
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lesion orientation of O4-alkylthymidine influences replication by human DNA polymerase η†
D. K.
O'Flaherty
a,
A.
Patra
b,
Y.
Su
b,
F. P.
Guengerich
b,
M.
Egli
*b and
C. J.
Wilds
*a
aDepartment of Chemistry and Biochemistry, Concordia University, Montréal, Québec H4B1R6, Canada. E-mail: chris.wilds@concordia.ca
bDepartment of Biochemistry, Vanderbilt Institute of Chemical Biology, Center for Structural Biology, School of Medicine, Vanderbilt University, Nashville, Tennessee 37232, USA. E-mail: martin.egli@vanderbilt.edu
First published on 26th April 2016
Abstract
DNA lesions that elude repair may undergo translesion synthesis catalyzed by Y-family DNA polymerases. O4-Alkylthymidines, persistent adducts that can result from carcinogenic agents, may be encountered by DNA polymerases. The influence of lesion orientation around the C4–O4 bond on processing by human DNA polymerase η (hPol η) was studied for oligonucleotides containing O4-methylthymidine (O4MedT), O4-ethylthymidine (O4EtdT), and analogs restricting the O4-methylene group in an anti-orientation. Primer extension assays revealed that the O4-alkyl orientation influences hPol η bypass. Crystal structures of hPol η·DNA·dNTP ternary complexes with O4MedT or O4EtdT in the template strand showed the nucleobase of the former lodged near the ceiling of the active site, with the syn-O4-methyl group engaged in extensive hydrophobic interactions. This unique arrangement for O4-methylthymidine with hPol η, inaccessible for the other analogs due to steric/conformational restriction, is consistent with differences observed for nucleotide incorporation and supports the concept that lesion conformation influences extension across DNA damage. Together, these results provide mechanistic insights on the mutagenicity of O4MedT and O4EtdT when acted upon by hPol η.
Introduction
DNA alkylation results from a variety of endogenous and/or exogenous agents that can interfere with vital cellular processes, i.e. replication and transcription.1 The addition of alkyl appendages on the DNA scaffold can have adverse consequences such as DNA polymerase (Pol) blockage, nucleotide misincorporation, chromosomal instability, and activation of the cellular apoptotic pathway.1,2 However, organisms have various repair pathways to restore damaged DNA. In the event that a lesion evades the process of DNA repair, translesion synthesis (TLS) by Y-family DNA Pols can occur, allowing bypass of the DNA lesion in an error-free or error-prone manner.3 Y-family DNA Pols are described as more “promiscuous” given their larger active sites when compared with replicative DNA Pols, which accounts for their ability to bypass damaged nucleotides that induce blockage. DNA Pol η in humans (hPol η) plays a pivotal role in the bypass of certain UV-induced DNA damage, which impedes DNA replication.4 hPol η activity has also been correlated with chemotherapeutic resistance to platinum-based agents such as cisplatin and the efficient bypass of the oxidative DNA lesion 7,8-dihydro-8-oxo-2′-deoxyguanosine.5,6The O4-position of thymidine is susceptible to alkylation by agents such as N-nitroso alkylamines in certain foods, water, air, and particularly tobacco products.7,8 Albeit a minor site of alkylation, lesions such as O4-methylthymidine (O4MedT) and O4-ethylthymidine (O4EtdT) are poorly processed by mammalian repair pathways, making them persistent in the genome.9,10 O4MedT and O4EtdT hinder high fidelity replicative DNA Pol activity, resulting in misinsertion of dGTP in the daughter DNA strands.10–12 Correlations between the mutagenicity of O4MedT and cancer have been established,13,14 highlighting the importance of investigating the structural properties and biological outcomes associated with this type of DNA damage.
The current understanding of the mechanism of Y-family DNA Pol misincorporation during TLS depends on a number of factors, including the nature of the DNA damage, the DNA Pol and the incoming nucleoside triphosphate. The geometrical array of the ternary complex formed (involving DNA, protein and nucleoside triphosphate) is the key characteristic that governs efficient bypass of a DNA lesion. Structural investigation by NMR and X-ray crystallography of duplexes containing an O4MedT insert has revealed that the methyl group preferentially adopts a syn conformation around the C4–O4 bond (Fig. 1).15,16 We hypothesized that the conformation of the O4-alkyl lesion could affect the base pair geometry during the primer extension reaction catalyzed by DNA Pol η. To address this possibility, we probed hPol η processivity with thymidine analogs that link the C5 and O4 atoms by a dimethylene or trimethylene group, which limits the O4-lesion to adopt an anti-conformation (Fig. 1) to relate the structural features of O4-alkylated dT with the bypass activity of hPol η. hPol η was selected as the model Y-family DNA polymerase, given previous studies concerning bypass of O4MedT and O4EtdT.17,18 Results of these studies indicated that Pol η, from yeast or human, were most efficient in extending across and past O4MedT17 and O4EtdT,18 respectively.
