and its derivatives are very important industrial chemicals and key structural motifs in various pharmaceuticals, agrochemicals, plastics, and resins.1 Annually, millions of tons of phenol are produced mainly by the well-established three-step cumene process starting from benzene (Scheme 1).2 However, this traditional cumene process suffers from several drawbacks, including the formation of acetone co-product and the involvement of an explosive cumene hydroperoxide intermediate.2 Although the current cumene process is the most competitive and widely employed commercial process for phenol, the development of new processes that avoid these problems is highly desirable. In this context, processes for the one-step direct oxygenation of benzene to phenol using various oxidants such as H2O2, N2O, and O2 have attracted considerable attention as promising alternatives for producing phenols.3 However, little success has been achieved in the field of direct selective hydroxylation of benzene to phenol, largely because of the inertness of C–H bonds in benzene and the more facile oxidation of phenol than benzene by oxidizing agents, leading to overoxidation.3 Dehydrogenative aromatization of a mixture of cyclohexanone and cyclohexanol—specifically, an unrefined mixture of cyclohexanone and cyclohexanol, known as ketone–alcohol (KA) oil—can provide an alternative approach to access phenol from benzene. KA oil is readily available through a well-developed two-step commercial process involving the hydrogenation of benzene to cyclohexane and aerobic autoxidation of cyclohexane (Scheme 1). | ||
| Scheme 1 Synthetic procedures for phenol. | ||
Recently, several homogeneous Pd-based catalytic systems for the dehydrogenation of cyclohexanones to phenols using O2 as the terminal oxidant under mild reaction conditions have been developed.4 These systems are highly valuable for the synthesis of phenols with various substitution patterns given (1) the limitations of traditional electrophilic substitution reactions for the regioselective introduction of substituents onto a phenol ring as a consequence of the strong electron-directing effects of hydroxy groups and (2) the easy accessibility of cyclohexanones with various substituents on cyclohexyl rings through a wide range of chemical transformations. Acceptorless dehydrogenation of cyclohexanones using a homogeneous Ir-based catalyst has also been developed.5 Despite the high efficiency of these homogeneous systems, they suffer several drawbacks, including the requirement of rather sophisticated ligands and/or high reaction temperatures and difficulties associated with the separation and reuse of the catalysts. Quite recently, a supported Pd-nanoparticle-catalyzed dehydrogenation of cyclohexanones has also been developed either using O2 (5 atm) as a hydrogen acceptor6a or under acceptorless conditions.6b However, in these cases, the need for large amounts of bases as additives and/or leaching of Pd species is problematic in terms of catalyst stability and reusability. Therefore, the development of novel catalytic systems using easily separable and reusable heterogeneous catalysts under ligand- and additive-free conditions is highly desirable from practical, economical, and environmental viewpoints. Despite remarkable progress in the dehydrogenation of cyclohexanones to phenols in recent years, as far as we know, the literature contains no reports of efficient catalytic systems that directly convert cyclohexanols to phenols with useful yields under relatively mild conditions. Therefore, the development of novel catalytic systems that facilitate dehydrogenation of cyclohexanols to phenols would not only enable the synthesis of phenol directly using KA oil as the feedstock without tedious isolation of cyclohexanone, but would also complement the aforementioned synthesis of substituted phenols via the dehydrogenation of cyclohexanones.
Over the past several decades, (supported) metal nanoparticles have attracted much attention as efficient catalysts for a wide variety of aerobic oxidation reactions.7 In particular, multi-component noble-metal nanoparticle catalysts have often shown catalytic activities superior to those of their monometallic counterparts because of the synergistic effects between different components.8 In recent years, Au-based multi-metallic nanoparticles have been among the most extensively studied catalysts.9 For example, Au–Pd bimetallic nanoparticle catalysts have shown significantly higher catalytic activity than Au or Pd monometallic nanoparticles catalysts for oxygen-related transformations such as the aerobic oxidations of CO to CO2,10 alcohols to carbonyl compounds,11 and H2 to H2O2.12 The high catalytic activity of multi-metallic nanoparticles is attributable to the ligand and ensemble effects, which can affect the local electronic and geometric structures of nanoparticles, thereby leading to enhancement of the catalytic performance.8 In the case of supported multi-metallic nanoparticles, in addition to their structures (e.g., core–shell, hetero, or alloyed structures), the choice of appropriate supports is also key to achieving high catalytic performance. The support can not only stabilize the highly dispersed metal nanoparticles, but also substantially promote the reaction through metal-support cooperation.13 Therefore, we envisage that Pd nanoparticles alloyed with other metals (e.g., Au) supported on suitable support materials could promote the aerobic oxidative dehydrogenation of cyclohexanols and cyclohexanones to phenols.
Herein, we report for the first time that Au–Pd bimetallic alloy nanoparticles supported on Mg–Al-layered double hydroxide14 (Au–Pd/LDH) can efficiently promote the aerobic oxidative dehydrogenation of various structurally diverse cyclohexanols to phenols via a consecutive triple-dehydrogenation process. Interestingly, whereas the bimetallic Au–Pd/LDH exhibits high catalytic performance, monometallic Au/LDH and Pd/LDH and their physical mixture exhibit almost no catalytic activity toward the dehydrogenation. The catalysis for the present dehydrogenative aromatization was truly heterogeneous, and the Au–Pd/LDH catalyst could be reused at least 10 times with retention of its high catalytic performance.
Results and discussion
Preparation and characterization of LDH-supported Au–Pd alloy nanoparticles
Initially, we prepared several Au–Pd/LDH catalysts with different Au/Pd ratios using the deposition–precipitation method. Specifically, Au and Pd species were co-precipitated onto the LDH surface as hydroxide species and these metal species were subsequently reduced using NaBH4 (for details on the preparation procedures for the Au–Pd/LDH catalysts, see the Experimental section). As revealed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis, the Au/Pd molar ratios of the Au–Pd/LDH catalysts slightly deviated from those of the initial metal precursors used to prepare the catalysts. Hereafter, Au–Pd/LDH catalysts with an initial solution Au/Pd molar ratio of x/y are designated as AuxPdy/LDH. The metal contents of AuxPdy/LDH are summarized in Table S1.†X-ray diffraction (XRD) patterns of the catalysts showed that the layered structure of LDH was well maintained after immobilization of the Au–Pd bimetallic nanoparticles onto the surface (Fig. S1†). Transmission electron microscopy (TEM) analysis revealed that the average Au–Pd bimetallic nanoparticle size of Au9Pd1/LDH was 3.2 nm (Fig. 1a and b; for the average metal nanoparticle sizes of other catalysts, see Table S1†). In addition, high-angle annular dark-field scanning TEM (HAADF-STEM) and energy dispersive X-ray spectroscopy (EDS) analyses of Au9Pd1/LDH indicated that Au–Pd alloy nanoparticles were formed on the surface of LDH.15 This result was also supported by the UV-vis spectrum of Au9Pd1/LDH; the intensity of the absorption band at approximately 520 nm, which is derived from plasmon resonance absorption of Au nanoparticles, was markedly decreased compared to that of monometallic Au/LDH (Fig. S2†).16 Similarly, UV-vis spectra of other supported bimetallic nanoparticle catalysts AuxPdy/LDH also indicated the formation of Au–Pd alloy nanoparticles (Fig. S2†).
