
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable helicity, stability and DNA-binding properties of short peptides with hybrid metal coordination motifs†
Sarah J.
Smith
,
Robert J.
Radford
,
Rohit H.
Subramanian
,
Brandon R.
Barnett
,
Joshua S.
Figueroa
and
F. Akif
Tezcan
*
Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr., La Jolla, USA. E-mail: tezcan@ucsd.edu
First published on 18th May 2016
Abstract
Given the prevalent role of α-helical motifs on protein surfaces in mediating protein–protein and protein–DNA interactions, there have been significant efforts to develop strategies to induce α-helicity in short, unstructured peptides to interrogate such interactions. Toward this goal, we have recently introduced hybrid metal coordination motifs (HCMs). HCMs combine a natural metal-binding amino acid side chain and a synthetic chelating group that are appropriately positioned in a peptide sequence to stabilize an α-helical conformation upon metal coordination. Here, we present a series of short peptides modified with HCMs consisting of a His and a phenanthroline group at i and i + 7 positions that can induce α-helicity in a metal-tunable fashion as well as direct the formation of discrete dimeric architectures for recognition of biological targets. We show that the induction of α-helicity can be further modulated by secondary sphere interactions between amino acids at the i + 4 position and the HCM. A frequently cited drawback of the use of peptides as therapeutics is their propensity to be quickly digested by proteases; here, we observe an enhancement of up to ∼100-fold in the half-lives of the metal-bound HCM-peptides in the presence of trypsin. Finally, we show that an HCM-bearing peptide sequence, which contains the DNA-recognition domain of a bZIP protein but is devoid of the obligate dimerization domain, can dimerize with the proper geometry and in an α-helical conformation to bind a cognate DNA sequence with high affinities (Kd ≥ 65 nM), again in a metal-tunable manner.
Introduction
Protein–protein1–4 and protein–DNA5–8 interactions are commonly mediated by α-helical domains on protein surfaces.9,10 Due to the prevalence of such α-helical interaction motifs, there has been considerable interest in designing small, synthetic systems that maintain or mimic the α-helical periodicity of such motifs and therefore can recapitulate their binding and recognition properties.11–13 One route toward this goal has been the design of entirely non-biological systems such as synthetic helix mimics14,15 or foldamers with incorporated β-amino acids.16,17 Another route is to design natural peptides with a propensity to form α-helical structures. Although they have the ideal size to be effective therapeutic agents and interact with an extended region on a protein or DNA surface,18 short peptides are often unstructured and prone to proteolytic digestion;19 these drawbacks have led to the pursuit of diverse strategies to stabilize peptides in α-helical conformations such as: covalent20–28 or metal-mediated29–32 stapling of helical turns, utilization of hydrogen-bond surrogates,33,34 incorporation of salt bridges in i and i + 4 positions,35 or inclusion of α-amino acids with restricted conformational availability.36As an alternative approach for helix induction in peptides, we have introduced hybrid metal coordination motifs (HCMs) that consist of a natural metal-binding side chain and a non-natural metal-chelating ligand, placed at i and i + 7 positions.37,38 Our motivation was that HCMs would not only impart α-helicity through metal-mediated crosslinking across two helix turns (Fig. 1a), but they would also provide stable coordination motifs whose metal binding properties can be modulated through the choice of metal, chelating ligand or the solution pH. This modularity can in turn be exploited for the incorporation of metal-based functions (e.g., luminescence and catalysis)39 or metal-mediated oligomerization (as we demonstrate in this report).
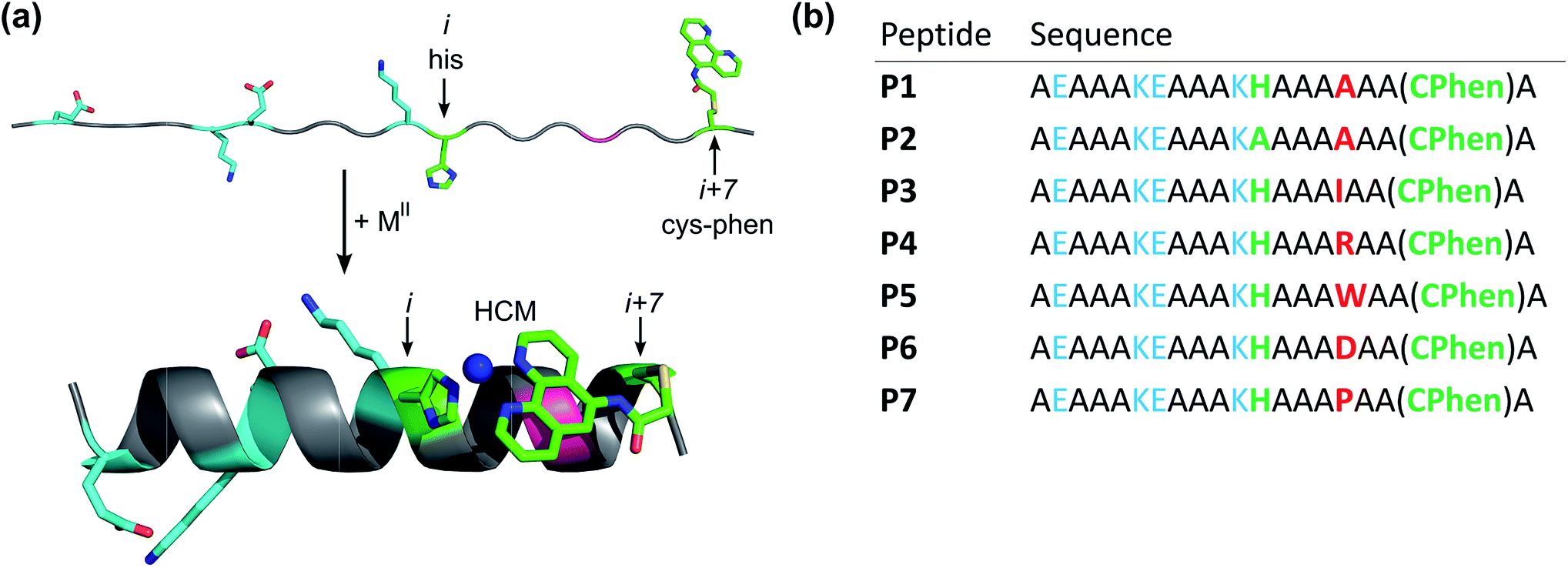 | ||
| Fig. 1 Design of peptides P1–P7. (a) Cartoon showing the proposed mode of helix induction by metal binding to the HCM. The peptides include two pairs of salt bridging side chains (cyan), the HCM motif with His at position i and Cys-Phen at position i + 7 (green), and various amino acids incorporated at the i + 4 position (red). (b) Sequences of P1–P7. | ||
Our early studies focused on the surface modification of a folded, α-helical protein (cyt cb562) with i/i + 7 HCMs that consisted of a His residue and the non-natural chelates 8-hydroxyquinoline (Quin), 1,10-phenanthroline (Phen) or 2,2′:6′,2′′-terpyridine (Tpy).37,38 These studies showed that metal coordination by these HCM motifs imparted significant chemical and thermal stability to cyt cb562 (due to helix crosslinking) in a way that could be tuned through the choice of the metal ion. It was also demonstrated that the oligomerization state and geometry of HCM-modified cyt cb562 could be controlled through the preferred coordination geometry of a metal ion. Notably, the addition of NiII to a His–Quin HCM-modified cyt cb562 at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 metal:protein stoichiometry led to the formation of a V-shaped protein dimer (see Fig. 4).37 This discrete protein arrangement was dictated by the octahedral coordination geometry of the Ni:(HCM)2 moiety, wherein the two Quin functionalities adopted the preferred cis orientation. A structural superposition of the surface helices in this dimer revealed a very close match (rmsd = 1.6 Å) with the α-helical DNA-binding domains of the homo-dimeric bZIP proteins (Fig. 4b) (PDB ID: 1JNM), suggesting that HCMs could direct the formation of helical, dimeric protein/peptide scaffolds that are structurally poised to recognize biological targets without the need for engineering extensive protein surfaces or peripheral oligomerization domains.
