
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Putting an ultrahigh concentration of amine groups into a metal–organic framework for CO2 capture at low pressures†
Pei-Qin
Liao
,
Xun-Wei
Chen
,
Si-Yang
Liu
,
Xu-Yu
Li
,
Yan-Tong
Xu
,
Minni
Tang
,
Zebao
Rui
,
Hongbing
Ji
,
Jie-Peng
Zhang
* and
Xiao-Ming
Chen
MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: zhangjp7@mail.sysu.edu.cn
First published on 13th July 2016
Abstract
Tremendous efforts have been devoted to increasing the CO2 capture performance of porous materials, especially for low CO2 concentration environments. Here, we report that hydrazine can be used as a diamine short enough to functionalize the small-pore metal–organic framework [Mg2(dobdc)] (H4dobdc = 2,5-dihydroxyl-1,4-benzenedicarboxylic acid). By virtue of the ultrahigh concentration of free amine groups (6.01 mmol g−1 or 7.08 mmol cm−3) capable of reversible carbamic acid formation, the new material [Mg2(dobdc)(N2H4)1.8] achieves a series of new records for CO2 capture, such as single-component isotherm uptakes of 3.89 mmol g−1 or 4.58 mmol cm−3 at the atmospheric CO2 concentration of 0.4 mbar at 298 K and 1.04 mmol g−1 or 1.22 mmol cm−3 at 328 K, as well as more than a 4.2 mmol g−1 or 4.9 mmol cm−3 adsorption/desorption working capacity under dynamic mixed-gas conditions with CO2 concentrations similar to those in flue gases and ambient air.
Introduction
Applications of porous materials for capturing CO2 from flue gas (ca. 150 mbar) and from air (ca. 0.4 mbar) have become an increasingly attractive area of research due to their importance in many applications related to the environment, energy and health.1–7 Compared with post-combustion capture, direct air capture (DAC) is more challenging, since the CO2 adsorption ability of most porous materials is low at trace CO2 concentrations.8Among the various types of adsorbents, porous coordination polymers (PCPs) or metal–organic frameworks (MOFs) with designable and modifiable pore surfaces are very attractive for CO2 capture.9–50 To increase the CO2 uptake at extremely low pressures, incorporating alkylamine groups on the pore surface is the most effective strategy so far,46–50 because of the chemisorption of CO2 associated with the formation of zwitterionic carbamate or carbamic acid. For instance, [Mg2(dobpdc)(eda)1.6] (eda-Mg2(dobpdc), eda = ethylenediamine, H4dobpdc = 4,4′-dihydroxy-(1,1′-biphenyl)-3,3′-dicarboxylic acid)48 holds the records for gravimetric and volumetric CO2 adsorption capacities with 2.85 mmol g−1 and 2.72 mmol cm−3, respectively, at 298 K and 0.4 mbar, this is because it has a high concentration of free amine groups at 3.86 mmol g−1 or 3.69 mmol cm−3. Obviously, the CO2 adsorption does not reach saturation at this condition, which is ascribed to the strong intermolecular hydrogen bonds between two adjacent amine groups that need to be opened by higher pressure CO2.47,51 As a consequence, its CO2 uptake at 0.4 mbar sharply drops to 0.120 mmol g−1 or 0.115 mmol cm−3 at a slightly higher temperature of 323 K. To overcome this problem, replacing the eda molecule with an even shorter diamine can be a good strategy, which not only reduces the intermolecular hydrogen bonding locks but also reduces the adsorbent weight.
Hydrazine (N2H4) can be considered as the smallest/shortest diamine, and its basicity is only slightly weaker than that of alkylamines (Table S1†).52 It should be noted that the long/large length/size of eda and its derivatives is the main reason for choosing the large-pore [Mg2(dobpdc)] instead of the simpler small-pore [Mg2(dobdc)] (H4dobdc = 2,5-dihydroxyl-1,4-benzenedicarboxylic acid) as a host framework.49 Nevertheless, to the best of our knowledge, an N2H4-grafted PCP has not been reported so far. Herein, we show that N2H4 can be used to functionalize the prototypical small-pore PCP [Mg2(dobdc)] without the formation of the intermolecular amine–amine interaction, achieving an ultrahigh concentration of useful amine groups in both the gravimetric and volumetric points of view, as well as a series of new benchmark CO2 capture performances, ranging from air to flue-gas conditions.