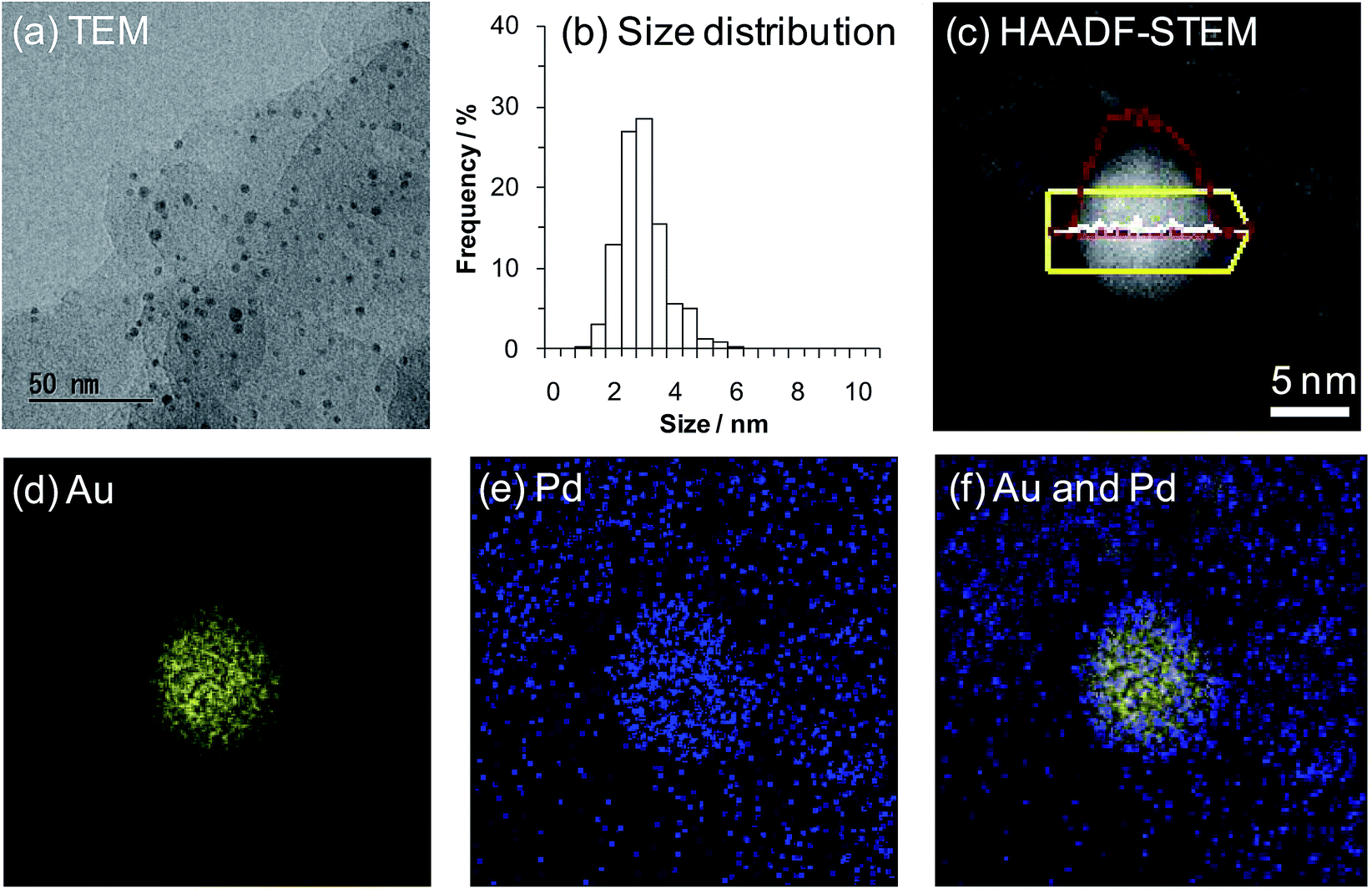 | ||
| Fig. 1 (a) TEM image of Au9Pd1/LDH. (b) The size distribution of Au9Pd1/LDH. (c) HAADF-STEM image of Au9Pd1/LDH. EDS intensity line profiles are shown in red (Au) and white (Pd) lines. (d) EDS elemental map of Au. (e) EDS elemental map of Pd. (f) Overlap of EDS elemental map of Au and Pd. | ||
Optimization of reaction conditions
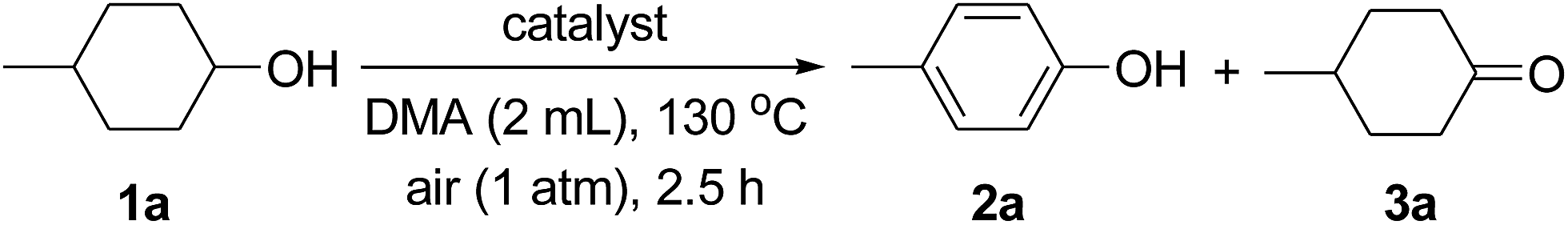
Next, various supported Au–Pd alloy nanoparticles were applied to the dehydrogenative aromatization of 4-methylcyclohexanol (1a) to 4-methylphenol (2a) (Table 1). The reaction was carried out in N,N-dimethylacetamide (DMA) at 130 °C under open-air conditions. Although monometallic Au/LDH and Pd/LDH promoted the oxidation of 1a to 4-methylcyclohexanone (3a), the desired 2a through the dehydrogenative aromatization of 3a was not obtained in either case (Table 1, entries 1 and 6). Surprisingly, when the reaction was carried out with Au9Pd1/LDH as the catalyst, 91% yield of 2a was obtained (Table 1, entry 2). The effect of the Au/Pd molar ratio on the present transformation was significant. The catalytic activity decreased with decreasing Au/Pd molar ratio (Table 1, entries 1–6), and Au1Pd1/LDH and Au1Pd4/LDH hardly gave 2a (Table 1, entries 4 and 5). The effect of the support on the present reaction was also critical. Au–Pd bimetallic nanoparticles supported on other materials such as Al2O3, TiO2, and CeO2 did not exhibit catalytic activity for the dehydrogenation; in these cases, neither 2a nor 3a was formed (Table 1, entries 7–9). Although the dehydrogenation of 1a gave 3a when using MgO as the support for Au–Pd alloy nanoparticles, 2a was not formed at all (Table 1, entry 10). Notably, the aerobic oxidation of 1a to 3a by Au4Pd1/Al2O3 was significantly promoted by addition of bases such as K2CO3 (stoichiometric amount, 0.5 mmol) and LDH (100 mg) to the reaction mixture (Table 1, entries 11–13). Thus, the basicity of the LDH support likely plays a key role in promoting the oxidation of 1a to 3a through its assistance of the abstraction of the proton from the hydroxy group of 1a.14 The dehydrogenative aromatization of 1a with a physical mixture of Au/LDH and Pd/LDH (where the Au/Pd ratio was the same as that of Au9Pd1/LDH) only gave a trace amount of 2a and a moderate yield of 3a (Table 1, entry 14), which indicates that formation of the bimetallic alloy is indispensable for the effective transformation of 3a to 2a. In addition, the reaction with a physical mixture of Au4Pd1/Al2O3 or Au9Pd1/TiO2 and LDH did not give 2a at all, thus suggesting that highly dispersed Au–Pd bimetallic alloy nanoparticles on the surface of LDH are the key to achieving highly efficient dehydrogenative aromatization of 3a to 2a (Table 1, entries 13 and 15). The reaction under an Ar atmosphere gave only a trace amount of 3a and did not give 2a, which suggests that O2 in air functioned as the terminal oxidant in the present system (Table 1, entry 16). In the absence of a catalyst, the reaction did not proceed (Table 1, entry 17). Among the various solvents examined, i.e., DMA, N,N-dimethylformamide (DMF), N-methylpyrrolidone (NMP), dimethylsulfoxide (DMSO), monochlorobenzene, and mesitylene, DMA best promoted the present dehydrogenation (Table S2†).17|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Conv. (%) | Yield (%) | Au/Pdb | |
| 2a | 3a | ||||
| a Reaction conditions: 1a (0.5 mmol), catalyst (total amount of metals: 3.6 mol%), DMA (2 mL), 130 °C, air (1 atm), 2.5 h. Conversion and yields were determined by GC analysis. DMA = N,N-dimethylacetamide. b Amount of metals (mol%). c K2CO3 (0.5 mmol). d LDH (100 mg). e A physical mixture of Au/LDH (3.1 mol%) and Pd/LDH (0.5 mol%). f Ar (1 atm). | |||||
| 1 | Au/LDH | 55 | nd | 55 | 3.6/0 |
| 2 | Au 9 Pd 1 /LDH | 94 | 91 | 2 | 3.1/0.5 |
| 3 | Au4Pd1/LDH | 87 | 79 | 4 | 2.6/1.0 |
| 4 | Au1Pd1/LDH | 52 | 2 | 29 | 1.5/2.1 |
| 5 | Au1Pd4/LDH | 59 | nd | 43 | 0.7/2.9 |
| 6 | Pd/LDH | 56 | nd | 45 | 0/3.6 |
| 7 | Au9Pd1/Al2O3 | <1 | nd | nd | 3.2/0.4 |
| 8 | Au9Pd1/TiO2 | <1 | nd | nd | 3.1/0.5 |
| 9 | Au9Pd1/CeO2 | <1 | nd | nd | 3.2/0.4 |
| 10 | Au9Pd1/MgO | 31 | nd | 21 | 2.9/0.7 |
| 11 | Au4Pd1/Al2O3 | 17 | nd | 11 | 2.8/0.8 |
| 12c | Au4Pd1/Al2O3 + K2CO3 | 60 | nd | 35 | 2.8/0.8 |
| 13d | Au4Pd1/Al2O3 + LDH | 32 | nd | 22 | 2.8/0.8 |
| 14e | Au/LDH + Pd/LDH | 73 | 5 | 53 | 3.1/0.5 |
| 15d | Au9Pd1/TiO2 + LDH | 2 | nd | 2 | 3.1/0.5 |
| 16f | Au9Pd1/LDH | 3 | nd | 3 | 3.1/0.5 |
| 17 | None | 21 | nd | nd | — |
The conditions for the reaction starting from 3a were also optimized. As shown in Table S3,† Au/LDH and Pd/LDH exhibited almost no catalytic activity toward the dehydrogenation of 3a (Table S3,† entries 1 and 6). For the reaction of 3a, Au9Pd1/LDH also exhibited the best performance among various LDH-supported Au–Pd alloy nanoparticle catalysts (Table S3,† entries 2–5). Au–Pd alloy nanoparticles supported on Al2O3, TiO2, CeO2, and MgO gave only trace amounts of 2a (Table S3,† entries 7–10). The dehydrogenation of 3a using Au4Pd1/Al2O3 as the catalyst was substantially promoted by the addition of K2CO3 or LDH (Table S3,† entries 11–13). These results strongly support the hypothesis that the basicity of LDH plays the key role in the dehydrogenative aromatization of 3a, likely via its assistance in the abstraction of α-H from 3a.14 The physical mixture of Au/LDH and Pd/LDH did not exhibit catalytic activity for the dehydrogenative aromatization (Table S3,† entry 14), and the physical mixture of Au9Pd1/TiO2 and LDH afforded a lower yield of 2a than Au9Pd1/LDH (Table S3,† entry 15), indicating the highly dispersed Au–Pd alloy nanoparticles on the surface of LDH are indispensable for the dehydrogenative aromatization of 3a. The transformation of 3a under an Ar atmosphere did not give 2a, which suggests that O2 in air is the terminal oxidant (Table S3,† entry 16). These results related to the transformation of 3a agree well with those obtained starting from 1a.