:2 metal:protein stoichiometry led to the formation of a V-shaped protein dimer (see Fig. 4).37 This discrete protein arrangement was dictated by the octahedral coordination geometry of the Ni:(HCM)2 moiety, wherein the two Quin functionalities adopted the preferred cis orientation. A structural superposition of the surface helices in this dimer revealed a very close match (rmsd = 1.6 Å) with the α-helical DNA-binding domains of the homo-dimeric bZIP proteins (Fig. 4b) (PDB ID: 1JNM), suggesting that HCMs could direct the formation of helical, dimeric protein/peptide scaffolds that are structurally poised to recognize biological targets without the need for engineering extensive protein surfaces or peripheral oligomerization domains.
More recently, we expanded our work from protein scaffolds to peptides and demonstrated that His–Quin HCMs could induce substantial helicity in short (10-amino acid) unstructured peptides in a metal-tunable fashion, while allowing the simultaneous generation of metal-based luminescence.39 In the current report, we build further upon this work by examining (1) whether His–Phen HCMs are also capable of helix induction in small peptides in order to expand the possible peptide-HCM ligand set, (2) the effects of secondary elements such as the identity of the i + 4 residue on HCM-mediated helix induction, and (3) the protection of HCM-stabilized peptides from proteolysis. Furthermore, motivated by our observations on the HCM-induced formation of discrete V-shaped protein dimers, we investigate (4) the ability of an HCM-bearing peptide scaffold to form α-helical dimers for selective DNA recognition and binding. Our findings highlight the remarkable modularity and versatility of HCMs in controlling the architecture and the biological recognition properties of small peptides.
Results and discussion
Design of HCM-peptides
A family of 20 amino-acid-long peptides (P1–P7; Fig. 1) were synthesized to test the ability of the His–Phen HCMs to induce metal-dependent α-helicity and examine the structural effects of secondary interactions between the HCM moiety and the side chain at i + 4 position. These Ala-rich peptides were obtained by solid-state synthesis and contain an amidated C-terminus and an acylated N-terminus. A His and a Cys residue were placed at positions 12 (i) and 19 (i + 7), respectively, and Cys 19 was post-synthetically modified with 5-iodoacetamido-1,10-phenanthroline (I-Phen) to form the desired tridentate His–Phen HCM (see Fig. S1† for peptide characterization). In this arrangement, the large aromatic Phen moiety spans directly over the side chain at the i + 4 position (position 16), suggesting that metal binding and helix induction by the HCM could be modulated through the interactions between Phen and the i + 4 residue. Thus, peptides P1 and P3–P7 were varied in terms of the amino acid functionalities at the i + 4 position (Fig. 2). In P2, His was replaced with Ala to generate a control peptide lacking a complete HCM. All sequences contained two Glu/Lys pairs to render helix formation more favourable through additional salt bridging interactions and to increase peptide solubility. | ||
| Fig. 2 (a) CD spectra showing the induction of helicity upon addition of various metal ions. (b) Model showing an Ile residue at the i + 4 (pink) position in contact distance with the Phen functionality in the metal-bound HCM. (c) Molar ellipticity of peptide variants (metal-free, bound to Ni, or in the presence of TFE) monitored at 222 nm. | ||
Metal-binding properties of HCM-peptides
Employing the parent peptide P1, we first determined the binding affinity of the His–Phen HCM for the late-first row transition metal ions CoII, NiII, CuII, and ZnII. Due to the expected high metal binding affinities, we used ethylene glycol tetraacetic acid (EGTA) and nitrilotriacetic acid (NTA) as competing ligands in our titrations (see ESI, Table S1†). Metal binding was monitored by the shift of the Phen π–π* absorption band from 264 nm to 276 nm (Fig. S2†). In the case of all metal ions, metal binding curves were best fit with a two-step equilibrium model that takes into account 1:1 HCM:metal (H:M) binding as well as the subsequent metal-mediated peptide dimerization (H:M:H) event (see Fig. S3, Schemes S2 and S3†). The dissociation constants of the metal–HCM complexes (Kd,M) range from 30 nM for CoII to 600 fM for CuII (Tables 1 and S2†), the affinities roughly following the Irving-Williams series: CoII < NiII < CuII ≫ ZnII, which was also observed in the case of His–Phen HCMs placed on the cyt cb562 surface and His–Quin HCMs (also tridentate) incorporated into 10-residue long peptides.37–39 The dissociation constants corresponding