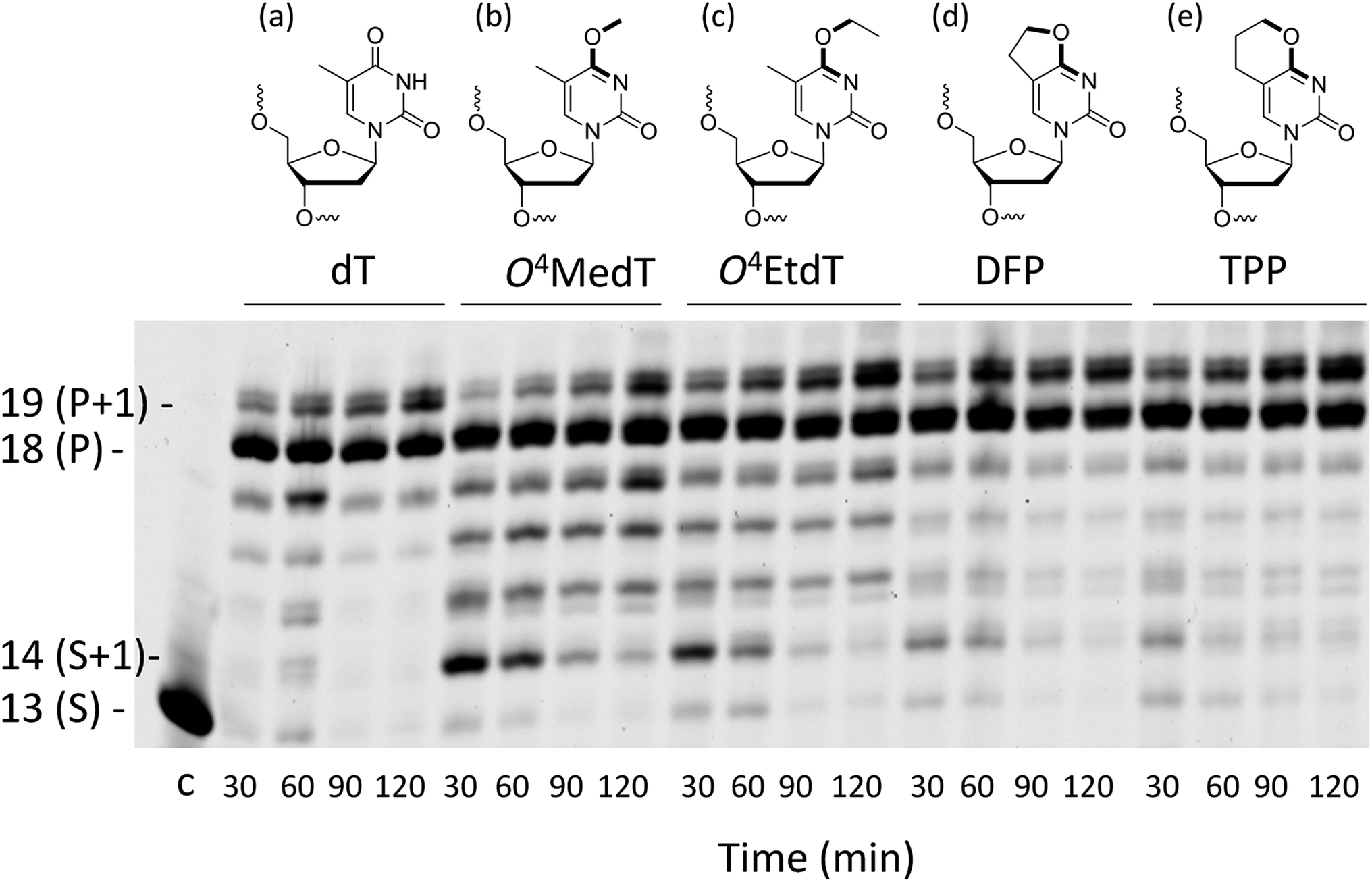 | ||
| Fig. 1 Structures of the (a) unmodified dT control, (b) O4MedT, (c) O4EtdT, (d) DFP and (e) TPP adducts. In bold are the bonds between the atoms shown to have a syn (b and c) or anti (d and e) orientation around the O4–C4 bond. Full primer extension assays for the control undamaged substrate (dT), O4MedT, O4EtdT, DFP, and TPP-bearing substrates with hPol η. The template strand sequence is 3′-AGCATTCGCAGTAXTACT-5′ where X denotes the modification and the 5′-FAM labeled primer strand sequence is 5′-TCGTAAGCGUCAT-3′. | ||
We investigated bypass profiles opposite all four lesions by hPol η (steady-state single nucleotide incorporation and LC-MS/MS analysis of full-length extension products). Crystal structures of ternary hPol η·DNA·dATP and hPol η·DNA·dGTP with template strands containing O4MedT or O4EtdT reveal a distinct orientation of the former lesion that stacks atop a tryptophan residue near the ceiling of the active site instead of pairing with the incoming nucleotide. Conversely, O4EtdT pairs with both incoming dA and dG nucleotides via bifurcated H-bonds in the insertion complexes and displays the same configuration opposite primer dG in the crystal structure of an extension complex adjacent to the nascent dG:dCTP pair. The structures provide a better understanding of the different behavior of the O4MedT or O4EtdT lesions in hPol η-catalyzed error-prone bypass reactions and suggests a unique intermediate step in the bypass of O4MedT.