Results and discussion
The hypothetical structure of [Mg2(dobdc)(N2H4)2], i.e. [Mg2(dobdc)] fully functionalized by one N2H4 molecule per Mg(II) ion, was simulated using molecular mechanics (MM) combined with periodic density functional theory (PDFT). During structural optimization, the host framework and N2H4 molecules were considered as rigid and flexible, respectively. The simulation results showed that the void ratio of [Mg2(dobdc)(N2H4)2] remains at 36.5%, and the smallest aperture diameter is ca. 4.8 Å (Fig. 1a), which should be enough for the diffusion and accommodation of CO2 molecules. Furthermore, the shortest possible intermolecular N⋯N separations between pairs of adjacent amine groups on the ab-plane and along the c-axis were simulated as 3.81 and 4.30 Å, respectively (Fig. 1b and c), which are much longer than the values for typical N–H⋯N hydrogen bonds. For comparison, we also simulated the structure of eda-Mg2(dobpdc), giving relatively short N⋯N separations of 3.06 and 3.07 Å on the ab-plane and along the c-axis, respectively (Fig. 1d and e), which can be assigned as a kind of weak hydrogen bonding.53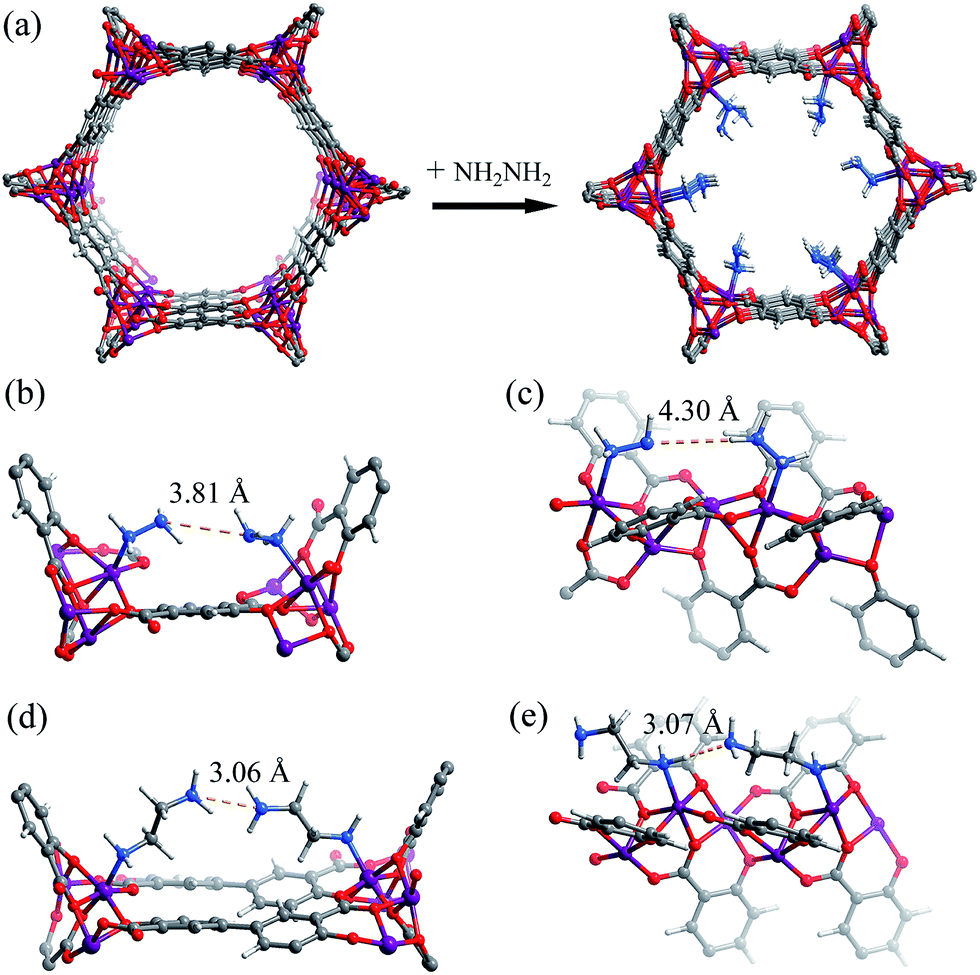 | ||
| Fig. 1 (a) Comparison of the structures of [Mg2(dobdc)] (drawn from the reported crystal structure) and [Mg2(dobdc)(N2H4)2] (simulated from PDFT). (b–e) Comparison of the shortest possible intermolecular hydrogen bonding in [Mg2(dobdc)(N2H4)2] and [Mg2(dobpdc)(eda)2] (obtained using a PDFT simulation, in which the flexibility of diamines was considered). (b) [Mg2(dobdc)(N2H4)2] on the a–b plane, (c) [Mg2(dobdc)(N2H4)2] along the c-axis, (d) [Mg2(dobpdc)(eda)2] on the a–b plane, and (e) [Mg2(dobpdc)(eda)2] along the c-axis. | ||
Yellow microcrystalline [Mg2(dobdc)] (1) was suspended in anhydrous toluene under N2 atmosphere at room temperature, and upon the addition of a toluene solution of anhydrous N2H4, the colour of the solid instantaneously changed to light yellow. The light yellow product was washed with toluene and then successively soaked in hexane and methanol to remove the excess N2H4. Elemental analysis gave a chemical formula of [Mg2(dobdc)(N2H4)1.8] (1a) for the light yellow product. Pawley refinements of the powder X-ray diffraction (PXRD) patterns showed similar unit-cell parameters for 1 and 1a and confirmed the retention of framework integrity after N2H4 grafting (Fig. S1†). PXRD and thermogravimetry-mass spectrometry analyses showed that 1a can be fully activated at 403 K (Fig. S1 and S2†), with negligible release of the N2H4 molecules. N2 sorption isotherms measured at 77 K gave apparent Langmuir surface areas of 1925 and 1012 m2 g−1 and pore volumes of 0.67 and 0.30 cm3 g−1, respectively, for 1 and 1a (Fig. S3†). Considering that their crystallographic pore volumes are 0.66 and 0.31 cm3 g−1, respectively, the N2 sorption data further confirmed the good sample crystallinity and purity, as well as the chemical formula of 1a derived from elemental analysis. 1a possesses a high concentration of surface-appended N2H4 molecules of 6.01 mmol g−1 or 7.08 mmol cm−3.