to the metal-mediated dimerization event (Kd,dimer) are all in the micromolar regime (1–200 μM), again similar to the dimerization constants of His–Phen modified cyt cb562 and His–Quin modified peptides.37–39 Importantly, the Kd,M values are ∼3-fold (for CoII) to >3 orders of magnitude (for CuII) higher than those determined for the free Phen ligand,40 and up to 6 orders of magnitude higher than those for i/i + 4 bis-His motifs incorporated into short peptides.32,41 Electrospray ionisation mass spectrometry (ESI-MS) measurements were carried out on HCM-bearing peptides after incubation with 1 or 2 equiv. of CoII, NiII, CuII, or ZnII. These measurements show the predominant formation of singly-metallated peptides (Fig. S4 and Table S3†); in the cases of CoII or ZnII, which display lower affinities for the His–Phen HCMs, metal-free peptides were also detected along with the singly metallated species. Conversely, no metallated peptide masses were detected with a control peptide lacking the Phen group (P4bare) (Fig. S5 and Table S4†). These observations confirm the formation of the desired His–Phen tridentate HCM and its ability to anchor various transition metal ions with high affinity.
| Metal | K d, His–Phen HCM (M) | K d, free Phen1 (M) | Calculated helicity (at 25 °C) | Percent of maximum helicity |
|---|---|---|---|---|
| Metal-free | N/A | N/A | 17% | 30% |
| CoII | 3(2) × 10−8 | 8.0 × 10−8 | 38% | 69% |
| NiII | 3(2) × 10−11 | 3.9 × 10−8 | 43% | 77% |
| CuII | 6(5) × 10−13 | 2.5 × 10−9 | 18% | 32% |
| ZnII | 1.7(7) × 10−8 | 3.7 × 10−7 | 34% | 62% |
Metal-dependent α-helix induction in HCM-peptides
Circular dichroism (CD) spectroscopy was used to examine the secondary structure of P1–P7 for determining the extent of metal-mediated α-helix induction and the structural influence of the i + 4 sidechain (Fig. 1b and 2, Table 1). In these experiments, we used a low concentration of peptide (∼15 μM) and a molar excess of at least 3-fold CoII, NiII, CuII, and ZnII so as to ensure the formation of monomeric, fully metalated peptides and to prevent metal-induced dimerization. The CD signal was measured between 190 and 260 nm at 4 and 25 °C, with attention to the emergence of double minima at 208 and 222 nm indicative of an α-helical structure.42 Methods of calculating helix percentages of short peptides can be unreliable.33 Therefore, we also measured the CD spectra of the peptides in 60% trifluoroethanol (TFE), a known α-helix inducer,43 and used these spectra as being representative of maximal helicity that could be attained for each peptide (Fig. S6–S7 and Table S5†).Several pertinent observations were made: (1) metal-mediated helix induction was observed in all peptides except the control peptide P2, indicating that the metal-bound His–Phen HCM indeed stabilizes an α-helical structure by crosslinking two turns of the helix. (2) Subtle differences in HCM coordination can have substantial effects on peptide structure. In all HCM-containing peptides utilizing Phen as a chelate, NiII binding induced the most helicity, CuII the least, CoII and ZnII to intermediate extents. Interestingly, this trend is quite distinct from that observed for the peptides bearing His–Quin HCMs, where CuII binding yielded the most helicity and NiII frequently the least.39 Although Phen and Quin are both planar bidentate ligands with similar bite angles, they differ in terms of the size of the aromatic moiety, composition of the donor atoms (N/N vs. N/O), and, potentially in the charge of the ligand upon metal coordination (0 vs. −1). These differences could result in Phen and Quin ligands directing the formation of alternate crosslinking geometries, which would change the effective length of the metal crosslink across i and i + 7 positions, and thereby modulate the structure of the metal-bound peptide backbone (vide infra). (3) Peptides P3, P4, and P5, which bear bulky, hydrophobic side chains (Ile, Arg and Trp) in the i + 4 position, display considerable helicity (47–65% at 25 °C with respect to TFE-containing samples) even in the absence of metal binding (Fig. 2 and Table S5†). Baldwin and others have previously documented the α-helix stabilizing effect of hydrophobic interactions between side chains in i and i + 4 positions as well as in i and i + 3 positions.44–46 It appears that this effect is amplified in the case of HCM-peptides due to the extensive Phen aromatic system and large hydrophobic side chains at i + 4 (i − 3 with respect to Phen). It is possible to envision how these bulky, hydrophobic amino acid side chains could stabilize the formation of a hydrophobic “pocket” formed by HCM-metal coordination (Fig. S6†). In contrast, peptide P1 with the small Ala side chain, P6 with the negatively charged Asp or P7 with the Pro residue, a known helix breaker,47 possess considerably less helicities (30–36% at 25 °C with respect to TFE-containing samples, Table S5†). (4) The increase in α-helicity upon NiII addition is sizeable in all cases (except P2), ranging from +47% (from 30% to 77% vs. TFE) for P1 to +12% (from 36% to 48% vs. TFE) for P7. The highest absolute helicity, ∼90%, is observed in the case of P3. In contrast, CuII coordination can actually lead to α-helix destabilization in some cases, for example, by as much as −10% in P3 and P7 (Fig. S7 and Table S5†).