Results
Synthesis and characterization of modified oligonucleotides
The structures of O4MedT, O4EtdT and the modified pyrimidyl nucleosides 3-(2′-deoxypentofuranosyl)-5,6-dihydrofuro[2,3-d]pyrimidin-2(3H)-one (DFP) and 3-(2′-deoxypentofuranosyl)-3,5,6,7-tetrahydro-2H-pyrano[2,3-d]pyrimidin-2-one (TPP) are shown in Fig. 1 (methods describing the preparation of nucleosides and oligonucleotides can be found in the ESI†). UV thermal denaturation studies of duplexes containing single inserts of the DFP or TPP modification revealed a comparable destabilizing effect to O4MedT and O4EtdT with complementary strands containing adenine or any mismatched base pairing partner (Fig. S38†). Circular dichroism spectra of duplexes containing the DFP or TPP inserts revealed little deviation from a B-form structure (see Fig. S39†).Steady-state kinetics
Steady-state kinetic assays of individual nucleotide incorporations opposite O4MedT, O4EtdT, DFP, TPP and unmodified dT were carried out with the catalytic core construct of hPol η (amino acids 1-432). In all cases, these pyrimidyl modifications blocked DNA synthesis by the polymerase relative to the unmodified control (Fig. 2a, values shown in Table S1†). Incorporation of the correct dAMP nucleotide by hPol η opposite to O4MedT, O4EtdT, DFP, and TPP was reduced approximately 6.5-, 12-, 4.5-, and 5-fold, respectively, relative to dT (see Fig. 2a).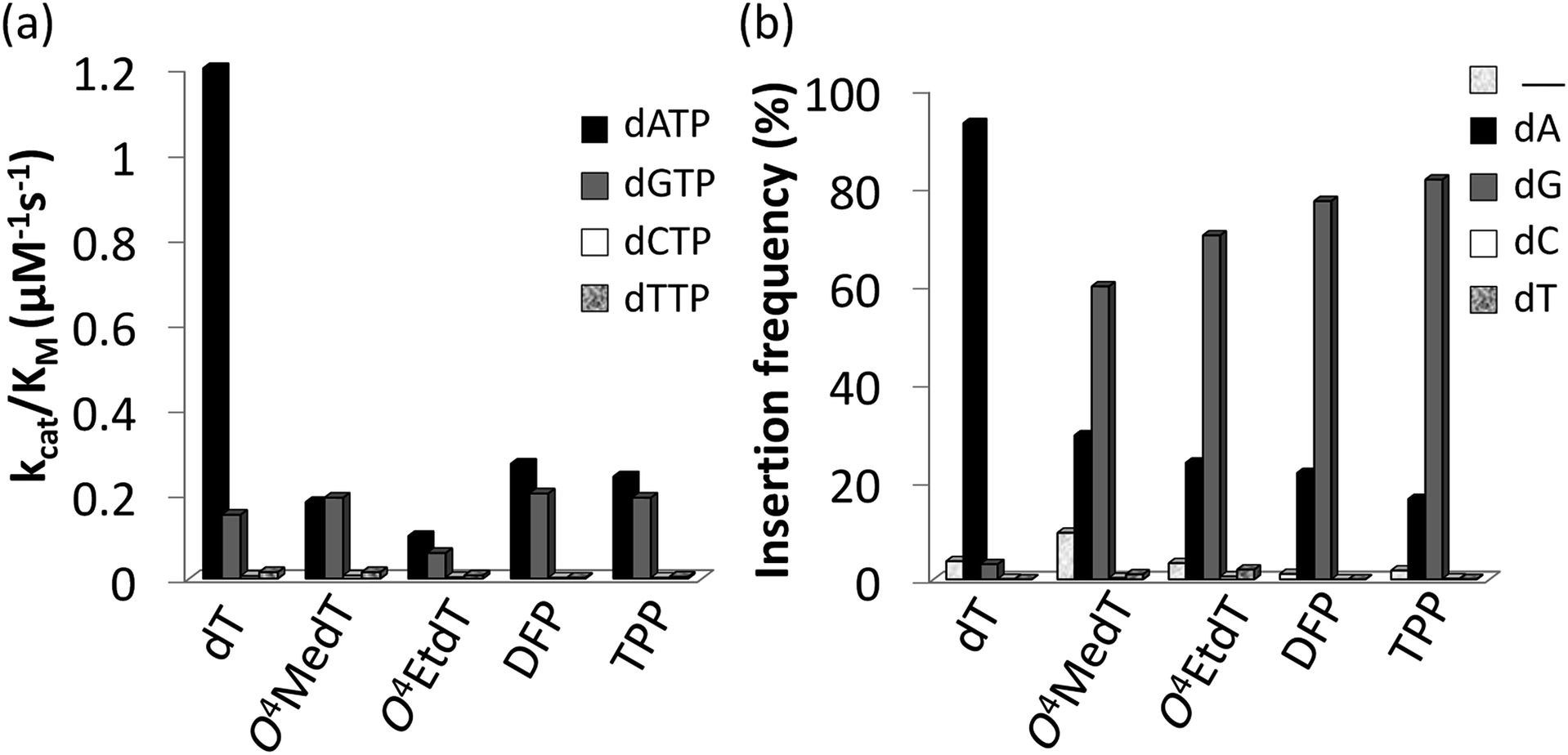 | ||
| Fig. 2 (a) Steady-state incorporation efficiencies opposite dT, O4MedT, O4EtdT, DFP, and TPP by hPol η with individual dNTPs. (b) Incorporation frequencies based on ESI-MS/MS analysis of primer extension products opposite the dT, O4MedT, O4EtdT, DPF, and TPP containing templates (“_” indicates frameshift adduct formation. Tabulated kinetic parameters and fragments identified by LC-MS/MS analysis of full-length extension products can be found in ESI†). | ||
hPol η incorporated dCMP and dTMP opposite all the pyrimidyl modifications and dT with similar catalytic efficiencies (ranging from approximately 0.002–0.015 μM−1 s−1). However, a strong preference for either dAMP or dGMP incorporation opposite the modified pyrimidines was observed. Other than O4MedT, hPol η preferentially incorporated the correct dAMP nucleotide opposite all the pyrimidyl modifications. The significant incorporation of dGMP when hPol η encountered these pyrimidyl modifications, compared to the unmodified control, indicates a clear loss in substrate specificity by the polymerase (Fig. 2a). In the case of O4MedT, dGMP was slightly preferred as the nucleotide incorporated by hPol η (0.19 ± 0.01 vs. 0.18 ± 0.03 μM−1 s−1 for dGMP and dAMP, respectively).