At 298 K and 1 bar (Fig. 2a), 1a showed a CO2 uptake of 5.51 mmol g−1 or 6.49 mmol cm−3, corresponding to 1.65 CO2 molecules per formula unit or 0.919 CO2 molecule per N2H4 molecule, which are lower than the values for 1 (8.04 mmol g−1 or 7.40 mmol cm−3) (Table S2†), this is because the relatively large pore volume of 1 can accommodate additional physically adsorbed CO2. At 0.15 bar, the gravimetric uptake of 1a (5.18 mmol g−1) is still lower than that of 1 (5.71 mmol g−1). However, the volumetric uptake of 1a (6.10 mmol cm−3) is 17% higher than that of 1 (5.25 mmol cm−3), illustrating the stronger CO2 binding affinity of the amine groups. At a much lower pressure of 0.4 mbar, corresponding to the CO2 concentration in air, 1a showed an exceptionally high CO2 uptake of 3.89 mmol g−1 or 4.58 mmol cm−3, which is not only a drastic improvement over 1 (0.09 mmol g−1 or 0.08 mmol cm−3), but also obviously higher than the previous records (Table S2†). The CO2 sorption kinetics of 1a were also evaluated using thermogravimetric analysis under dry air at 298 K. A CO2 uptake of 3.66 mmol g−1 or 16.1 wt% (3.71 mmol g−1 or 16.3 wt% for the single-component CO2 adsorption isotherm at 0.4 mbar and 298 K) was observed after ca. 950 min by switching the atmosphere from pure N2 at 403 K to dry air at 298 K. The equilibrium time is much longer than that at higher CO2 partial pressures, being similar to known examples such as eda-Mg2(dobpdc) (1000 min, Fig. S4†).12,48 Interestingly, even at a higher temperature of 328 K, 1a still exhibits a relatively high CO2 uptake of 1.04 mmol g−1 or 1.22 mmol cm−3 at 0.4 mbar, which is 2–3 times higher than the previous records (Table S3†).
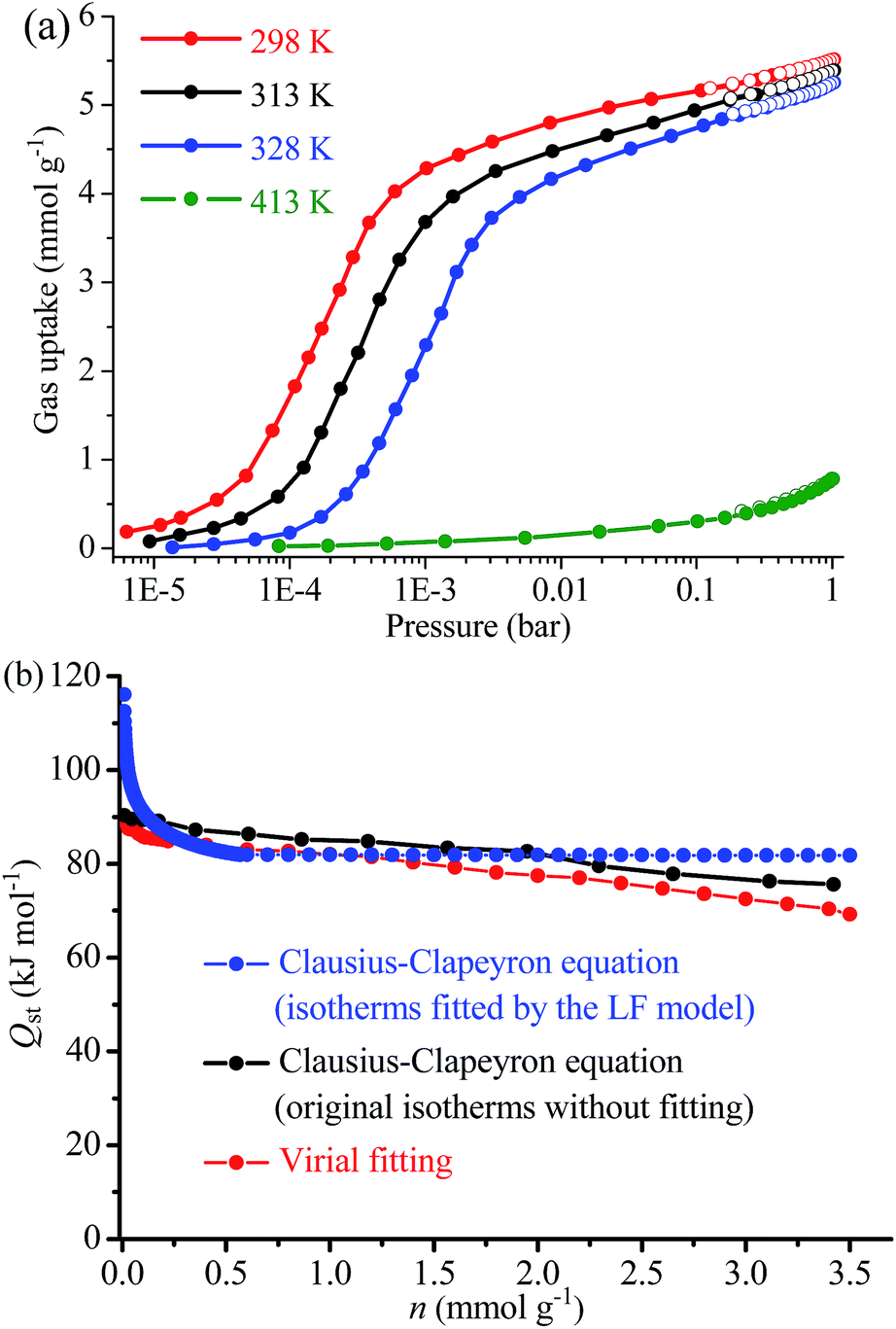 | ||
| Fig. 2 (a) CO2 adsorption (solid) and desorption (open) isotherms measured at 298, 313, 328 and 413 K and (b) the coverage-dependent CO2 adsorption enthalpy of 1a. | ||
The coverage-dependent CO2 adsorption enthalpy (Qst) of 1a was calculated using the Clausius–Clapeyron equation and the Virial fitting method using isotherms measured at 298, 313, and 328 K (Fig. 2b, S5 and S6†). Among various isotherm models, only the Langmuir–Freundlich (LF) one gave a fair fitting. Based on the Clausius–Clapeyron equation, the Langmuir–Freundlich isotherms gave a very high Qst of 118 kJ mol−1 at zero-coverage, while the original isotherms (without data fitting) gave near-zero-coverage Qst of 90 kJ mol−1. These Qst values are higher than the values of most reported materials (Table S2†) and indicative of chemisorption. On the other hand, the Virial fitting gave similar results as the Clausius–Clapeyron equation using original isotherms. In situ infrared (IR) spectra of 1a showed that the primary amine peaks at 3330 