Density functional theory (DFT) calculations were performed with simplified models (4-methylimidazole, Phen or Quin containing 5-formamyl substitution and aquo ligands) in order to better understand how inner-sphere metal coordination geometries could affect helix induction in His–Phen and His–Quin HCMs (Fig. S8–S14†). On previous α-helical protein systems, NiII was found to provide the highest stabilizing effect with both the His–Quin37 and His–Phen HCMs,38 and CuII the least, in agreement with the results presented here. The DFT calculations (OLYP functional) indicate that NiII forms the expected octahedral geometries with the fac orientation of the 4-methylimidazole and the Quin or Phen chelates (Fig. S9 and S10†). This orientation places the peptide attachment points of the imidazole (the methyl group) and the Phen or Quin functionalities (the amine group) at a distance of 9.3 or 8.9 Å, respectively (Fig. S8–S10†). These distances agree well with the corresponding distance of 9.5 Å observed in the crystal structure of the Ni-bound His–Quin motif on the helical surface of cyt cb562,37 and suggest an optimal helix induction effect for the NiII/His–Phen combination. Compared to NiII, CuII coordination forces the HCM functionalities into a much more planar orientation, leading to an increase in the distance of their peptide attachment points to 11.6 Å (for His–Phen) and 11.0 Å (for His–Quin) (Fig. S11 and S12†); these crosslinking distances are likely incompatible with a helical peptide fold. While these CuII–HCM geometries are in accord with the helix-destabilizing effect of CuII in the present work, they are inconsistent with its helix-stabilizing effect in the case of the His–Quin peptide system.39 Additional DFT calculations, in which the phenolate group of Quin (pKa of the free ligand ≈ 9.9) was not deprotonated, favour a significantly more non-planar orientation of the imidazole and Quin ligands with CuII coordination, while no significant changes are observed in the case of NiII coordination (Fig. S8, S13 and S14†). While it is tempting to suggest that the protonation/deprotonation of the Quin hydroxyl group may be the underlying cause for the contrasting effects of CuII with His–Quin and His–Phen HCMs, the contributing factors to helix formation are likely very complex and their delineation will require extensive quantum mechanics/molecular mechanics (QM/MM) calculations that take into account both the metal coordination geometry and the structure of the peptide.
Regardless, taken together with our previous studies on His–Quin systems,39 these observations on His–Phen peptides highlight the modularity of HCMs in controlling peptide structure through the choice of the metal ion and the metal chelating functionality as well as through the amino acid side chains that make secondary contacts with the HCM motif.
Thermal and proteolytic stability of HCM-peptides
We next examined whether His–Phen HCMs confer stability onto the peptide scaffolds upon metal coordination. Thermal unfolding of P1–P7 was monitored by the disappearance of the CD signal at 222 nm as the temperature was raised from 10–90 °C (Fig. S15†). As expected from their short sequences, all peptides display relatively low thermal denaturation points (Tm)48 and shallow denaturation transitions, indicative of low cooperativity.49 In general, the trends seen in the metal-mediated α-helix induction are also observed in the thermal stabilities of the peptides, with NiII binding providing the greatest stabilization and CuII the least. In accordance with their highest helical contents, NiII-bound P1, P3, P4 and P5 possess the highest Tm values, ranging from 28 °C for P4 and P5 to 39 °C for P3. The largest metal-induced increases in Tm are seen for P1 (up to +15 °C for NiII) which is consistent with the greatest induction of helicity for this peptide. The control peptide P2 does not display any metal-dependent changes in stability, consistent with the lack of an HCM (Fig. S15, Tables S6 and S7†).Perhaps a more practically relevant measure of peptide stability is resistance to proteolytic cleavage. A commonly cited drawback to using peptides for biological targeting is their propensity to be quickly digested in the presence of proteases.50 Since it has been established that proteases bind their substrates in an unfolded, extended fashion, one way of protecting the peptide backbone is by promoting a stable, folded peptide structure.50 In order to measure the ability of the HCM to confer protease resistance, we chose P3 as a test case because it possessed the highest extent of helicity in the presence of NiII among all peptides. We incubated both metal-bound and metal-free P3 (1.5 mM) with trypsin (0.3 mg mL−1 or 12.9 μM), which specifically cleaves peptides on the carboxy end of Lys or Arg residues,51 of which there are two in P3 (Lys6 and Lys11). The extent of digestion at 4 °C was determined at various time points by HPLC (Fig. 3 and Table 2, see also Fig. S16 and S17†) through monitoring the decrease in the intensity of the intact peptide peak. The cleavage products were identified by additional LC-MS experiments (Fig. S18†). Under pseudo-first order reaction rate kinetics, apo-P3 was efficiently digested by trypsin with a kdigest = 1.6 × 10−3 s−1; the proteolytic digestion reached completion after approx. 1 h (Fig. 3 and Table 2). Upon metal binding, P3 becomes considerably more resistant to digestion, following the same trend (NiII > CoII ≈ ZnII > CuII) observed for helix induction. In fact, NiII-bound P3 is cleaved by only 15% at 150 min, with kdigest = 1.7 × 10−5 s−1, nearly 100-fold slower than the apo-peptide. As a comparison, a half-life enhancement of 82-fold was reported for the chymotryptic digestion of a 36-residue peptide that was stabilized through two i/i + 4 covalent staples near its N- and C-termini.52 CoII- and ZnII-bound P3 are ∼30-40-fold more resistant to cleavage, and CuII enhances proteolytic stability by only ∼8-fold, consistent with its inability to induce significant α-helicity. At 25 °C, the digestion rates are uniformly faster for all species and follow the same trends (Table 2), but the overall stabilization effect of metal binding is dampened as expected from lower absolute helicities combined with elevated enzymatic activity at this temperature. Stabilization by NiII coordination at 25 °C is now 45-fold over the apo-peptide and that by CuII binding only 2-fold. As a control, we used a variant of P3 (P3bare) that was not labelled with Phen at Cys19, and therefore is devoid of the HCM and incapable of metal-mediated α-helix induction. In the absence of any metal or in the presence of NiII, P3bare was cleaved by trypsin with essentially the same kinetics as apo-P3 (Table 2 and Fig. S17†). These observations confirm that the metal-bound HCM is necessary for increased resistance to digestion and that the observed stabilization effects are not due to a possible inhibition of trypsin due to free metal ions.