Heterogeneous catalysis and reusability of Au9Pd1/LDH
To verify whether the observed catalysis was truly heterogeneous, the Au9Pd1/LDH catalyst was removed by hot filtration when the conversion of 1a reached approximately 40%, and the reaction was repeated using the filtrate under the same reaction conditions. As shown in Fig. 2, the reaction was completely stopped by the removal of the catalyst. Furthermore, the ICP-AES analysis results confirmed that Au and Pd species were hardly present in the filtrate (below the instrumental detection limits: Au < 0.01%, Pd < 0.38%). On the basis of these experimental results, we ruled out the possibility that the catalysis originated from metal species leached from the catalyst; thus, the observed catalysis was truly heterogeneous.18 | ||
| Fig. 2 Effect of removal of the Au9Pd1/LDH catalyst on the present oxidative dehydrogenation. The reaction conditions were the same as those described in Table 1. GC yields are shown here. Closed squares and circles indicate the yields of 2a and 3a, respectively, without removal of the catalyst; open squares and circles indicate the yields of 2a and 3a, respectively, after removal of the catalyst. | ||
After the dehydrogenation of 1a to 2a was completed, Au9Pd1/LDH was easily retrieved from the reaction mixture by simple filtration, with greater than 95% recovery, for each reuse experiment. The retrieved catalyst could be reused at least 10 times without substantial loss of its high catalytic performance. Even at the 10th reuse experiment, 85% yield of 2a was still obtained (Fig. 3). The catalytic performance could be restored by simply washing the used catalyst with a 0.1 M NaOH solution in water/ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). XRD and TEM analyses of the fresh as-prepared Au9Pd1/LDH catalyst and the catalyst after the 10th reuse experiment show that the LDH structure and the average size of the Au–Pd bimetallic alloy nanoparticles were almost unchanged (fresh: 3.2 nm, after the 10th reuse experiment: 3.3 nm) (Fig. S3 and S4†). The difference UV-vis spectrum between fresh Au9Pd1/LDH and Au9Pd1/LDH after the 10th reuse experiment revealed a new absorption band centered at approximately 250 nm, which is attributable to aromatic compounds (possibly 2a) (Fig. S5†). These results indicate that the gradual decrease of the catalytic activity is likely caused by the absorption of acidic phenol onto the surface of the catalyst. Thus, the catalytic ability (basicity) of the LDH support was readily recovered by washing with the NaOH solution, thereby restoring the catalytic activity of Au9Pd1/LDH.
:1). XRD and TEM analyses of the fresh as-prepared Au9Pd1/LDH catalyst and the catalyst after the 10th reuse experiment show that the LDH structure and the average size of the Au–Pd bimetallic alloy nanoparticles were almost unchanged (fresh: 3.2 nm, after the 10th reuse experiment: 3.3 nm) (Fig. S3 and S4†). The difference UV-vis spectrum between fresh Au9Pd1/LDH and Au9Pd1/LDH after the 10th reuse experiment revealed a new absorption band centered at approximately 250 nm, which is attributable to aromatic compounds (possibly 2a) (Fig. S5†). These results indicate that the gradual decrease of the catalytic activity is likely caused by the absorption of acidic phenol onto the surface of the catalyst. Thus, the catalytic ability (basicity) of the LDH support was readily recovered by washing with the NaOH solution, thereby restoring the catalytic activity of Au9Pd1/LDH.
 | ||
| Fig. 3 Au9Pd1/LDH reuse experiments. The reaction conditions were the same as those described in Table 1. GC yields are shown here. Generally, the retrieved catalyst was washed with water and ethanol. However, the retrieved catalysts after 5th, 8th, and 9th reuse experiments were washed with a 0.1 M NaOH solution in water/EtOH (1:1). | ||
Substrate scope
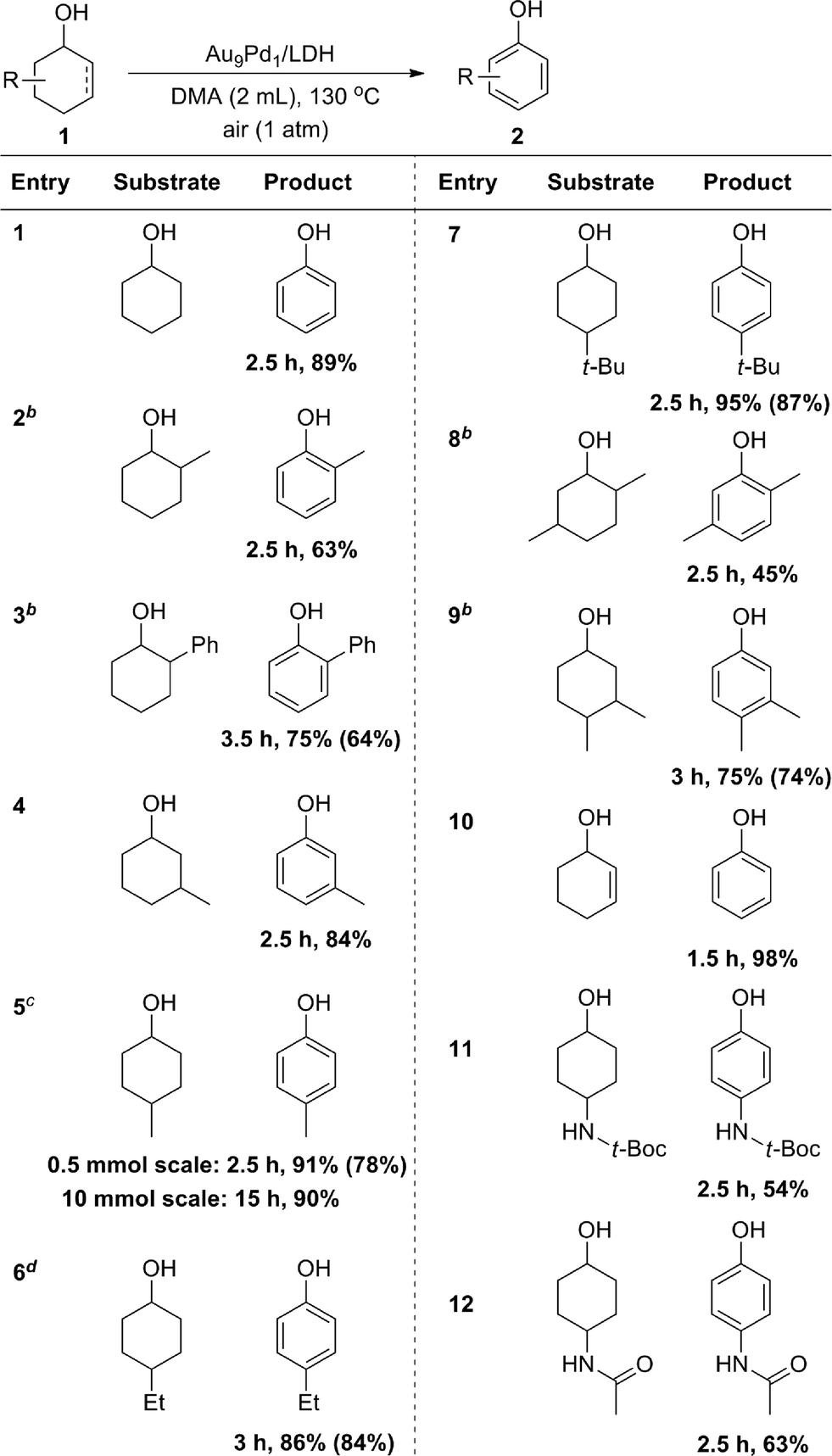
With the optimized reaction conditions in hand, we next investigated the scope of the present Au9Pd1/LDH-catalyzed dehydrogenative aromatization of cyclohexanols. Under the optimized reaction conditions, various structurally diverse cyclohexanols were efficiently converted into the corresponding phenols. The phenol products were readily isolated; their yields are summarized in Table 2. The dehydrogenation of simple cyclohexanol afforded phenol in 89% yield (Table 2, entry 1). The dehydrogenation of cyclohexanols with various substituents at 2-, 3-, and 4-positions smoothly proceeded (Table 2, entries 2–12). 2-Substituted cyclohexanols were not as reactive as 3- and 4-substituted ones, suggesting that steric effects on the dehydrogenation are substantial (Table 2, entries 2, 3, and 8). Various disubstituted cyclohexanols were also suitable substrates for the present reaction (Table 2, entries 8 and 9). 2-Cyclohexen-1-ol also reacted well to give phenol in excellent yield (Table 2, entry 10). Amide groups on the 4-positions remained intact under the reaction conditions (Table 2, entries 11 and 12). The present dehydrogenation could also be scaled up; for the 10 mmol-scale dehydrogenation of 1a, the amount of the catalyst could be reduced to 1 mol%, and 2a was obtained in 90% isolated yield (Table 2, entry 5).|