and 3287 cm−1 weakened under CO2 atmosphere. The residue primary amine groups can be assigned to those anchoring on the Mg(II) sites. Meanwhile, two new absorption peaks appeared at 3397 and 1250 cm−1. The former new peak may arise from the stretching band of the N–H bond and/or carboxylic O–H bond, while the latter can be assigned to the stretching band of the C–N bond of carbamic acid. The peak for the asymmetric deformation of the ammonium group at ∼2200 cm−1 was invisible, indicating the absence of proton transfer, which is consistent with the relatively long intermolecular separation of hydrazine molecules (Fig. S7†).54,55 After being heated under vacuum at 403 K, this peak disappeared, indicating the chemisorption of CO2 is reversible. The formation of carbamic acid was further confirmed via13C NMR measurements with the observation of a broad resonance peak centered at δ = 162 ppm (Fig. 3).22
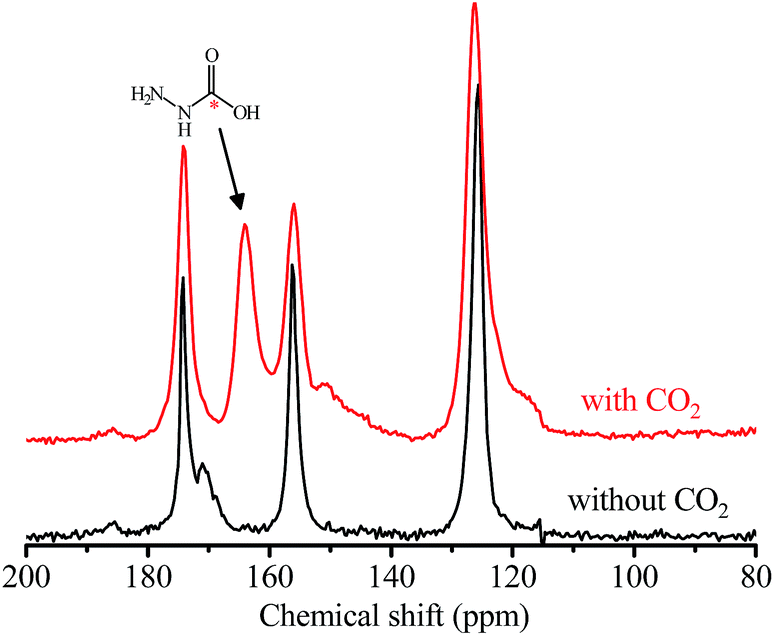 | ||
| Fig. 3 Solid-state 13C NMR spectra of 1a with and without adsorbed CO2. An asterisk (*) marks the carbon atom of CO2. | ||
The CO2 adsorption and desorption behaviours of 1a under mixed-gas and kinetic conditions were analyzed via thermogravimetry (Fig. 4), in which the sample was blown repeatedly under conditions between a 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 CO2/N2 (v/v) mixture at 313 K (the typical flue-gas environment) and a pure N2 flow (a typical regeneration method for temperature-vacuum swing adsorption (TVSA) like process) at 403 K. It should be noted that the regeneration temperature is lower than for other alkylamine functionalized PCPs. By switching from pure N2 at 403 K to the mixed gas and allowing the temperature to automatically decrease to 313 K after 17 min, a weight increase of 17.0% was observed, corresponding to volumetric CO2 uptake of 3.86 mmol g−1 or 4.55 mmol cm−3, which is 78% of the single-component isotherm uptake at 313 K and 0.15 bar. The CO2 uptake further increased to 18.7% (4.25 mmol g−1 or 5.01 mmol cm−3, i.e., 87% of the isotherm value) by maintaining the temperature at 313 K for an additional 23 min. Although the adsorption and desorption processes for 1a were not allowed to reach equilibrium, as its relatively small pores are not beneficial for fast guest diffusion, the CO2 adsorption/desorption working capacities obtained here are much higher than all the reported values for other materials under similar conditions and time scales (Table S4†). After 5 such adsorption–desorption cycles, there was nearly no change in the adsorption capacity, illustrating the high thermal stability and good CO2 adsorption–desorption reversibility of 1a, as indicated by the thermogravimetry-mass spectrometry analyses (Fig. S2†).