LC-MS/MS analysis of full-length extension products produced by hPol η
Analysis of single insertions by a DNA polymerase is useful for kinetic evaluation but may not reflect incorporation fidelity in the presence of all four dNTPs across the damage and beyond this site. The fidelity of hPol η and its processivity past the damage site was addressed by the use of a full extension assay coupled with LC-MS/MS analysis.5,19,20The optimal reaction times to observe the full extension products from the template strands containing the modifications and the unmodified control were evaluated (Fig. 1). Full extension was achieved for the unmodified control at 30 min, whereas templates containing the modifications required longer reaction times (60–90 min). The time course assay revealed that hPol η had difficulty in extending past O4MedT and O4EtdT and displayed a significant “S + 1” band at reaction times of 30 and 60 min. UPLC separation of the cleaved products and mass spectrometry analysis of their sequence identities revealed that dGMP was incorporated most efficiently opposite all the modifications (see Fig. 2b).
The incorporation frequency opposite dT, O4MedT, O4EtdT, DFP, and TPP for the full extension products was evaluated (see Fig. 2b and Table S2†). The presence of O4MedT increased the level of frameshift formation by hPol η relative to the control (9.5 vs. 3.5%). Comparable levels of frameshifts were observed opposite O4EtdT and the dT control. However, the templates containing the bicyclic pyrimidine adducts did not induce a similar increase in frameshift formation with levels that were approximately one-half, relative to the control. The correct dAMP nucleotide was incorporated by hPol η at a frequency of approximately 30, 24, 22, 16, and 93% opposite O4MedT, O4EtdT, DFP, TPP, and dT, respectively. Out of the lesions investigated, hPol η exhibited the highest fidelity opposite O4MedT and lowest opposite the TPP. Incorporation of dGMP was observed to occur in the extension products with overall frequencies of 60, 70, 72, 82, 3% opposite O4MedT, O4EtdT, DFP, TPP, and dT, respectively.
The accuracy of bypass varied for the O4-alkylthymidine modifications by approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in favor of dGMP opposite O4MedT to 5:1 in favor of dGMP opposite TPP. An increased adduct size, from O4MedT to O4EtdT and DFP to TPP, resulted in a 10% increase of dGMP misinsertion at the expense of a 10% decrease of the correct dAMP incorporation. Similarly, the conformationally restrained analogues (DFP and TPP) induced an increase in dGMP misinsertion (10%) by hPol η compared to O4MedT and O4EtdT, respectively.