a Reaction conditions: 1 (0.5 mmol), Au9Pd1/LDH (total amount of metals: 3.6 mol%), DMA (2 mL), 130 °C, air (1 atm). Conversion and yields were determined by GC analysis. The values in the parentheses are the isolated yields. bAu9Pd1/LDH (total amount of metals: 7.2 mol%). cFor 10 mmol-scale synthesis, reaction conditions: Au9Pd1/LDH (total amount of metals: 1 mol%), DMA (10 mL), 130 °C, air (1 atm). The isolated yield is shown. dMesitylene/DMA (1:1, 2 mL). |
|---|
|
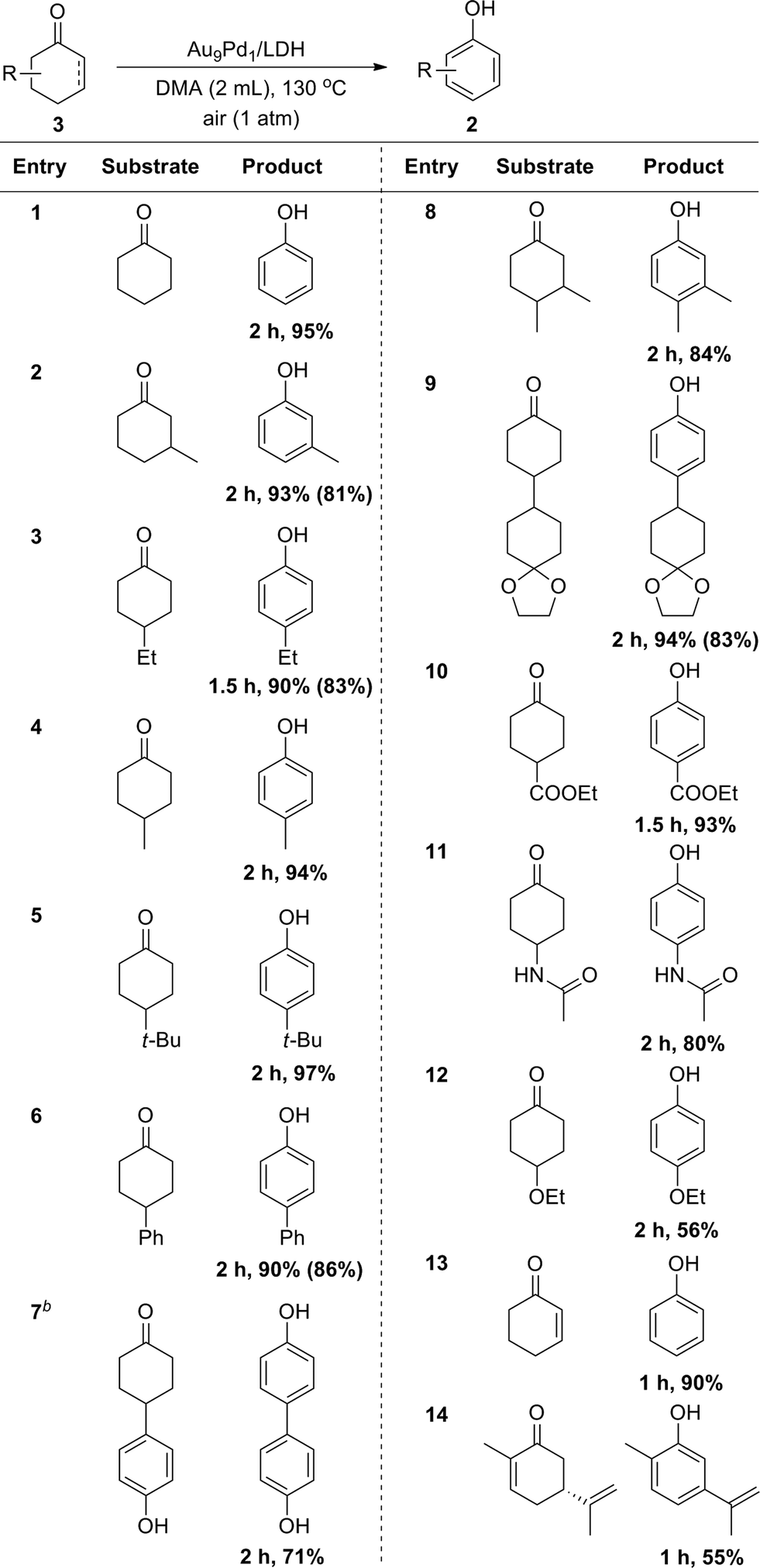
Cyclohexanones could also be utilized as starting materials for the synthesis of phenols, and simple cyclohexanone and its derivatives with various substituents at 2-, 3- and 4-positions were all suitable substrates (Table 3). Disubstituted phenols could also be synthesized starting from the corresponding disubstituted cyclohexanones (Table 3, entry 8). In the case of a glycol-protected cyclohexanone, the dehydrogenation proceeded without deprotection of the carbonyl group (Table 3, entry 9). In addition, cyclohexanones substituted with an ester, amide, or alkoxy group were dehydrogenated efficiently without hydrolytic decomposition of these functional groups (Table 3, entries 10–12). Enones, such as 2-cyclohexen-1-one and carvone, could also be utilized as substrates for the present dehydrogenation (Table 3, entries 13 and 14).
|
a Reaction conditions: 3 (0.5 mmol), Au9Pd1/LDH (total amount of metals: 3.6 mol%), DMA (2 mL), 130 °C, air (1 atm). Conversion and yields were determined by GC analysis. The values in the parentheses are the isolated yields. bAu4Pd1/LDH (total amount of metals: 3.6 mol%). |
|---|
|
Recently, Pd-catalyzed dehydrogenative aromatization has emerged as an attractive method for the synthesis of N-substituted anilines from cyclohexanones and amines.19 Encouraged by the unique activity of Au9Pd1/LDH for the dehydrogenative aromatization of cyclohexanols and cyclohexanones, we also synthesized various N-substituted anilines using the dehydrogenative aromatization strategy. Under reaction conditions similar to those used for the aforementioned dehydrogenation, cyclohexanones with various substituents on the cyclohexyl rings were efficiently reacted with various alkyl and benzylamines to give the corresponding N-substituted anilines (Table 4).


To further demonstrate the potential practical applicability of the present Au9Pd1/LDH-catalyzed dehydrogenative aromatization, we attempted to synthesize phenol and anilines directly from KA oil. When a 1:1 mixture of cyclohexanol and cyclohexanone was used as the starting material, 96% yield of phenol was obtained (Scheme 2a). In addition, KA oil was reacted with n-octylamine to give N-octylaniline in high yield (Scheme 2b).
 | ||
| Scheme 2 Syntheses of phenol and N-octylaniline starting from KA oil. Reaction conditions: (a) 1b (0.5 mmol), 3b (0.5 mmol), DMA (2 mL), 130 °C, air (1 atm), 4 h. (b) 1b (0.5 mmol), 3b (0.5 mmol), n-octylamine (0.5 mmol), DMA (1 mL), mesitylene (1 mL), 130 °C, air (1 atm), 3 h. Yields were determined by GC analysis. The parenthetical value is the isolated yield. | ||
Mechanistic studies
The reaction profile for the Au9Pd1/LDH-catalyzed oxidative dehydrogenation of cyclohexanol (1b) showed that cyclohexanone (3b) was initially produced, followed by the dehydrogenation of 3b to 2-cyclohexen-1-one (4b); the further dehydrogenation of 4b gave phenol (2b) as the final product (Fig. 4a). The dehydrogenation starting from 3b also gave 2b in high yield, which suggests the reaction proceeded through 3b as the intermediate (Fig. 4b). The reaction profile for the dehydrogenation starting from 4b revealed that 2b was produced through the disproportionation of 4b (Fig. 4c). Furthermore, the time course of the dehydrogenation starting from 3b showed that only a trace amount of 4b was detected throughout the reaction, which indicates that the disproportionation of 4b is much faster than the dehydrogenation of 3b to 4b (Fig. 4b).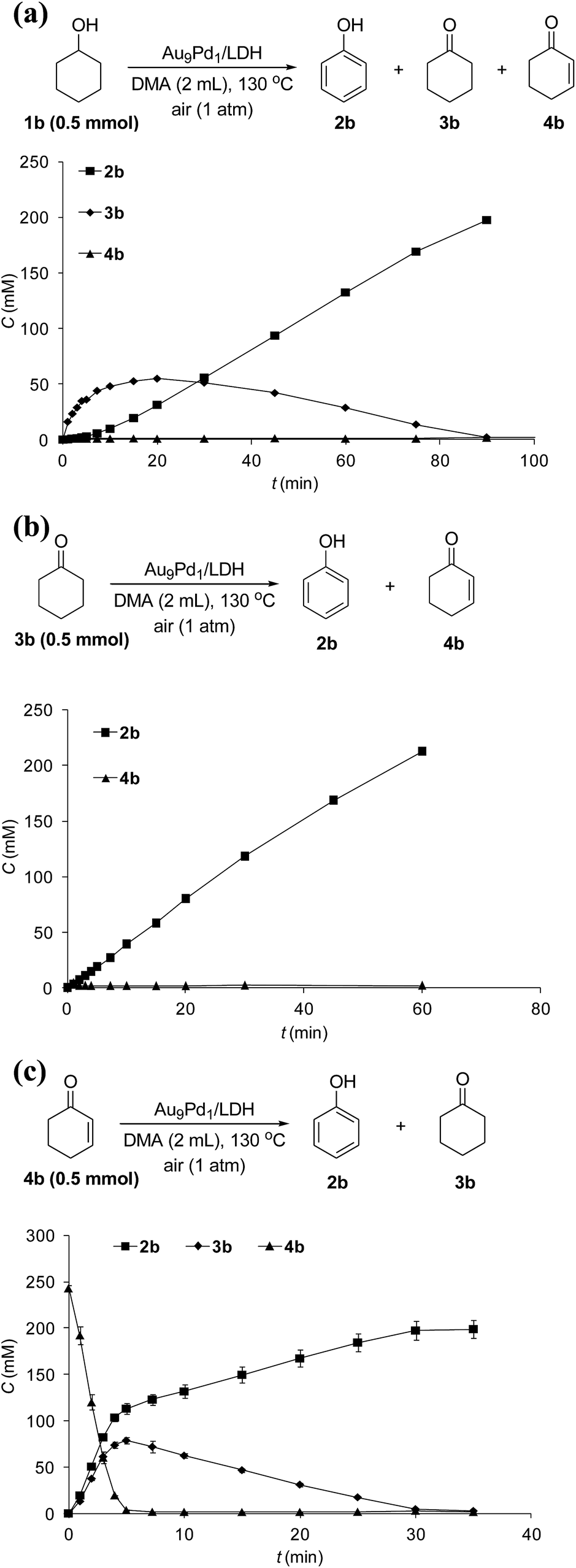 | ||
| Fig. 4 Reaction profiles of dehydrogenations starting from (a) 1b, (b) 3b, and (c) 4b. Reaction conditions: substrates (0.5 mmol), Au9Pd1/LDH (total amount of metals: 3.6 mol%), DMA (2 mL), 130 °C, air (1 atm). GC yields are shown here. | ||