:85 CO2/N2 (v/v) mixture at 313 K (the typical flue-gas environment) and a pure N2 flow (a typical regeneration method for temperature-vacuum swing adsorption (TVSA) like process) at 403 K. It should be noted that the regeneration temperature is lower than for other alkylamine functionalized PCPs. By switching from pure N2 at 403 K to the mixed gas and allowing the temperature to automatically decrease to 313 K after 17 min, a weight increase of 17.0% was observed, corresponding to volumetric CO2 uptake of 3.86 mmol g−1 or 4.55 mmol cm−3, which is 78% of the single-component isotherm uptake at 313 K and 0.15 bar. The CO2 uptake further increased to 18.7% (4.25 mmol g−1 or 5.01 mmol cm−3, i.e., 87% of the isotherm value) by maintaining the temperature at 313 K for an additional 23 min. Although the adsorption and desorption processes for 1a were not allowed to reach equilibrium, as its relatively small pores are not beneficial for fast guest diffusion, the CO2 adsorption/desorption working capacities obtained here are much higher than all the reported values for other materials under similar conditions and time scales (Table S4†). After 5 such adsorption–desorption cycles, there was nearly no change in the adsorption capacity, illustrating the high thermal stability and good CO2 adsorption–desorption reversibility of 1a, as indicated by the thermogravimetry-mass spectrometry analyses (Fig. S2†).
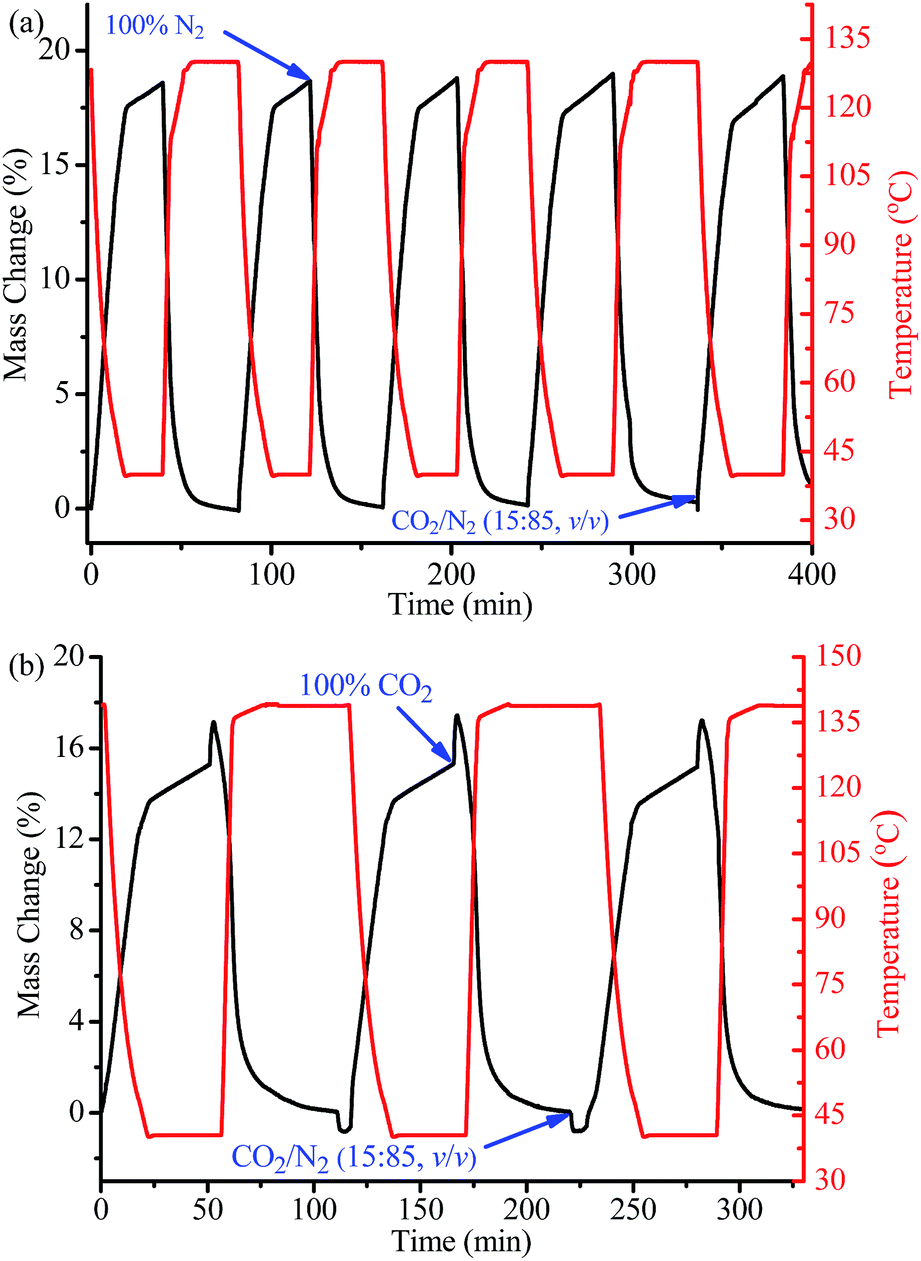 | ||
| Fig. 4 Repeated adsorption–desorption kinetics for 1a (a) between a 15:85 CO2/N2 (v/v) flow at 313 K and a pure N2 flow at 403 K and (b) between a 15:85 CO2/N2 (v/v) flow at 313 K and a pure CO2 flow at 413 K. | ||
A pure temperature swing adsorption (TSA) process for 1a was further carried out, and a working capacity of 3.52 mmol g−1 or 4.15 mmol cm−3 was obtained between the adsorption in a 15:85 CO2/N2 (v/v) mixture at 313 K for 48 min and the desorption in a pure CO2 flow at 413 K (optimized) for 54 min. It should be noted that, even though it was obtained in a relatively short time which did not allow the adsorption/desorption to reach equilibration, this working capacity is higher than previously reported values obtained in similar and longer adsorption/desorption times (Table S4†). To evaluate the regeneration energy for CO2 desorption, the heat capacity of 1a was quantified via differential scanning calorimetry. About −200 J g−1 was evolved as the material was cooled from 413 to 313 K (Fig. S8†). With these data (see calculation method in the ESI†), approximately 3.02 MJ of energy would be required to regenerate 1 kg of CO2 from 1a (Fig. 4b).49,56 The regeneration energy for CO2 desorption from 1a (3.02 MJ kg−1) is higher than those of mmeda-Mg2(dobpdc) (2.34 MJ kg−1) and state-of-the-art amine-based solutions (2.6 MJ kg−1), but lower than that of monoethanolamine (3.5 MJ kg−1).49