:1 in favor of dGMP opposite O4MedT to 5:1 in favor of dGMP opposite TPP. An increased adduct size, from O4MedT to O4EtdT and DFP to TPP, resulted in a 10% increase of dGMP misinsertion at the expense of a 10% decrease of the correct dAMP incorporation. Similarly, the conformationally restrained analogues (DFP and TPP) induced an increase in dGMP misinsertion (10%) by hPol η compared to O4MedT and O4EtdT, respectively.
Pre-steady-state kinetics
The pre-steady-state kinetic assays of dATP and dGTP incorporations opposite O4MedT, O4EtdT, DFP, and TPP, and dATP incorporation opposite unmodified dT were carried out with the catalytic core of hPol η. The burst rates for dATP insertion were 3.1-, 4.2-, and 1.8-fold higher compared to dGTP opposite O4MedT, DFP, and TPP, respectively (Fig. S48 and Table S3†). The burst rates were low in the case of dATP and dGTP opposite O4EtdT. The burst amplitudes for the extensions were 15–35% opposite O4MedT, O4EtdT, DPF, and TPP, which may indicate the presence of multiple non-productive ternary complexes.Crystal structures of ternary hPol η·DNA·dNTP complexes with templates containing O4MedT or O4EtdT at the insertion stage
To visualize the O4MedT and O4EtdT lesions at the active site of hPol η trapped at the insertion stage, we determined four crystal structures of ternary complexes with the Pol bound to a 12mer template strand with the incorporated lesion and paired to an 8mer primer and incoming purine nucleoside triphosphate. For details regarding the crystallization, data collection and structure determination and refinement procedures please see the ESI.† Selected crystal data, data collection and refinement parameters and examples of the quality of the final electron density for all structures are summarized and depicted in the Table S4.† The two complexes with O4MedT-containing templates and incoming dATP or dGMPNPP reveal similar orientations of the lesion (Fig. 3, PDB ID codes 5DLF and 5DLG, respectively). Instead of pairing with the incoming nucleotide, O4MedT is lodged near the ceiling of the active site. Thus, its base portion is nestled against Trp-64 (base stacking interaction), Met-63 and Ser-62 (hydrophobic contacts between O4Me and both Cα and C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) from the two residues) from a loop region in the finger domain and Gly-46 from an adjacent β-strand that together form the roof of the active site (Fig. 3a and c). In addition, O2 of O4MedT and the amino group of Asn-38 are H-bonded (Fig. 3b and d). This position of the O4MedT lesion at the entrance of the active site places it quite far away from the incoming nucleotide triphosphate. The distance between its O4 atom and N6 of dATP is 9 Å (8.2 Å between O4 of O4MedT and O6 of dGMPNPP).
O) from the two residues) from a loop region in the finger domain and Gly-46 from an adjacent β-strand that together form the roof of the active site (Fig. 3a and c). In addition, O2 of O4MedT and the amino group of Asn-38 are H-bonded (Fig. 3b and d). This position of the O4MedT lesion at the entrance of the active site places it quite far away from the incoming nucleotide triphosphate. The distance between its O4 atom and N6 of dATP is 9 Å (8.2 Å between O4 of O4MedT and O6 of dGMPNPP).
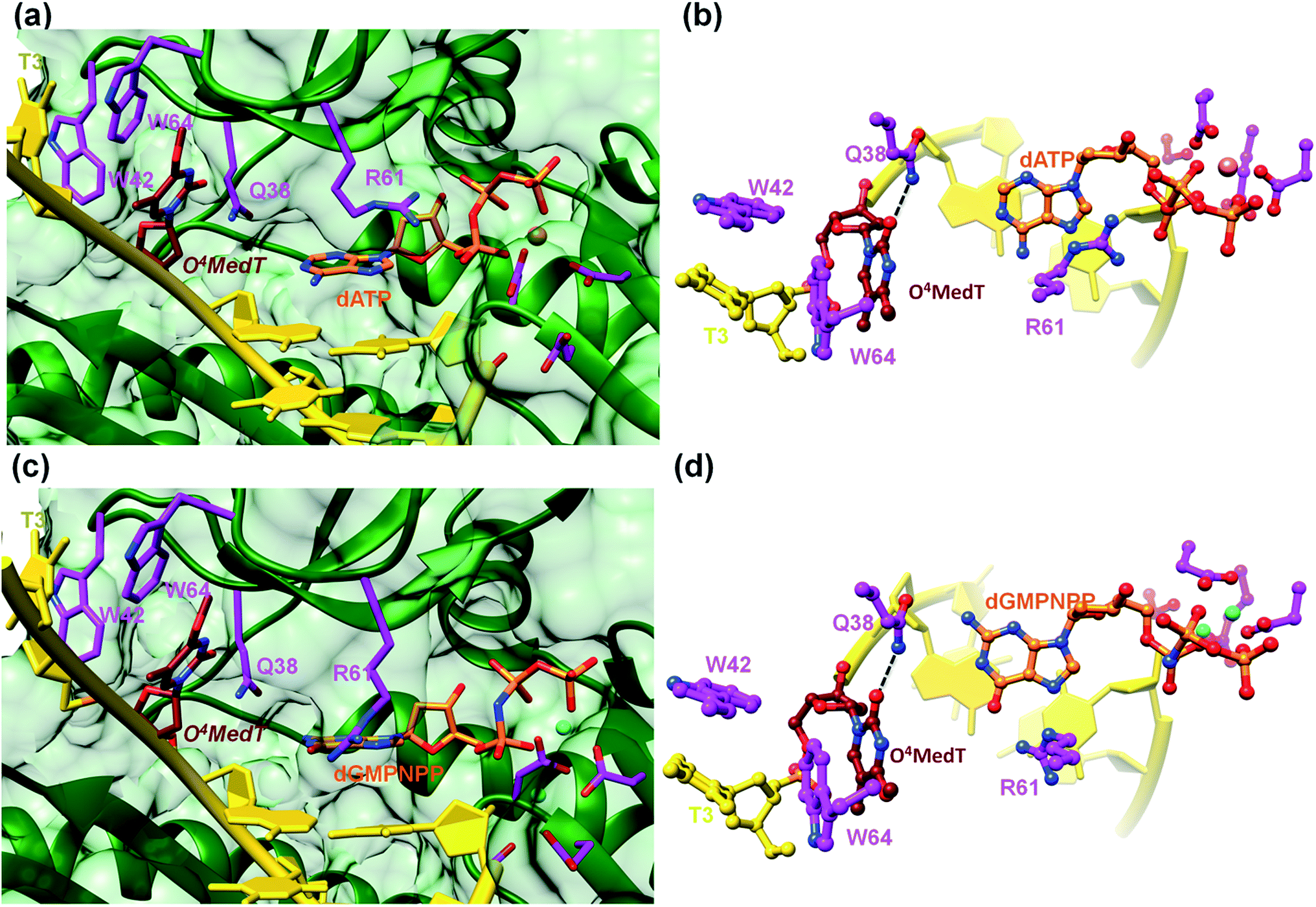 | ||