To verify whether the dehydrogenation proceeded through radical intermediates, the Au9Pd1/LDH-catalyzed dehydrogenation of 1b was carried out in the presence of a stoichiometric amount of a radical scavenger, 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO). In this case, the reaction was not suppressed, but was substantially promoted by the addition of TEMPO and the formation of 1-hydroxy-2,2,6,6-tetramethylpiperidine (TEMPOH) was observed (Fig. 5a). Similarly, the dehydrogenations starting from 3b and 4b were both accelerated in the presence of TEMPO, with the concomitant formation of TEMPOH (Fig. 5b and c). Furthermore, control experiments showed that TEMPO only or LDH together with TEMPO (in the absence of catalysts) could not promote the dehydrogenation of 3b (Fig. 5b). All these results indicate that the involvement of radical intermediates is unlikely in the present transformation. TEMPO has been reported to act as a one-electron oxidant to abstract a hydrogen atom from metal hydride species, where TEMPO itself is reduced to TEMPOH.20,21 Therefore, the aforementioned experimental results strongly support the involvement of metal hydride species such as Au–H20 and Pd–H21 in the consecutive dehydrogenations; the reactions were promoted by the oxidation of these metal hydride species by TEMPO. In particular, for the initial stage of the Au9Pd1/LDH-catalyzed disproportionation of 4b, the formation of 3b was substantially suppressed by the addition of TEMPO compared to the reaction in the absence of TEMPO (Fig. 5c, Table S5,† entries 3 and 4). These results suggest that the ability for the oxidation of the metal hydride species (Pd–H) increases in the order O2 < 4b < TEMPO.
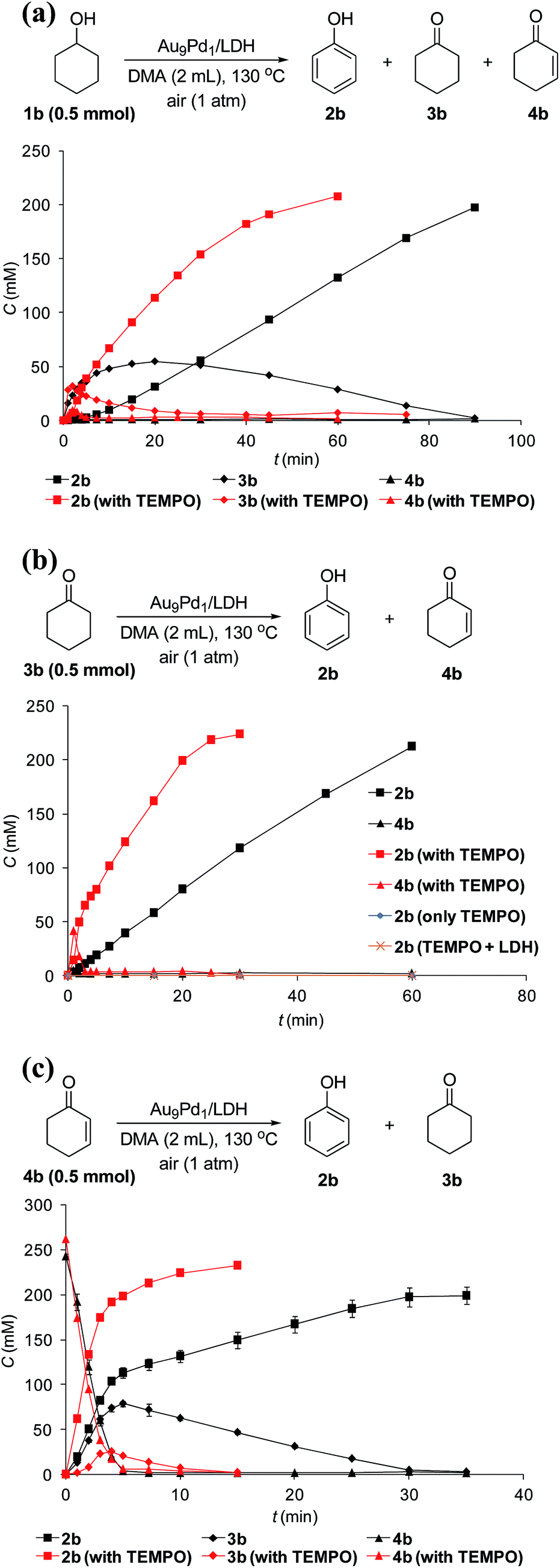 | ||
| Fig. 5 Effects of TEMPO on dehydrogenations starting from (a) 1b, (b) 3b, and (c) 4b. Reaction conditions: substrates (0.5 mmol), Au9Pd1/LDH (total amount of metals: 3.6 mol%), DMA (2 mL), 130 °C, air (1 atm). GC yields are shown here. | ||
As shown in Table 1, for the oxidative dehydrogenation of 1a, either Au/LDH or Pd/LDH could promote the dehydrogenation of 1a to 3a (Table 1, entries 1 and 6); however, their substrate conversions were much lower than that achieved when Au9Pd1/LDH was used as the catalyst (Table 1, entry 2). These results indicate that both Pd and Au species in Au9Pd1/LDH intrinsically possess catalytic activity toward the dehydrogenation of 1a to 3a and that the dehydrogenation ability was improved by alloying Au and Pd; this observation is in good agreement with results previously reported by other research groups.11
Although Au/LDH did not show catalytic activity for the dehydrogenative aromatization of 3b, Pd/LDH could promote the transformation into 2b, albeit in low yield (Fig. S6†). In addition, with respect to the disproportionation of 4b, Pd/LDH exhibited high catalytic activity, whereas Au/LDH afforded only a small amount of 2b (Fig. S7†). These results suggest that Pd species in the Au–Pd alloy nanoparticles likely play the main role in the dehydrogenation of 3b to 4b and the successive disproportionation of 4b. In addition, the disproportionation of 4b using Pd/LDH as the catalyst was promoted by the addition of TEMPO, which suggests the involvement of the Pd–H species in the disproportionation (Table S5,† entries 5 and 6).
On the basis of the aforementioned experimental results, we herein propose a plausible reaction mechanism for the dehydrogenation of cyclohexanols to phenols. For the oxidation of cyclohexanols to cyclohexanones (Scheme 3, cycle 1), deprotonative coordination of cyclohexanols to the metal species (either Au or Pd) promoted by LDH generates metal alkoxy species (Scheme 3, cycle 1, step 1), followed by β-H elimination to give cyclohexanones with the concomitant formation of metal hydride species (either Au–H or Pd–H) (Scheme 3, cycle 1, step 2). The metal hydride species can then be oxidized by O2 (Scheme 3, cycle 1, step 3). For the dehydrogenation of cyclohexanones to cyclohexenones (Scheme 3, cycle 2), deprotonative coordination of cyclohexanones to Pd with the assistance of basic LDH affords Pd-enolate species (Scheme 3, cycle 2, step 4) followed by β-H elimination to give cyclohexenones (Scheme 3, cycle 2, step 5). The oxidation of Pd–H can then regenerate the catalyst (Scheme 3, cycle 2, step 6).22 Finally, the disproportionation of cyclohexenones proceeds through a catalytic cycle similar to that of the dehydrogenation of cyclohexanones to cyclohexenones (Scheme 3, cycle 3). Specifically, LDH-assisted α-C–H cleavage generates Pd-enolate species (Scheme 3, cycle 3, step 7) followed by β-H elimination to give the final phenol products with the concomitant formation of the Pd–H species (Scheme 3, cycle 3, step 8). In the cycle 3, the hydrogen acceptor is cyclohexenones rather than O2 (Scheme 3, cycle 3, step 9).
 | ||
| Scheme 3 A plausible reaction mechanism. M = Au or Pd. | ||
Because of the mechanism of alcohol oxidation by supported Au–Pd alloy nanoparticles has been extensively investigated by other research groups,11 we herein mainly focused on the elucidation of the detailed roles of Au–Pd alloy nanoparticles in the dehydrogenation of cyclohexanones to phenols. As previously mentioned, Pd/LDH exhibited significant catalytic activity for the oxidation of 1a to 3a (Table 1, entry 6) but did not exhibit catalytic activity for the dehydrogenative aromatization of 3a to 2a (Table S2,† entry 6). Furthermore, the disproportionation of 4b was efficiently promoted by Pd/LDH (Fig. S7†). These results suggest that the Au atoms in Au–Pd alloy nanoparticles play an important assisting role on the dehydrogenation of cyclohexanones to cyclohexenones by Pd (Scheme 3, cycle 2).