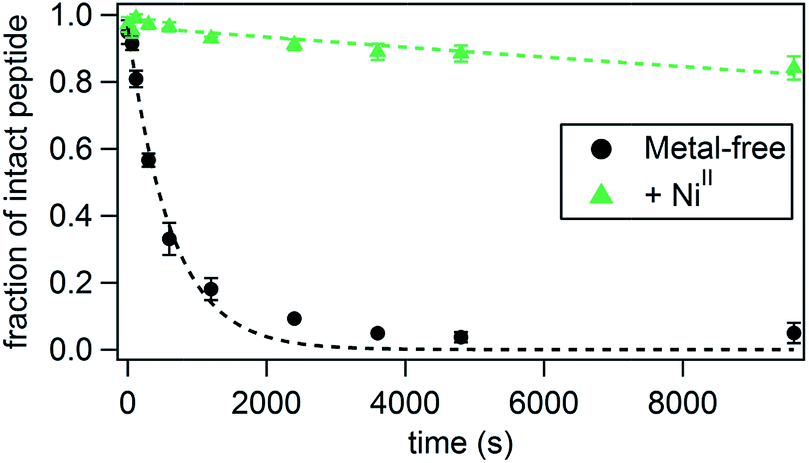 | ||
| Fig. 3 Kinetics of the tryptic digestion of P3 in the presence or absence of NiII. Additional relevant data (effects of other metal ions, LC-MS analysis) are shown in Fig. S16–S18.† | ||
| Metal | Rate (s−1) | Half-life (min) | Enhancement over metal-free HCM |
|---|---|---|---|
| 4 °C | |||
| Metal-free:P3bare | 2.0(1) × 10−3 | 6 | 0.8 |
| NiII:P3bare | 1.7(3) × 10−3 | 7 | 0.9 |
| Metal-free | 1.6(2) × 10−3 | 7 | 1 |
| NiII | 1.7(2) × 10−5 | 700 | 96.7 |
| CuII | 1.9(2) × 10−4 | 60 | 8.4 |
| ZnII | 4.1(6) × 10−5 | 280 | 38.8 |
|
|||
| 25 °C | |||
| Metal-free | 5.0(6) × 10−3 | 2 | 1 |
| NiII | 1.1(1) × 10−4 | 106 | 53 |
| CuII | 2.3(2) × 10−3 | 5 | 2.5 |
DNA binding and recognition by HCM-peptides
Having established the metal coordination and tunable α-helix induction properties of HCM-peptides, we next investigated whether they could be leveraged for binding and recognition of biomolecular targets, specifically DNA. As a model, we chose basic leucine zipper (bZIP) proteins which are dimeric gene transcription factors that consist of a dimerization domain characterized by a heptad repeat of leucine residues and a DNA binding domain (basic domain) containing basic and hydrophobic residues.6 Extensive structural and biochemical studies have revealed that in the activated (DNA-bound) dimeric form of bZIP proteins, the basic domains assume an α-helical conformation and adopt a distinct scissor-shaped geometry, which allows them to form extensive interactions with two adjacent DNA major grooves.6 The α-helix induction in the basic domains (which are not appreciably helical in isolation),53 their dimerization54 and their proper orientation55 were found to be crucial for efficient and specific DNA binding.56 In earlier studies, we observed that NiII coordination by His–Quin HCMs can direct the formation of protein dimers with a discrete, rigid V-shaped architecture that closely resembles the orientation of the basic domains of bZIP proteins (Fig. 4a and b).37 We thus reasoned that HCMs could provide a means to regulate DNA binding by peptide sequences simultaneously through metal-tunable α-helix induction and through metal-directed dimerization and orientation. | ||
| Fig. 4 Design of the DNA-binding peptide P8. (a) The V-shaped cyt cb562 dimer dictated by NiII coordination to the His/Quin HCMs (green) (PDB ID: 3L1M). Adapted from ref. 39. (b) Backbone superposition of the Helix3 domains of HCM-modified cyt cb562 (black) onto the basic domain of Jun bZip homodimer (magenta) complexed with cAMP responsive element (CRE) (brown) (PDB ID = 1JNM). (c) Overall architecture and structural components of P8, and its proposed Ni-induced dimerization geometry based on the structural model in (b). | ||
We designed the 33-residue peptide P8 based on GCN4, a bZIP protein that specifically binds the CRE DNA sequence.57 The P8 sequence contains the N-terminal cap of GCN4 (Asp1 to Leu5, in our numbering) and the entire 16-residue long basic domain of GCN4 (Lys6 to Lys21) without alteration. It also contains the 4-residue long linker region and an 8-residue portion of the dimerization domain, which provides an extension of sufficient length for the insertion of an i/i + 7 His–Phen HCM. To install the HCM, the last residue of the linker region (Met25) was converted into a Cys for the attachment of Phen and Val32 into His. Additionally, Leu28 and Asp29 in the i + 3 and i + 4 positions were converted into Ala to prevent any possible clashes of the side chains with the HCM motif.