| Fig. 3 Detached arrangement of incoming purine nucleotide triphosphate and O4MedT in two hPol η insertion-stage complexes. (a) Active site conformation in the complex with dATP opposite O4MedT viewed into the DNA major groove, and (b), rotated by 90° and viewed perpendicular to the adenine plane. (c) Active site conformation in the complex with dGMPNPP opposite O4MedT, viewed into the DNA major groove, and (d), rotated by 90° and viewed perpendicular to the guanine plane. Carbon atoms of O4MedT, the incoming nucleotide triphosphate, and selected hPol η amino acid side chains are colored in maroon, orange and magenta, respectively. Mg2+ and Ca2+ ions are cyan and pink spheres, respectively, and selected H-bonds are shown as dashed lines. | ||
Closer inspection of the positions of the incoming nucleotides shows that adenine stacks on the adjacent t(emplate)A:p(rimer)T pair, with the side chain of Arg-61 from the finger domain hovering closely above the adenine plane and engaged in H-bonds to the α- and β-phosphates of dATP via its guanidino moiety (Fig. 3b). By comparison, guanine is shifted into the minor groove and the stacking interaction with the adjacent tA:pT pair is slightly less favorable. The shift is likely a consequence of the altered orientation of the Arg-61 side chain that is extended in the structure of the complex with dGMPNPP, resulting in formation of H-bonds between its guanidino moiety and guanine O6 and N7 (Fig. 3d). The particular orientation of the incoming dG brings it closer to Asn-38 from the finger domain, but the distance of 3.64 Å between N2 of the former and the Oε1 oxygen of asparagine is slightly too long for formation of a H-bond.
A surface rendering of the hPol η active site in the O4MedT insertion-stage complex with the incoming dATP indicates that the nucleobase moiety of the lesion fits snugly into the gap between Trp-64 and Ser-62 (Fig. S49†). It is clear that O4EtdT (with the ethyl group in the syn conformation) cannot be accommodated in the same fashion, as the longer substituent would clash with residues from the finger domain. Instead, the O4EtdT lesion has been pulled inside the active site and pairs with incoming dAMPNPP or dGMPNPP via bifurcated H-bonds in the two crystal structures of insertion-stage complexes with this lesion (Fig. 4a–d, PDB ID codes 5DQG and 5DQH, respectively). As in the complexes with O4MedT, Asn-38 forms a H-bond to O2 of O4EtdT in the minor groove. However, unlike in the O4MedT complex with incoming dGMPNPP, the side chain of Arg-61 in the corresponding complex with O4EtdT does not adopt an extended conformation to contact the major groove edge of guanine. As can be seen in Fig. 4, Arg-61 is directed toward the triphosphate moiety and forms a salt bridge with the α-phosphate group in both insertion-stage complexes. The methylene group (C1) of the O4 substituent in O4EtdT adopts an anti conformation in the complex with dAMPNPP (torsion angle C1–O4–C4–N3 = −142°) and a syn conformation in the complex with dGMPNPP (torsion angle C1–O4–C4–N3 = +62°). This is a clear difference to the structures of complexes with O4MedT, both of which show the lesion adopting a syn conformation (torsion angles C1–O4–C4–N3 of +33° and +25° in the dATP and dGMPNPP complexes, respectively). A further difference between the O4MedT and O4EtdT complexes is constituted by the orientations of template nucleotides 5′-adjacent to the lesions. In the former, A2 and T3 form a stack with Trp-42 outside the active site (Fig. 3). In the O4EtdT complexes, A2 is located outside the active site and forms a stacking interaction with Trp-42. However, T3 sits inside the active site and stacks onto O4EtdT (dAMPNPP complex; Fig. 4a). In the complex with dGMPNPP, T3 juts into the major groove (Fig. 4c). Thus, neither orientation adopted by T3 in these complexes resembles that of O4MedT, lodged near the ceiling of the active site and stacked onto Trp-64.