To simulate more practical CO2 capture applications, we also measured the column breakthrough curves of 1a using a binary 10:90 CO2/N2 (v/v) mixture at 313 K and 1 bar. As shown in Fig. 5a and S9a,† the breakthrough point (outlet concentration > detection limit) of CO2 was observed at a mixture injection amount of 44.3 mmol g−1 or 52.2 mmol cm−3, exceeding all the reported values even measured at a lower temperature of 298 K (Table S5,† note that a lower temperature generally leads to a higher adsorption amount, at the same pressure), including 42.2 mmol g−1 or 38.8 mmol cm−3 for 1 and 25.5 mmol g−1 or 50.2 mmol cm−3 for the recently discovered microporous copper silicate SGU-29.7 After that, the outlet CO2 concentration rose gradually to the inlet value at 51.6 mmol g−1 or 60.8 mmol cm−3. The capability of 1a for trace CO2 capture was investigated by column breakthrough tests using a 1:999 CO2/N2 (i.e. 1000 ppm CO2) mixture at 298 K (Fig. 5b). The CO2 breakthrough and saturation points were observed at 4210 and 4400 mmol g−1, or 4960 and 5180 mmol cm−3, respectively.
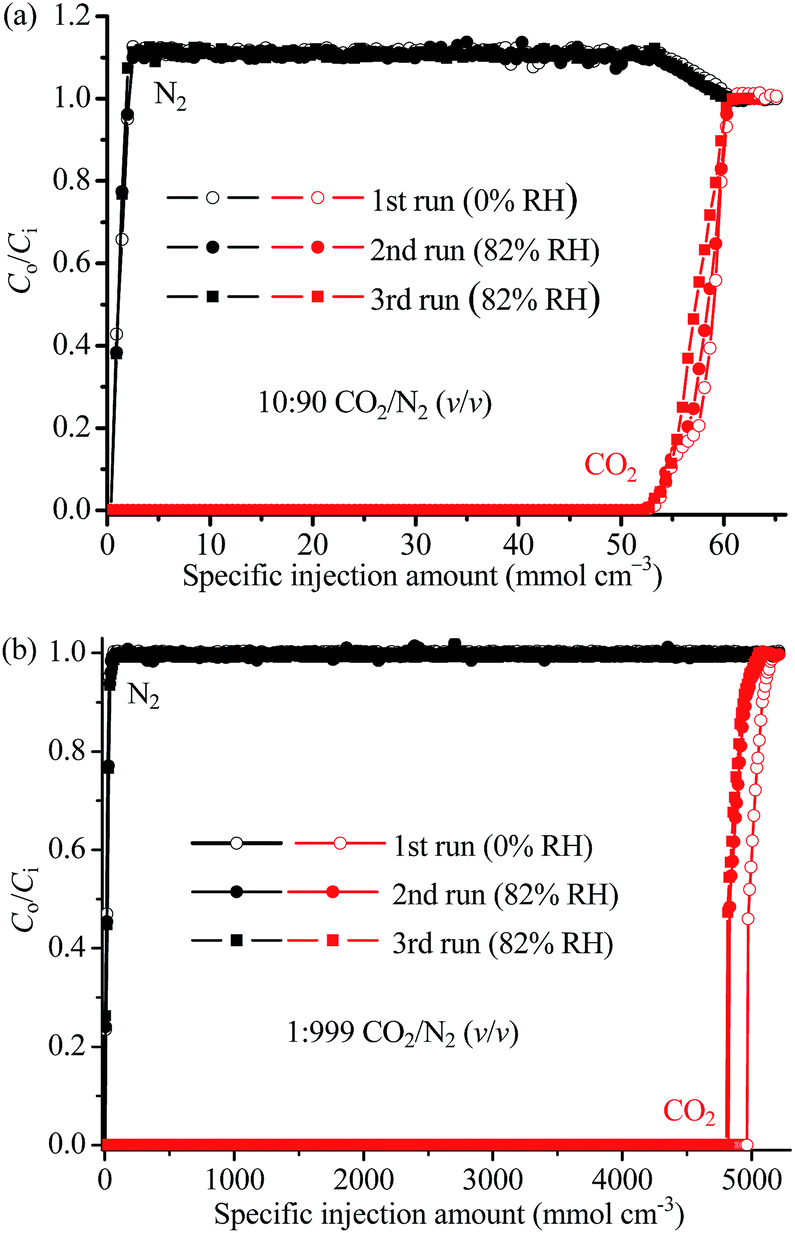 | ||
| Fig. 5 Repeated breakthrough curves of the 1a column operated at 1 bar. (a) 10:90 CO2/N2 (v/v) at 313 K. (b) 1:999 CO2/N2 (v/v) mixture at 298 K. Lines are drawn to guide eyes. Ci and Co are the concentrations of each gas at the inlet and outlet, respectively. | ||
The CO2 uptake of the adsorbent 1a in the columns can be calculated (see ESI† for calculation method) as 4.89 and 4.22 mmol g−1, or 5.76 and 4.97 mmol cm−3, for the 10:90 and 1:999 CO2/N2 mixtures, respectively, which are both ca. 98% of the values obtained from the single-component adsorption isotherms (4.97 mmol g−1 or 5.85 mmol cm−3 at 313 K and 0.1 bar, 4.30 mmol g−1 or 5.07 mmol cm−3 at 298 K and 1 mbar). Suffering from weak CO2/N2 selectivity and/or low adsorption rate, many adsorbents show a much lower CO2 uptake under mixed-gas conditions compared to single-component adsorption isotherms (Table S5†). The exceptionally high adsorption efficiency of 1a implies that its CO2 diffusion rate and CO2/N2 selectivity are high enough under such mixed-gas breakthrough conditions.