In addition, the dehydrogenation of 3b with Pd/LDH was not accelerated by TEMPO even though TEMPO is known to promote the oxidation of Pd–H species (Table S4,† entries 5 and 6);21 this result suggests that the low activity of Pd/LDH for the dehydrogenation of 3b is not likely due to the inertness of Pd–H species toward oxidation by O2 (Scheme 3, cycle 2, step 6) but is instead due to slow β-H elimination (Scheme 3, cycle 2, step 5). The Au species in Au–Pd alloy nanoparticles possibly promote β-H elimination by Pd, likely via electronic ligand effects.9–12 The greater electronegativity of Au compared to that of Pd has been reported to result in a net electron transfer from Pd to Au; thus, the Au species become more electron-rich and the Pd species become more electron-poor.9–12 Therefore, the β-H elimination step in the dehydrogenation of 3b to 4b likely become more favorable with increasing Au/Pd molar ratio of the catalysts. Consequently, the catalytic activity of the AuxPdy/LDH catalysts possibly increased with increasing Au/Pd molar ratio, and thus Au9Pd1/LDH exhibits much higher catalytic activity than other catalysts toward the present dehydrogenation (Tables 1 and S3†).
Kinetic studies on the Au9Pd1/LDH-catalyzed dehydrogenation of 3b showed the saturation kinetics of the reaction rate based on the concentration of 3b (22.9–478.9 mM, Fig. 6a), which indicates that a pre-equilibrium exists between Pd species and Pd-enolate species for cleavage of an α-C–H bond. The dehydrogenation of cyclohexanone-2,2,6,6-d4 (97% deuterium labeling at the 2- and 6-positions) gave phenol with 65% deuterium labeling at the 2- and 6-positions (Scheme S1†); this deuterium loss also supports the existence of the pre-equilibrium. In addition, the kinetic studies revealed a first-order dependence of the reaction rate on the partial pressure of O2 (0.1–1.0 atm, Fig. 6b), which suggests that the oxidation of the Pd–H species is included in the rate-determining step for the dehydrogenation of 3b. The aforementioned significant promotion effects of TEMPO also suggest that the oxidation of the Pd–H species is included in the rate-determining step.
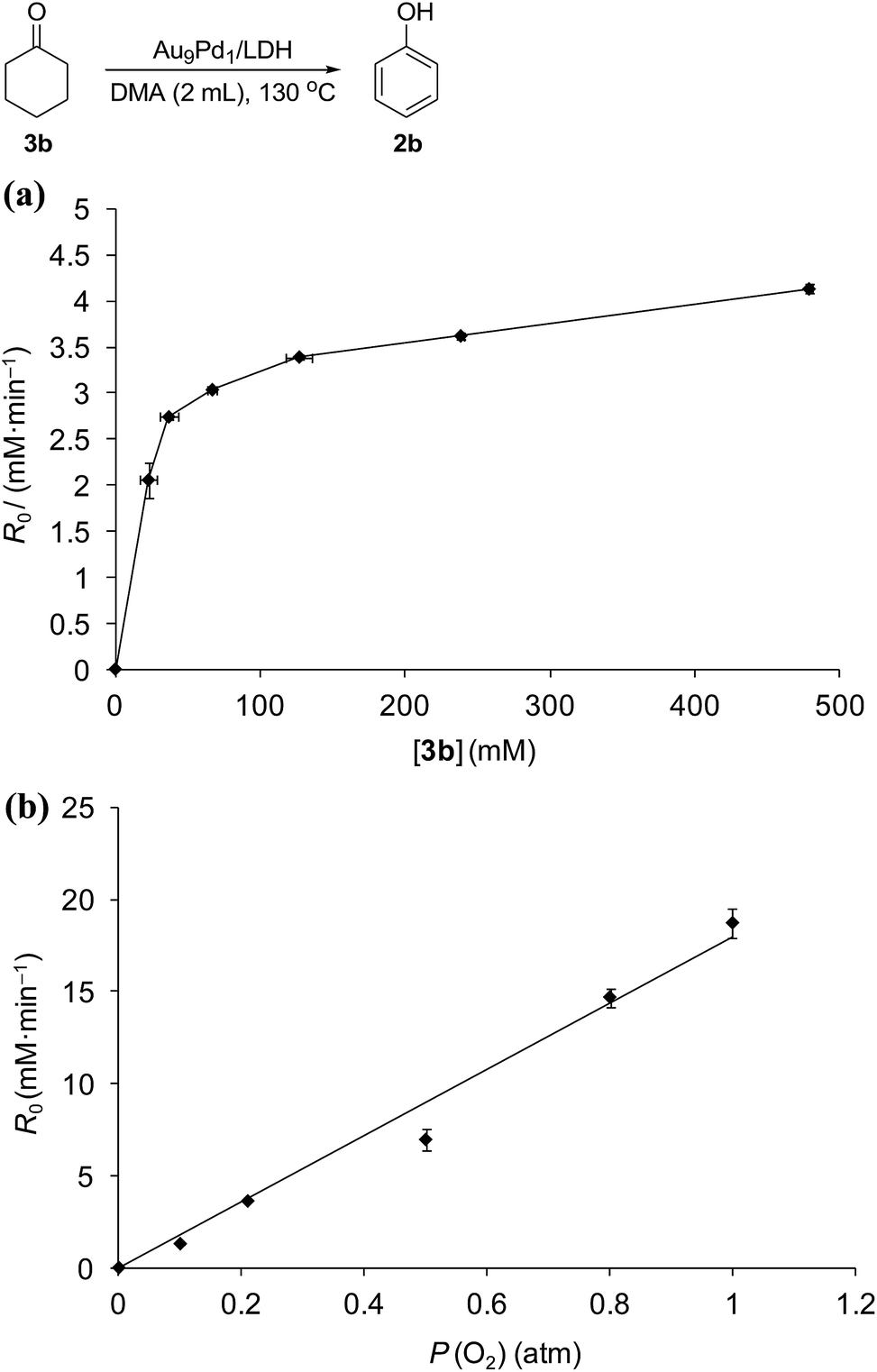 | ||
| Fig. 6 Dependence of reaction rates of the dehydrogenation of 3b on (a) the concentration of 3b and (b) the partial pressure of O2, line fit: R0 = 17.9P (O2), R2 = 0.98. Reaction conditions: (a) 3b (22.9–478.9 mM), Au9Pd1/LDH (total amount of metals: 3.6 mol%), DMA (2 mL), 130 °C, air (1 atm); (b) 3b (250 mM), Au9Pd1/LDH (total amount of metals: 3.6 mol%), DMA (2 mL), 130 °C, O2 (0.1–1 atm). GC yields are shown here. | ||
Conclusion
We successfully developed a novel heterogeneously-catalyzed oxidative dehydrogenation reaction for the conversion of cyclohexanols and cyclohexanones into phenols using Au9Pd1/LDH. Monometallic Au or Pd supported on LDH exhibited almost no activity toward the dehydrogenation, whereas marked enhancement of the catalytic activity was observed for alloyed Au and Pd. Mechanistic studies showed that β-H elimination from Pd-enolate species is accelerated by Au species, likely via electronic ligand effects; this acceleration is the key to achieve the high activity of Au9Pd1/LDH. The basicity of LDH also plays a deterministic role in the present dehydrogenation through its assistance in the deprotonation steps. Au9Pd1/LDH functioned as a truly heterogeneous catalyst and could be reused at least 10 times with retention of its high catalytic performance. The present dehydrogenation utilizes O2 in air as the terminal oxidant and generates water as the only by-product, which highlights the environmentally benign nature of the present transformation. The substrate scope was very broad with respect to both cyclohexanols and cyclohexanones, and N-substituted anilines could also be synthesized starting from cyclohexanones and amines. Furthermore, the present catalytic system could be scaled up and applied to the direct conversion of KA oil to phenol. Thus, we demonstrated the potential practical applicability of the present catalyst system for the synthesis of phenol from benzene in industrial processes.Experimental section
Instruments and reagents
Gas chromatography (GC) analyses were performed on a Shimadzu GC-2014 equipped with a flame ionization detector (FID) and an InertCap-5 or a TC-WAX capillary column. GC mass spectrometry (GC-MS) spectra were recorded on a Shimadzu GCMS-QP2010 equipped with an InertCap 5 capillary column at an ionization voltage of 70 eV. Liquid-state NMR spectra were recorded on a JEOL JNM-ECA-500 spectrometer. 1H and 13C NMR spectra were measured at 495.1 and 124.5 MHz, respectively, using tetramethylsilane (TMS) as an internal reference (δ = 0 ppm). ICP-AES analyses were performed on a Shimadzu ICPS-8100. TEM observations were performed on JEOL JEM-2010HC. HAADF-STEM and EDS images were obtained using JEOL JEM-ARM 200F operating at 200 kV. XRD patterns were collected on a Rigaku SmartLab diffractometer (CuKα, λ = 1.5405 Å, 45 kV, 200 mA). UV-Vis diffuse reflectance spectra were recorded on a Jasco V-570DS. Mg–Al LDH (Mg6Al2(OH)16CO3·4H2O, BET surface area: 51 m2 g−1, Tomita Pharmaceutical Co., Ltd.), CeO2 (BET surface area: 111 m2 g−1, Cat. no. 544841-25G, Aldrich), Al2O3 (BET surface area: 160 m2 g−1, Cat. no. KHS-24, Sumitomo Chemical), MgO (BET surface area: 28 m2 g−1, Cat. No. P0082, Ube Industries Ltd.), and TiO2 (BET surface area: 316 m2 g−1, Cat. No. ST-01, Ishihara Sangyo Kaisya) were acquired from commercial sources. Solvents and substrates were obtained from Kanto Chemical, TCI, Wako, or Aldrich (reagent grade) and were purified before use (if required).23Preparation of catalysts
Au–Pd/LDH with different Au/Pd ratios, such as Au9Pd1/LDH, was typically prepared as follows. First, Mg–Al LDH (2.0 g) was added to 60 mL of an aqueous solution of HAuCl4·4H2O (7.50 mM), PdCl2 (0.83 mM), and KCl (2 equiv. with respect to PdCl2, 1.67 mM). The resulting slurry was then stirred vigorously at room temperature for 12 h. The solid was then filtered off, washed with water (3 L), and dried in vacuo to afford the supported hydroxide precursor. The hydroxide precursor was redispersed in 50 mL water and reduced with NaBH4 (70 mg). The resulting slurry was then stirred vigorously at room temperature for 2 h. The solid was then filtered off, washed with water (2 L), and dried in vacuo overnight, giving Au9Pd1/LDH as a dark-gray powder (Au content: 0.148 mmol g−1, Pd content: 0.024 mmol g−1). Au4Pd1/LDH (Au content: 0.134 mmol g−1, Pd content: 0.051 mmol g−1), Au1Pd1/LDH (Au content: 0.091 mmol g−1, Pd content: 0.122 mmol g−1), and Au1Pd4/LDH (Au content: 0.044 mmol g−1, Pd content: 0.192 mmol g−1) were prepared via the same method used to prepare Au9Pd1/LDH. Preparation methods for other catalysts are described in the ESI.†Catalytic dehydrogenation