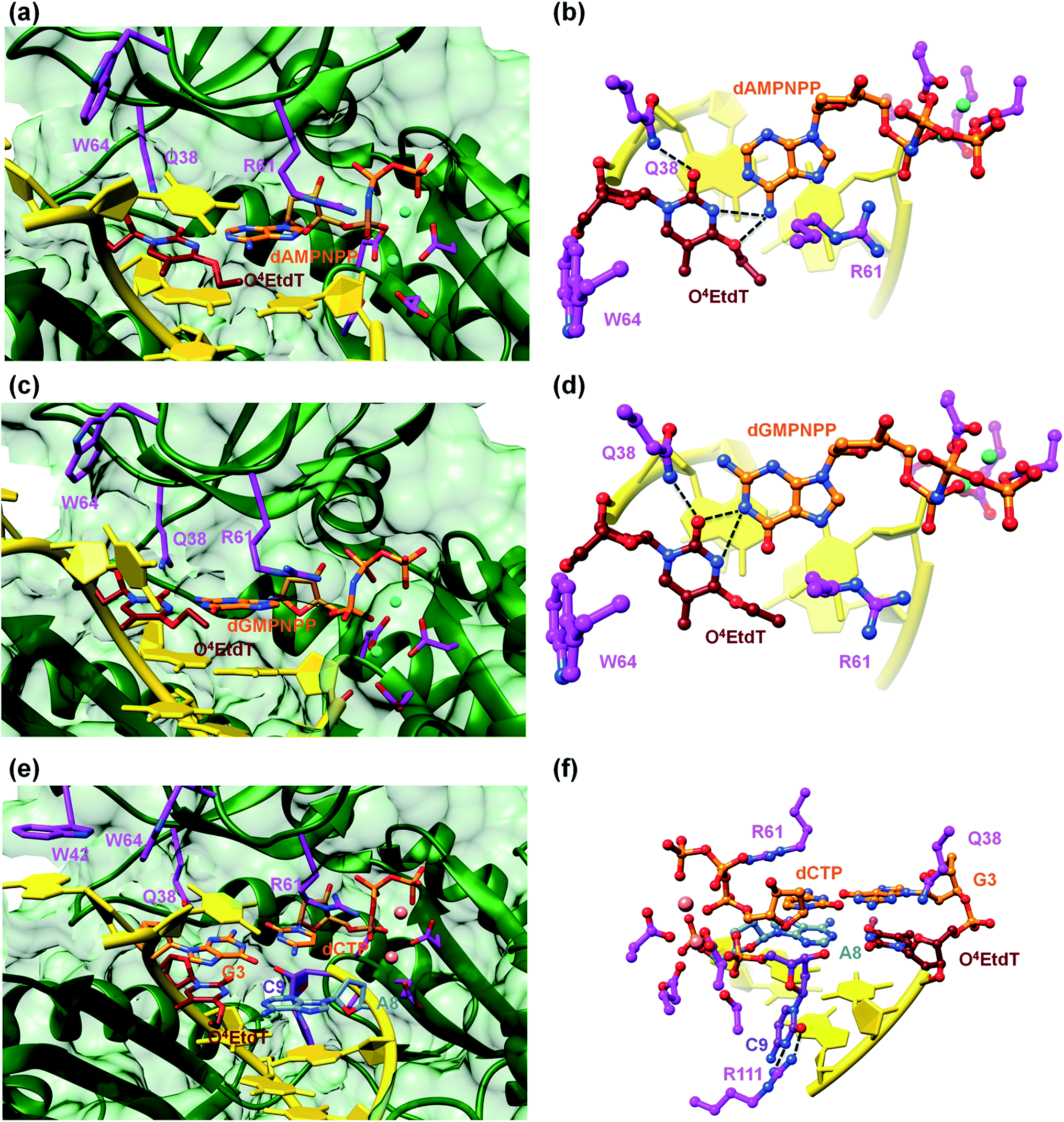 | ||
| Fig. 4 Pairing between incoming purine nucleotide triphosphate and O4EtdT in two hPol η insertion-stage complexes (a–d). (a) Major groove view of the Watson–Crick pair between dAMPNPP and O4EtdT with formation of bifurcated H-bonds. (b) The active site rotated by 90° relative to A, with the base pair viewed perpendicular to the adenine plane. (c) Major groove view of the sheared configuration between dGMPNPP and O4EtdT, with formation of bifurcated H-bonds. (d) The active site rotated by 90° relative to C, with the base pair viewed perpendicular to the guanine plane. Carbon atoms of O4EtdT, the incoming nucleotide triphosphate, and selected hPol η amino acid side chains are colored in maroon, orange and magenta, respectively. Mg2+ ions are cyan spheres and selected H-bonds are shown as dashed lines. Active site configuration in a ternary hPol η extension-step complex with O4EtdT opposite primer dA, followed by template dG opposite incoming dCTP (e and f). (e) Major groove view of the active site, with carbon atoms of O4EtdT and the paired dA colored in maroon and light blue, respectively. Carbon atoms of the nascent dG:dCTP pair are colored in orange, carbon atoms of selected hPol η amino acid side chains are colored in magenta, and those of the 3′-terminal primer dC are colored in purple. (f) The active site rotated by 180° relative to A and viewed into the minor groove. | ||
Crystal structure of a ternary hPol η·DNA·dCTP extension-stage complex with O4EtdT paired opposite primer dA
The structure of a complex with O4EtdT paired to dA at the −1 position followed by template dG opposite incoming dCTP was determined at 2.05 Å resolution (Fig. 4e and f, PDB ID codes 5DQI). The geometry of the O4EtdT:dA pair replicates that seen in the insertion complex with O4EtdT opposite incoming dAMPNPP (Fig. 4a, b, e and f). As in the case of the latter, the ethyl group has moved outside the thymine plane and adopts an anti orientation (torsion angle C1–O4–C4–N3 = −135°). The base pair itself adopts a Watson–Crick like geometry with formation of a single H-bond; the adjacent dG:dCTP pair displays a standard geometry with three H-bonds. Arg-61 is directed toward the phosphate moieties of the incoming nucleotide and forms two salt bridges with the α- and β-phosphate groups, and Asn-38 is engaged in two H-bonds with N3 and O4′ of template dG.The most unusual feature of the extension-stage structure is the presence of an additional nucleotide at the 3′-end of the primer (Fig. 