Using CD spectroscopy, we first confirmed that P8 undergoes a metal-induced increase in helicity, following the same trend (NiII > ZnII > CuII) observed in P1–P7 (Fig. 5a). As before, this effect is eliminated in P8bare, which lacks the Phen functionality (Fig. S19†). Titrations of metal ions into a 15 μM solution of P8 indicated that the maximal helicity (measured by CD at 222 nm) and the formation of fully metallated HCMs (measured by UV-vis, λmax at 276 nm) are achieved at a ratio of approximately 0.4–0.5 NiII:P8, indicating the formation of the desired metal-directed peptide dimers (NiII:P82) (Fig. 5b, S20 and S21†). We note that it is possible that the NiII:P83 complex may also be present in solution through the [Ni(phen)3]2+ coordination mode, which could account for the inflection point in Fig. 5b to be below 0.5. In order to more directly measure the formation of the metal-directed P8 dimers, we conducted analytical ultracentrifugation (AUC) experiments. Metal-free P8 exhibits a maximal sedimentation coefficient at 0.75 S. The sedimentation peak shifts to 0.8 S upon addition of 0.25 equiv. of NiII and reaches a maximum of 0.95 S at 0.5 equiv. of NiII, again consistent with the Ni:P82 stoichiometry. Addition of any further NiII leads to the enrichment of the solution with fully metallated P8 species (Ni:P8) which cannot dimerize, leading to the shift of the sedimentation peak to lower values (Fig. 5c).
 | ||
| Fig. 5 (a) Induction of helicity upon metal binding in P8. (b) Ni-binding titration of P8 monitored by changes in the absorption spectrum of Phen at 280 nm. The dotted black line indicates saturation of binding at 0.5 equiv. of NiII per P8, while the small absorbance change in the shaded area is attributed to a transient tris-Phen species. (c) Sedimentation velocity data for P8 in the presence of different amounts of NiII determined by AUC. | ||
In order to probe interactions of P8 with DNA (i.e., the CRE sequence), which should lead to increased helicity in the basic domain through structural templating, we carried out CD experiments (Fig. S22†). These experiments were conducted with sufficiently high P8 concentrations (5 μM) to ensure the formation of the metal-mediated P8 dimer. The addition of an equimolar amount of double-stranded (ds) CRE led to a significant increase in the helicity of apo-P8 (at both 4 and 25 °C). The addition of 0.5 equiv. of NiII or CuII to the peptide–DNA solution led to a further increase in helicity, indicative of the formation of quaternary M:P82:CRE complex. At 4 °C, the NiII-bound P8–DNA adduct displayed a higher helicity compared to the CuII-bound form, although at 25 °C, this difference was minimized. While this observation is surprising in the light of our previous finding that NiII is a considerably better helix inducer than CuII for peptides with His–Phen HCMs, it suggests that there is indeed an interplay between DNA-binding interactions and metal-mediated peptide dimerization/helix induction.
In order to quantitatively characterize P8–DNA interactions and to determine their specificity and metal-dependence, we conducted gel-shift assays with radiolabeled DNA sequences. Due to the expected dissociation constants of basic domain-CRE interactions in the nM range,53 we used low concentrations of ds-DNA (2 nM) and P8 (1–500 nM), in addition to 0.5 equiv. of MII (with respect to P8 concentration). Based on the affinities of the His–Phen HCM's for various metal ions (in the nM range) and the metal-mediated dimerization constants (in the μM range), we would expect the P8 peptides to be mostly metal-bound, but in a monomeric state in the absence of DNA interactions.
As shown in Fig. 6, the binding of P8 to CRE occurs in a metal-dependent fashion, whereby the final product has the desired M:P82:CRE stoichiometry (see also Fig. S23 and S24†). Interestingly, small amounts of monomeric M:P8:CRE species are also observed (Fig. 6b), which is consistent with a model where the monomeric, metal-bound P8 also interacts with CRE, likely as an intermediate en route to the dimeric P8–DNA complex (Fig. 7). These observations agree with previous findings that the dimerization of certain bZIP peptides occurs on the target DNA, rather than DNA binding by preformed dimers.58–60 In the absence of metal ions, no DNA binding by P8 is observed. As expected from our structural model, NiII coordination yields the highest affinity (Kd,DNA = 65 nM) for CRE, followed by ZnII (Kd,DNA = 84 nM) and CuII (Kd,DNA = 139 nM) (Table 3); this range of affinities compare well with those obtained for other synthetically dimerized bZIP-peptide constructs.54 Importantly, our results show that peptide–DNA interactions can be finely-tuned in a way that is dependent on the choice of the metal ion. Finally, in the presence of NiII, no binding by P8 to the non-cognate DNA sequence SCR is observed, confirming the sequence specificity of metal-directed P8–DNA interactions.