In a typical dehydrogenation procedure, Au–Pd/LDH (105 mg, total amount of metals: 3.6 mol%), cyclohexanone (0.5 mmol), DMA (2.0 mL), and a Teflon-coated magnetic stir bar were successively placed in a Pyrex glass reactor (volume: ca. 20 mL) and the reaction mixture was vigorously stirred at 50 °C under 1 atm of air. After the reaction was completed, an internal standard (diphenyl) was added to the reaction mixture and the conversion of cyclohexanone and the yield of phenol were determined by GC analysis. In cases where phenol products were isolated, an internal standard was not added. After the reaction, the catalyst was filtered off (>95% recovery). EtOAc (15 mL) and n-hexane (5 mL) were then added to the filtrate, which was washed with brine (10 mL) three times. The organic phase was collected and evaporated to remove solvents. The crude product was subjected to column chromatography on silica gel (for phenols, n-hexane/acetone was typically used as an eluent; for N-octylaniline, n-hexane/ethylacetate was used), giving the pure phenols. The products were identified by GC-MS and NMR (1H and 13C) analyses. The recovered catalyst was washed with water and ethanol (or 0.1 M NaOH in 1:1 water/ethanol) before being used in the reuse experiment.
Acknowledgements
This work was financially supported by Grant-in-Aid for Scientific Research on Innovative Area “Precisely Designed Catalysts with Customized Scaffolding” (15H05797) and Research Activity start-up (15H06143) from MEXT, Japan.Notes and references
- (a) Z. Rappoport, The Chemistry of Phenols, Wiley-VCH, Weinheim, 2003 CrossRef; (b) J. H. P. Tyman, Synthetic and Natural Phenols, Elsevier, New York, 1996 Search PubMed; (c) H. Fiege, H.-W. Voges, T. Hamamoto, S. Umemura, T. Iwata, H. Miki, Y. Fujita, H.-J. Buysch, D. Garbe and W. Paulus, Phenol Derivatives: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed.
- (a) M. Weber, M. Weber and M. Kleine-Boymann, Phenol: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed; (b) J. Wallace, Phenol: Kirk-Othmer Encyclopedia of Chemical Technology, Wiley-VCH, Weinheim, 2005 Search PubMed.
- (a) E. Klemm, A. Reitzmann and B. Vogel, Direct Ring Oxidation of Aromatics to Phenols: Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) T. Jiang, W. Wang and B. Han, New J. Chem., 2013, 37, 1654 RSC; (c) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561 RSC.
- (a) Y. Izawa, D. Pun and S. S. Stahl, Science, 2011, 333, 209 CrossRef CAS PubMed; (b) D. Pun, T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8213 CrossRef CAS PubMed.
- S. Kusumoto, M. Akiyama and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 18726 CrossRef CAS PubMed.
- (a) Z. Zhang, T. Hashiguchi, T. Ishida, A. Hamasaki, T. Honma, H. Ohashi, T. Yokoyama and M. Tokunaga, Org. Chem. Front., 2015, 2, 654 RSC; (b) J. Zhang, Q. Jiang, D. Yang, X. Zhao, Y. Dong and R. Liu, Chem. Sci., 2015, 6, 4674 RSC.
- (a) N. Mizuno, Modern Heterogeneous Oxidation Catalysis, Wiley-VCH, Weinheim, 2009 Search PubMed; (b) C. Louis and O. Pluchery, Gold Nanoparticles for Physics, Chemistry and Biology, Imperial College Press, London, 2013 Search PubMed; (c) G. C. Bond, C. Louis and D. T. Thompson, Catalysis by Gold, Imperial College Press, London, 2006 Search PubMed; (d) M. Boronat, A. Leyva-Pérez and A. Corma, Acc. Chem. Res., 2014, 47, 834 CrossRef CAS PubMed; (e) K. J. Stowers, R. J. Madix and C. M. Friend, J. Catal., 2013, 308, 131 CrossRef CAS; (f) M. S. Ide and R. J. Davis, Acc. Chem. Res., 2014, 47, 825 CrossRef CAS PubMed; (g) L. Prati and A. Villa, Acc. Chem. Res., 2014, 47, 855 CrossRef CAS PubMed; (h) I. X. Green, W. Tang, N. Neurock and J. T. Yates Jr, Acc. Chem. Res., 2014, 47, 805 CrossRef CAS PubMed; (i) Y. Mikami, A. Dhakshinamoorthy, M. Alvaro and H. García, Catal. Sci. Technol., 2013, 3, 58 RSC; (j) S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC; (k) L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825 CrossRef CAS PubMed; (l) N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Chem. Sci., 2012, 3, 20 RSC; (m) H. Cong and J. A. Porco Jr, ACS Catal., 2012, 2, 65 CrossRef CAS PubMed.
- (a) K. Kusada and H. Kitagawa, Adv. Mater., 2016, 28, 1129 CrossRef CAS PubMed; (b) Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 2013, 46, 1816 CrossRef CAS PubMed; (c) A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652 CrossRef CAS.
- (a) J. K. Edwards, S. J. Freakley, A. F. Carley, C. J. Kiely and G. J. Hutchings, Acc. Chem. Res., 2014, 47, 845 CrossRef CAS PubMed; (b) G. J. Hutchings, Catal. Today, 2014, 238, 69 CrossRef CAS; (c) A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403 CrossRef CAS PubMed; (d) M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099 RSC; (e) H. Zhang and N. Toshima, Catal. Sci. Technol., 2013, 3, 268 RSC; (f) G. J. Hutchings and C. J. Kiely, Acc. Chem. Res., 2013, 46, 1759 CrossRef CAS PubMed; (g) H. Miyamura and S. Kobayashi, Acc. Chem. Res., 2014, 47, 1054 CrossRef CAS PubMed; (h) A. Wang, X. Y. Liu, C.-Y. Mou and T. Zhang, J. Catal., 2013, 308, 258 CrossRef CAS; (i) F. Gao and D. W. Goodman, Chem. Soc. Rev., 2012, 41, 8009 RSC.
- For selected examples, see: (a) R. W. J. Scott, C. Sivadinarayana, O. M. Wilson, Z. Yan, D. W. Goodman and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 1380 CrossRef CAS PubMed; (b) G. Glaspell, L. Fuoco and M. S. El-Shall, J. Phys. Chem. B, 2005, 109, 17350 CrossRef CAS PubMed; (c) Z. Suo, C. Ma, M. Jin, T. He and L. An, Catal. Commun., 2008, 9, 2187 CrossRef CAS; (d) J. Zhang, H. Jin, M. B. Sullivan, F. C. H. Lim and P. Wu, Phys. Chem. Chem. Phys., 2009, 11, 1441 RSC; (e) Z. Li, F. Gao and W. T. Tysoe, J. Phys. Chem. C, 2010, 114, 16909 CrossRef CAS; (f) J. Xu, T. White, P. Li, C. He, J. Yu, W. Yuan and Y.-F. Han, J. Am. Chem. Soc., 2010, 132, 10398 CrossRef CAS PubMed; (g) D. Cheng, H. Xu and A. Fortunelli, J. Catal., 2014, 314, 47 CrossRef CAS; (h) T. Ward, L. Delannoy, R. Hahn, S. Kendell, C. J. Pursell, C. Louis and B. D. Chandler, ACS Catal., 2013, 3, 2644 CrossRef CAS; (i) H. Y. Kim and G. Henkelman, ACS Catal., 2013, 3, 2541 CrossRef CAS.