We also tested the CO2 capture performances and stabilities of the columns under high humidity.9–13 After the breakthrough curves reached equilibrium under the above mentioned dry conditions (0% RH, RH = relative humidity), the columns were activated under He flow at 403 K, cooled down to the measurement temperature of 313 K, saturated using humid He (82% RH), and then switched to humid CO2/N2 mixtures (82% RH, no change to other parameters) to start a new breakthrough experiment. Remarkably, the breakthrough curve for the humid 10:90 CO2/N2 mixture almost overlapped with that measured at the dry condition (Fig. 5a). For the humid 1:999 CO2/N2 mixture, the CO2 breakthrough point slightly (2.6% compared with that observed for the dry mixture) shortened to 4100 mmol g−1 or 4830 mmol cm−3 (Fig. 5b and S9b†). These observations indicated that water has little effect on the CO2 adsorption capacity of 1a, even when the CO2 concentration is extremely low. It should be noted that H2O can strongly compete with CO2 for the adsorption sites in most porous materials, and only a handful of examples have demonstrated such a high waterproof CO2 capture performance (Table S5†).
As shown in Fig. 5, the same activation/breakthrough procedures were applied to the columns once again, and the breakthrough curves almost overlapped with each other, indicating the high stability of the adsorbent 1a and the column even under high humidity. This also indicates that 1a is particularly selective for CO2 over water, and confirms that the grafted N2H4 molecules were not replaced by water molecules.
Conclusions
In summary, we demonstrated that the classical small-pore PCP [Mg2(dobdc)] can be modified by the shortest diamine N2H4 to obtain a very powerful CO2 adsorbent [Mg2(dobdc)(N2H4)1.8]. Thanks to the ultrahigh concentration of amine groups and the chemisorption of CO2 associated with carbamic acid formation, this material exhibited exceptionally high CO2 adsorption capacity at low pressures (from 0.4 mbar to 150 mbar) and a wide range of temperatures (from 298 to 328 K), well exceeding previous records. More importantly, these CO2 adsorption performances can be maintained under mixed-gas kinetic conditions, even in the presence of high humidity.Acknowledgements
This work was supported by the “973 Project” (2014CB845602 and 2012CB821706) and NSFC (21225105 and 21473260).Notes and references
- S. U. Rege, R. T. Yang and M. A. Buzanowski, Chem. Eng. Sci., 2000, 55, 4827–4838 CrossRef CAS.
- S. U. Rege, R. T. Yang, K. Y. Qian and M. A. Buzanowski, Chem. Eng. Sci., 2001, 56, 2745–2759 CrossRef CAS.
- J. C. Santos, F. D. Magalhaes and A. Mendes, Ind. Eng. Chem. Res., 2008, 47, 6197–6203 CrossRef CAS.
- Y. Belmabkhout, R. Serna-Guerrero and A. Sayari, Chem. Eng. Sci., 2010, 65, 3695–3698 CrossRef CAS.
- S. Sung and M. P. Suh, J. Mater. Chem. A, 2014, 2, 13245–13249 CAS.
- Y. Lin, Q. Yan, C. Kong and L. Chen, Sci. Rep., 2013, 3, 1859 Search PubMed.
- S. J. Datta, C. Khumnoon, Z. H. Lee, W. K. Moon, S. Docao, T. H. Nguyen, I. C. Hwang, D. Moon, P. Oleynikov, O. Terasaki and K. B. Yoon, Science, 2015, 350, 302–306 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, G. K. Surya Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 CAS.
- A. Kumar, D. G. Madden, M. Lusi, K.-J. Chen, E. A. Daniels, T. Curtin, J. J. Perry and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed.
- J. A. Mason, T. M. McDonald, T.-H. Bae, J. E. Bachman, K. Sumida, J. J. Dutton, S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2015, 137, 4787–4803 CrossRef CAS PubMed.
- W. R. Lee, H. Jo, L.-M. Yang, H. Lee, D. W. Ryu, K. S. Lim, J. H. Song, D. Y. Min, S. S. Han, J. G. Seo, Y. K. Park, D. Moon and C. S. Hong, Chem. Sci., 2015, 6, 3697–3705 RSC.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CAS.
- N. T. T. Nguyen, H. Furukawa, F. Gándara, H. T. Nguyen, K. E. Cordova and O. M. Yaghi, Angew. Chem., Int. Ed., 2014, 53, 10645–10648 CrossRef CAS PubMed.