 | ||
| Fig. 6 Electrophoretic mobility shift assay for monitoring P8-CRE binding in the absence (a) and the presence (b) of 0.5 equiv. of NiII. Lane Q contains CRE without any added peptide; DNA concentration is kept constant at 1 nM while the P8 concentrations varies: (A) 1 nM, (B) 2 nM, (C) 5 nM, (D) 10 nM, (E) 15 nM, (F) 20 nM, (G) 25 nM, (H) 30 nM, (I) 40 nM, (J) 50 nM, (K) 75 nM, (L) 100 nM, (M) 150 nM, (N) 200 nM, (O) 250 nM, (P) 500 nM. The intensities of the radioactively labelled CRE bands were measured by phosphorimaging. (c) Effects of different metal ions (0.5 equiv.) on CRE binding by P8, determined by electrophoretic mobility shift assays. See Fig. S23 and S24† for additional data. (d) DNA sequence specificity of P8 binding. | ||
 | ||
| Fig. 7 Scheme for the possible modes of P8 binding to DNA. In this case, the DNA can act as a template for dimerization. | ||
| Metal | DNA sequence | K d,apparent |
|---|---|---|
| NiII | SCR | >350 nM |
| NiII | CRE | 65 ± 4 nM |
| CuII | CRE | 139 ± 7 nM |
| ZnII | CRE | 84 ± 5 nM |
Conclusions
Our results demonstrate the versatility of HCMs in tuning the structural and biochemical properties of peptides in a metal-dependent fashion. First, the HCMs offer a readily accessible and functionally flexible alternative to other types of peptide-crosslinking or helix induction strategies19 in that they are reversible and the extent of helix induction is easily tuned through the choice of metal identity. Helix induction on peptide–HCM platforms is further augmented by exploiting secondary interactions of appropriately positioned amino acid sidechains with the organic component of the HCM. Second, the resulting increase in stability with respect to chemical or thermal denaturation or enzymatic hydrolysis is comparable to that which is achieved by covalent stapling approaches.52 Third, under proper experimental conditions, HCMs enable the dimerization of peptides with distinct geometries that are dictated by the inner-sphere coordination preferences of the metal ions. In this study, we exploited this feature to demonstrate the ability of HCM-bearing peptides to recognize and bind a biological target. Starting with the pioneering work by Kim,54,61 several synthetic strategies have been developed for the dimerization of bZIP peptides,55,62–64 including disulfide bonding,54 metal coordination,65–68 and crosslinking with metal-tunable69 or photo-switchable linkers.70–72 These studies have collectively shown the importance of the dimeric organization of the basic domains as well as their proper orientation for DNA binding and recognition. The HCM-based approach that we have described here is complementary to these previously described strategies. Yet, it is also distinct in the sense that the HCM motif is an integral part of the peptide scaffold due to its two-point attachment to the backbone, while in all other strategies the dimerization units are attached to the peptide chains through a single point. As a result, HCMs can simultaneously induce peptide helicity through metal coordination and lead to the formation of discrete (i.e., unique) peptide dimerization geometries, which can be modulated in combination to optimize DNA binding and recognition.Acknowledgements
The authors would like to thank Professor Nathan Gianneschi for the use of a peptide synthesizer, Professor Elizabeth Komives for the use of a peptide synthesizer and HPLC, Dr Yongxuan Su for assistance with mass spectrometry and CD, Professor Daniel Donoghue for help with gel shift assays, Professor Ulrich Muller for help with radiolabelling DNA, and Dr Richard Cochran for assistance with LC-MS. This work was supported by the US Department of Energy (DOE) (Division of Materials Sciences, Office of Basic Energy Sciences, Award DE-FG02-10ER46677 to F. A. T. for materials synthesis and characterization), the National Science Foundation (CHE1306646 to F. A. T. for data analysis and a Graduate Research Fellowship to B. R. B), the National Institutes of Health (Molecular Biophysics traineeship to S. J. S., Grant T32 GM008326), and the W. M. Keck Foundation for the use of computing resources at the W. M. Keck Laboratory for Integrated Biology II.Notes and references
- P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 11, 82–88 CrossRef CAS PubMed.
- A. N. Lupas and M. Gruber, in Advances in Protein Chemistry, Academic Press, 2005, vol. 70, pp. 37–38 Search PubMed.
- R. J. Youle and A. Strasser, Nat. Rev. Mol. Cell Biol., 2008, 9, 47–59 CrossRef CAS PubMed.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS PubMed.
- S. J. Busch and P. Sassone-Corsi, Trends Genet., 1990, 6, 36–40 CrossRef CAS PubMed.
- T. E. Ellenberger, C. J. Brandl, K. Struhl and S. C. Harrison, Cell, 1992, 71, 1223–1237 CrossRef CAS PubMed.
- J. Miller, A. D. McLachlan and A. Klug, EMBO J., 1985, 4, 1609–1614 CAS.
- C. O. Pabo, E. Peisach and R. A. Grant, Annu. Rev. Biochem., 2001, 70, 313–340 CrossRef CAS PubMed.
- W. E. Stites, Chem. Rev., 1997, 97, 1233–1250 CrossRef CAS PubMed.
- B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- T. Edwards and A. Wilson, Amino Acids, 2011, 41, 743–754 CrossRef CAS PubMed.
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301–317 CrossRef CAS PubMed.
- B. P. Orner, J. T. Ernst and A. D. Hamilton, J. Am. Chem. Soc., 2001, 123, 5382–5383 CrossRef CAS PubMed.
- J. M. Davis, L. K. Tsou and A. D. Hamilton, Chem. Soc. Rev., 2007, 36, 326–334 RSC.
- W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399–1408 CrossRef CAS PubMed.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- A. Whitty and G. Kumaravel, Nat. Chem. Biol., 2006, 2, 112–118 CrossRef CAS PubMed.
- K. Estieu-Gionnet and G. Guichard, Expert Opin. Drug Discovery, 2011, 6, 937–963 CrossRef CAS PubMed.
- D. Y. Jackson, D. S. King, J. Chmielewski, S. Singh and P. G. Schultz, J. Am. Chem. Soc., 1991, 113, 9391–9392 CrossRef CAS.
- S. E. Miller, N. R. Kallenbach and P. S. Arora, Tetrahedron, 2012, 68, 4434–4437 CrossRef CAS PubMed.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS.
- K. Fujimoto, M. Kajino and M. Inouye, Chem.–Eur. J., 2008, 14, 857–863 CrossRef CAS PubMed.
- J. W. Taylor, Biopolymers, 2002, 66, 49–75 CrossRef CAS PubMed.
- J. C. Phelan, N. J. Skelton, A. C. Braisted and R. S. McDowell, J. Am. Chem. Soc., 1997, 119, 455–460 CrossRef CAS.
- J. W. Taylor, Biopolymers, 2002, 66, 49–75 CrossRef CAS PubMed.
- F. Ruan, Y. Chen and P. B. Hopkins, J. Am. Chem. Soc., 1990, 112, 9403–9404 CrossRef CAS.
- M. T. Ma, H. N. Hoang, C. C. G. Scully, T. G. Appleton and D. P. Fairlie, J. Am. Chem. Soc., 2009, 131, 4505–4512 CrossRef CAS PubMed.
- A. N. Zaykov, B. V. Popp and Z. T. Ball, Chem.–Eur. J., 2010, 16, 6651–6659 CrossRef CAS PubMed.
- M. R. Ghadiri and C. Choi, J. Am. Chem. Soc., 1990, 112, 1630–1632 CrossRef CAS.