- For selected examples, see: (a) J. Wang, S. A. Kondrat, Y. Wang, G. L. Brett, C. Giles, J. K. Bartley, L. Lu, Q. Liu, C. J. Kiely and G. J. Hutchings, ACS Catal., 2015, 5, 3575 CrossRef CAS; (b) T. A. G. Silva, E. Teixeira-Neto, N. López and L. M. Rossi, Sci. Rep., 2014, 4, 5766 CAS; (c) T. Jiang, C. Jia, L. Zhang, S. He, Y. Sang, H. Li, Y. Li, X. Xu and H. Liu, Nanoscale, 2015, 7, 209 RSC; (d) X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175 CrossRef CAS; (e) S. Sarina, S. Bai, Y. Huang, C. Chen, J. Jia, E. Jaatinen, G. A. Ayoko, Z. Bao and H. Zhu, Green Chem., 2014, 16, 331 RSC; (f) S. Sarina, H. Zhu, E. Jaatinen, Q. Xiao, H. Liu, J. Jia, C. Chen and J. Zhao, J. Am. Chem. Soc., 2013, 135, 5793 CrossRef CAS PubMed; (g) S. Nishimura, Y. Yakita, M. Katayama, K. Higashimine and K. Ebitani, Catal. Sci. Technol., 2013, 3, 351 RSC; (h) S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519 CrossRef CAS; (i) W.-J. Yoo, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 3095 CrossRef CAS PubMed; (j) D. Wang, A. Villa, P. Spontoni, D. S. Su and L. Prati, Chem.–Eur. J., 2010, 16, 10007 CrossRef CAS PubMed; (k) N. Dimitratos, J. A. Lopez-Sanchez, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely, D. Bethell and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 5142 RSC; (l) W. Hou, N. A. Dehm and R. W. J. Scott, J. Catal., 2008, 253, 22 CrossRef CAS; (m) N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113 CrossRef CAS; (n) D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS PubMed.
- For selected examples, see: (a) P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058 RSC; (b) J. Pritchard, L. Kesavan, M. Piccinini, Q. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Langmuir, 2010, 26, 16568 CrossRef CAS PubMed; (c) J. Pritchard, M. Piccinini, R. Tiruvalam, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, D. J. Morgan, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 308 RSC; (d) B. E. Solsona, J. K. Edwards, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, Chem. Mater., 2006, 18, 2689 CrossRef CAS; (e) J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69 CrossRef CAS; (f) Y.-F. Han, Z. Zhong, K. Ramesh, F. Chen, L. Chen, T. White, Q. Tay, S. N. Yaakub and Z. Wang, J. Phys. Chem. C, 2007, 111, 8410 CrossRef CAS; (g) Y. Nomura, T. Ishihara, Y. Hata, K. Kitawaki, K. Kaneko and H. Matsumoto, ChemSusChem, 2008, 1, 619 CrossRef CAS PubMed.
- (a) Z. Ma and S. Dai, ACS Catal., 2011, 1, 805 CrossRef CAS; (b) T. Mitsudome and K. Kaneda, Green Chem., 2013, 15, 2636 RSC; (c) M. Flytzani-Stephanopoulos, Acc. Chem. Res., 2014, 47, 783 CrossRef CAS PubMed; (d) T. Oishi, K. Yamaguchi and N. Mizuno, ACS Catal., 2011, 1, 1351 CrossRef CAS; (e) K. Yamaguchi, Y. Wang, T. Oishi and N. Mizuno, Angew. Chem., Int. Ed., 2013, 52, 5627 CrossRef CAS PubMed; (f) X. Jin, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 455 CrossRef CAS PubMed; (g) T. Yatabe, X. Jin, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2015, 54, 13302 CrossRef CAS PubMed.
- For selected examples of utilizing LDH as a basic support for functional group transformations, see: (a) K. Motokura, D. Nishimura, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 5662 CrossRef CAS PubMed; (b) K. Ebitani, K. Motokura, T. Mizugaki and K. Kaneda, Angew. Chem., Int. Ed., 2005, 44, 3423 CrossRef CAS PubMed; (c) T. Mitsudome, Y. Mikami, H. Funai, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 138 CrossRef CAS PubMed; (d) T. Mitsudome, S. Arita, H. Mori, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 7938 CrossRef CAS PubMed; (e) T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green Chem., 2009, 11, 793 RSC; (f) T. Mitsudome, A. Noujima, Y. Mikami, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2010, 49, 5545 CrossRef CAS PubMed; (g) A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2011, 50, 2986 CrossRef CAS PubMed; (h) A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2012, 48, 6723 RSC; (i) T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2012, 48, 11733 RSC; (j) T. Mizugaki, T. Yamakawa, Y. Nagatsu, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, ACS Sustainable Chem. Eng., 2014, 2, 2243 CrossRef CAS; (k) X. Liu, H.-Q. Li, S. Ye, Y.-M. Liu, H.-Y. He and Y. Cao, Angew. Chem., Int. Ed., 2014, 53, 7624 CrossRef CAS PubMed.
- One referee pointed out from Fig. 1 that some fragmentary-dispersed Pd clusters or nanoparticles also form on the LDH surface, and we cannot exclude the possibility at this time. We reason from the ineffectiveness of Pd/LDH (see entry 6, Table 1; entry 6, Table S3†) that even if such Pd species exist on the LDH surface, they are not active for the dehydrogenative aromatization.
- R. W. J. Scott, O. M. Wilson, S.-K. Oh, E. A. Kenik and R. M. Crooks, J. Am. Chem. Soc., 2004, 126, 15583 CrossRef CAS PubMed.
- Kaneda and co-workers carefully examined the solvent effect on the oxidation of Pd species for the Wacker oxidation. They demonstrated by cyclic-voltammetric analyses of Pd species in various solvents that DMA gave a more negative potential value, thus showing that DMA is an effective solvent for the oxidation of Pd species. In the present Au–Pd/LDH-catalyzed transformation, the oxidation of Pd–H species is included in the rate-determining step. Thus, DMA is the best solvent for the present transformation, and the role of DMA is possibly to promote the oxidation of Pd–H species. See: T. Mitsudome, T. Umetani, N. Nosaka, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2006, 45, 481 CrossRef CAS PubMed.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- (a) S. A. Girard, X. Hu, T. Knauber, F. Zhou, M.-O. Simon, G.-J. Deng and C.-J. Li, Org. Lett., 2012, 14, 5606 CrossRef CAS PubMed; (b) A. Hajra, Y. Wei and N. Yoshikai, Org. Lett., 2012, 14, 5488 CrossRef CAS PubMed; (c) M. T. Barros, S. S. Dey, C. D. Maycock and P. Rodrigues, Chem. Commun., 2012, 48, 10901 RSC; (d) Y. Xie, S. Liu, Y. Liu, Y. Wen and G.-J. Deng, Org. Lett., 2012, 14, 1692 CrossRef CAS PubMed; (e) J. Zhao, H. Huang, W. Wu, H. Chen and H. Jiang, Org. Lett., 2013, 15, 2604 CrossRef CAS PubMed; (f) M. Sutter, M.-C. Duclos, B. Guicheret, Y. Raoul, E. Métay and M. Lemaire, ACS Sustainable Chem. Eng., 2013, 1, 1463 CrossRef CAS; (g) Y. Liao, P. Jiang, S. Chen, F. Xiao and G.-J. Deng, RSC Adv., 2013, 3, 18605 RSC; (h) X. Cao, Y. Bai, Y. Xie and G.-J. Deng, J. Mol. Catal. A: Chem., 2014, 383–384, 94 CrossRef CAS; (i) K. Taniguchi, X. Jin, K. Yamaguchi and N. Mizuno, Chem. Commun., 2015, 51, 14969 RSC; (j) K. Taniguchi, X. Jin, K. Yamaguchi and N. Mizuno, Catal. Sci. Technol., 2016 DOI:10. 1039/c5cy01908g.
- For TEMPO as a Au–H trap, see: M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, J. Am. Chem. Soc., 2009, 131, 7189 CrossRef CAS PubMed ; also see ref. 13g.
- For TEMPO as a Pd–H trap, see: (a) A. C. Albéniz, P. Espinet, R. López-Fernández and A. Sen, J. Am. Chem. Soc., 2002, 124, 11278 CrossRef; (b) E. H. P. Tan, G. C. Lloyd-Jones, J. N. Harvey, A. J. J. Lennox and B. M. Mills, Angew. Chem., Int. Ed., 2011, 50, 9602 CrossRef CAS PubMed; (c) D. J. Vyas, E. Larionov, C. Besnard, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2013, 135, 6177 CrossRef CAS PubMed.
- For selected examples of oxidation of Pd–H species by O2, see: (a) M. C. Denney, N. A. Smythe, K. L. Cetto, R. A. Kemp and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 2508 CrossRef CAS PubMed; (b) M. M. Konnick, B. A. Gandhi, I. A. Guzei and S. S. Stahl, Angew. Chem., Int. Ed., 2006, 45, 2904 CrossRef CAS PubMed; (c) B. V. Popp and S. S. Stahl, J. Am. Chem. Soc., 2007, 129, 4410 CrossRef CAS PubMed; (d) K. M. Gligorich and M. S. Sigman, Angew. Chem., Int. Ed., 2006, 45, 6612 CrossRef CAS PubMed; (e) J. M. Keith, R. J. Nielsen, J. Oxgaard and W. A. Goddard III, J. Am. Chem. Soc., 2005, 127, 13172 CrossRef CAS PubMed; (f) J. M. Keith, W. A. Goddard III and J. Oxgaard, J. Am. Chem. Soc., 2007, 129, 10361 CrossRef CAS PubMed; (g) M. S. Sigman and D. R. Jensen, Acc. Chem. Res., 2006, 39, 221 CrossRef CAS PubMed.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 3rd edn, 1988 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc00874g |
| This journal is © The Royal Society of Chemistry 2016 |