- S. Choi, T. Watanabe, T.-H. Bae, D. S. Sholl and C. W. Jones, J. Phys. Chem. Lett., 2012, 3, 1136–1141 CrossRef CAS PubMed.
- P.-Q. Liao, A.-X. Zhu, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 6350 CrossRef CAS PubMed.
- P.-Q. Liao, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 8697 CrossRef PubMed.
- P.-Q. Liao, H. Chen, D.-D. Zhou, S.-Y. Liu, C.-T. He, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Energy Environ. Sci., 2015, 8, 1011–1016 CAS.
- P. K. Thallapally, R. K. Motkuri, C. A. Fernandez, B. P. McGrail and G. S. Behrooz, Inorg. Chem., 2010, 49, 4909–4915 CrossRef CAS PubMed.
- J. Liu, J. Tian, P. K. Thallapally and B. P. McGrail, J. Phys. Chem. C, 2012, 116, 9575–9581 CAS.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gandara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS PubMed.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS PubMed.
- B. Li, Z. Zhang, Y. Li, K. Yao, Y. Zhu, Z. Deng, F. Yang, X. Zhou, G. Li, H. Wu, N. Nijem, Y. J. Chabal, Z. Lai, Y. Han, Z. Shi, S. Feng and J. Li, Angew. Chem., Int. Ed., 2012, 51, 1412–1415 CrossRef CAS PubMed.
- J.-R. Li, J. Yu, W. Lu, L.-B. Sun, J. Sculley, P. B. Balbuena and H.-C. Zhou, Nat. Commun., 2013, 4, 1538 CrossRef PubMed.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- Y. He, W. Zhou, R. Krishna and B. Chen, Chem. Commun., 2012, 48, 11813–11831 RSC.
- T. Fukushima, S. Horike, Y. Inubushi, K. Nakagawa, Y. Kubota, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 4820–4824 CrossRef CAS PubMed.
- B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2010, 133, 748–751 CrossRef PubMed.
- R. Vaidhyanathan, S. S. Iremonger, G. K. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- M. Wriedt, J. P. Sculley, A. A. Yakovenko, Y. G. Ma, G. J. Halder, P. B. Balbuena and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 9804–9808 CrossRef CAS PubMed.
- P.-Q. Liao, D.-D. Zhou, A.-X. Zhu, L. Jiang, R.-B. Lin, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2012, 134, 17380–17383 CrossRef CAS PubMed.
- J. A. Johnson, S. Chen, T. C. Reeson, Y. S. Chen, X. C. Zeng and J. Zhang, Chem.–Eur. J., 2014, 20, 7632–7637 CrossRef CAS PubMed.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 132, 38–39 CrossRef PubMed.
- T. Panda, P. Pachfule, Y. Chen, J. Jiang and R. Banerjee, Chem. Commun., 2011, 47, 2011–2013 RSC.
- T. Devic, F. Salles, S. Bourrelly, B. Moulin, G. Maurin, P. Horcajada, C. Serre, A. Vimont, J.-C. Lavalley, H. Leclerc, G. Clet, M. Daturi, P. L. Llewellyn, Y. Filinchuk and G. Férey, J. Mater. Chem., 2012, 22, 10266–10273 RSC.
- C. R. Wade and M. Dincă, Dalton Trans., 2012, 41, 7931–7938 RSC.
- C. Montoro, E. Garcia, S. Calero, M. A. Perez-Fernandez, A. L. Lopez, E. Barea and J. A. R. Navarro, J. Mater. Chem., 2012, 22, 10155–10158 RSC.
- H. J. Park and M. P. Suh, Chem. Sci., 2013, 4, 685–690 RSC.
- L.-H. Xie and M. P. Suh, Chem.–Eur. J., 2013, 19, 11590–11597 CrossRef CAS PubMed.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed.
- P. Wang, Y. Lu Yi, Q. Liu and W.-Y. Sun, Sci. Sin.: Chim., 2013, 43, 1288–1296 CrossRef CAS.
- S. S. Nagarkar, A. K. Chaudhari and S. K. Ghosh, Inorg. Chem., 2011, 51, 572–576 CrossRef PubMed.
- A. Khutia and C. Janiak, Dalton Trans., 2014, 43, 1338–1347 RSC.
- L.-J. Li, P.-Q. Liao, C.-T. He, Y.-S. Wei, H.-L. Zhou, J.-M. Lin, X.-Y. Li and J.-P. Zhang, J. Mater. Chem. A, 2015, 3, 21849–21855 CAS.
- T. M. McDonald, D. M. D'Alessandro, R. Krishna and J. R. Long, Chem. Sci., 2011, 2, 2022–2028 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- W. R. ChangLee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744–751 Search PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed.
- N. Planas, A. L. Dzubak, R. Poloni, L.-C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 CrossRef CAS PubMed.
- B. Lee, S. H. Kang, D. Kang, K. H. Lee, J. Cho, W. Nam, O. H. Han and N. H. Hur, Chem. Commun., 2011, 47, 11219–11221 RSC.
- J. Emsley, Chem. Soc. Rev., 1980, 9, 91–124 RSC.
- N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3311–3319 CrossRef CAS.
- A. Danon, P. C. Stair and E. Weitz, J. Phys. Chem. C, 2011, 115, 11540–11549 CAS.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, methods, additional discussions, thermogravimetry curves, PXRD patterns, and spectroscopy characterizations, as well as additional isotherms. See DOI: 10.1039/c6sc00836d |
| This journal is © The Royal Society of Chemistry 2016 |