- A. Patgiri, A. L. Jochim and P. S. Arora, Acc. Chem. Res., 2008, 41, 1289–1300 CrossRef CAS PubMed.
- E. Cabezas and A. C. Satterthwait, J. Am. Chem. Soc., 1999, 121, 3862–3875 CrossRef CAS.
- S. Marqusee and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8898–8902 CrossRef CAS.
- C. Toniolo, M. Crisma, F. Formaggio and C. Peggion, Biopolymers, 2001, 60, 396–419 CrossRef CAS PubMed.
- R. J. Radford, P. C. Nguyen, T. B. Ditri, J. S. Figueroa and F. A. Tezcan, Inorg. Chem., 2010, 49, 4362–4369 CrossRef CAS PubMed.
- R. J. Radford, P. C. Nguyen and F. A. Tezcan, Inorg. Chem., 2010, 2010, 7106–7115 CrossRef PubMed.
- S. J. Smith, K. Du, R. J. Radford and F. A. Tezcan, Chem. Sci., 2013, 4, 3740–3747 RSC.
- A. E. Martell and R. M. Smith, Critical Stability Constants, Plenum Press, New York, 1974 Search PubMed.
- B. A. Krantz and T. R. Sosnick, Nat. Struct. Mol. Biol., 2001, 8, 1042–1047 CAS.
- D.-H. Chin, R. W. Woody, C. A. Rohl and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15416–15421 CrossRef CAS PubMed.
- P. Luo and R. L. Baldwin, Biochemistry, 1997, 36, 8413–8421 CrossRef CAS PubMed.
- S. Padmanabhan and R. L. Baldwin, J. Mol. Biol., 1994, 241, 706–713 CrossRef CAS PubMed.
- S. Padmanabhan and R. L. Baldwin, Protein Sci., 1994, 2, 1992–1997 CrossRef PubMed.
- T. P. Creamer and G. D. Rose, Protein Sci., 1995, 4, 1305–1314 CrossRef CAS PubMed.
- P. Y. Chou and G. D. Fasman, Biochemistry, 1974, 13, 211–222 CrossRef CAS PubMed.
- D. M. John and K. M. Weeks, Protein Sci., 2000, 9, 1416–1419 CrossRef CAS PubMed.
- H. S. Chan, S. Bromberg and K. A. Dill, Philos. Trans. R. Soc., B, 1995, 348, 61–70 CrossRef CAS PubMed.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- J. V. Olsen, S.-E. Ong and M. Mann, Mol. Cell. Proteomics, 2004, 3, 608–614 CAS.
- G. H. Bird, N. Madani, A. F. Perry, A. M. Princiotto, J. G. Supko, X. He, E. Gavathiotis, J. G. Sodroski and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14093–14098 CrossRef CAS PubMed.
- M. A. Weiss, T. Ellenberger, C. R. Wobbe, J. P. Lee, S. C. Harrison and K. Struhl, Nature, 1990, 347, 575–578 CrossRef CAS PubMed.
- R. Talanian, C. McKnight and P. Kim, Science, 1990, 249, 769–771 CAS.
- T. Morii, M. Simomura, S. Morimoto and I. Saito, J. Am. Chem. Soc., 1993, 115, 1150–1151 CrossRef CAS.
- J. J. Hollenbeck, D. L. McClain and M. G. Oakley, Protein Sci., 2002, 11, 2740–2747 CrossRef CAS PubMed.
- I. A. Hope and K. Struhl, Cell, 1986, 46, 885–894 CrossRef CAS PubMed.
- S. Cranz, C. Berger, A. Baici, I. Jelesarov and H. R. Bosshard, Biochemistry, 2004, 43, 718–727 CrossRef CAS PubMed.
- S. J. Metallo and A. Schepartz, Nat. Struct. Mol. Biol., 1997, 4, 115–117 CAS.
- J. J. Kohler, S. J. Metallo, T. L. Schneider and A. Schepartz, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11735–11739 CrossRef CAS.
- R. V. Talanian, C. J. McKnight, R. Rutkowski and P. S. Kim, Biochemistry, 1992, 31, 6871–6875 CrossRef CAS PubMed.
- M. Ueno, A. Murakami, K. Makino and T. Morii, J. Am. Chem. Soc., 1993, 115, 12575–12576 CrossRef CAS.
- Y. Aizawa, Y. Sugiura and T. Morii, Biochemistry, 1999, 38, 1626–1632 CrossRef CAS PubMed.
- M. Pellegrini and R. H. Ebright, J. Am. Chem. Soc., 1996, 118, 5831–5835 CrossRef CAS.
- E. Oheix and A. F. A. Peacock, Chem.–Eur. J., 2014, 20, 2829–2839 CrossRef CAS PubMed.
- B. Cuenoud and A. Schepartz, Science, 1993, 259, 510–513 CAS.
- B. Cuenoud and A. Schepartz, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1154–1159 CrossRef CAS.
- C. R. Palmer, L. S. Sloan, J. C. Adrian, B. Cuenoud, D. N. Paolella and A. Schepartz, J. Am. Chem. Soc., 1995, 117, 8899–8907 CrossRef CAS.
- J. Mosquera, A. Jiménez-Balsa, V. I. Dodero, M. E. Vázquez and J. L. Mascareñas, Nat. Commun., 2013, 4, 1874 CrossRef PubMed.
- G. A. Bullen, J. H. R. Tucker and A. F. A. Peacock, Chem. Commun., 2015, 51, 8130–8133 RSC.
- A. M. Caamaño, M. E. Vázquez, J. Martínez-Costas, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2000, 39, 3104–3107 CrossRef.
- G. A. Woolley, A. S. I. Jaikaran, M. Berezovski, J. P. Calarco, S. N. Krylov, O. S. Smart and J. R. Kumita, Biochemistry, 2006, 45, 6075–6084 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, figures and tables. See DOI: 10.1039/c6sc00826g |
| This journal is © The Royal Society of Chemistry 2016 |