4f). Because the crystallization solutions contained dCTP and the residual electron density is consistent with a pyrimidine, we extended the primer by dC (Fig. S50†). We suspect that hPol η possesses weak catalytic activity with Ca2+ as the cofactor or that traces of Mg2+ present in the crystallizations led to primer extension in situ (even a very low activity could result in extension by a single nucleotide over the course of two weeks). We showed earlier that the translesion DNA polymerase Dpo4 from Sulfolobus solfataricus is able to catalyze nucleotide insertion with Ca2+, although the activity is far below that seen with Mg2+ as the prosthetic group.21 The additional dC stacks against the backbone of the template strand in the minor groove and its position is further stabilized by two H-bonds between N3 and O2 and the guanidino moiety of Arg-111 (Fig. 4f).
Discussion
The known toxicity of alkylated adducts at the O4-position of thymidine prompted us to explore the influence of restricting orientation of the alkyl group around the C4–O4 bond to an anti conformation in translesion synthesis catalyzed by hPol η. In these studies, the bicyclic pyrimidine analogs DFP and TTP, which link the C5 and O4 atoms with a di- or trimethylene linker, were evaluated in addition to O4MedT and the bulkier O4EtdT lesion. Conformationally locked analogs of damage that can occur at the nucleobase have been previously synthesized and employed in studies which have provided insights into the requirements for DNA repair processes.22,23UV thermal denaturation studies of oligomers containing DFP and TTP revealed similar influences on duplex stability to both complementary and mismatched nucleobases compared to O4MedT and O4EtdT. The most stable pairing of either DFP or TTP was with dG, also observed with O4MedT and O4EtdT. NMR studies of a duplex containing an O4MedT·dG pair revealed, in addition to the O4-methyl group adopting a syn-conformation, that the base formed a Watson–Crick “like” pairing with a single hydrogen bond.24 In this structure, the syn-orientation of the O4–Me group influences the hydrogen bond between the imino proton of dG and the N3 atom of O4MedT by increasing the distance between the O6 and O4 atoms of dG and O4MedT, respectively. Limiting the orientation of the methylene group at the O4-atom to the anti-conformation, in the case of the DFP and TTP modifications, appears to have a minimal impact on the interaction with dG and duplex stability. In pairing with dA, a similar drop in duplex stability compared with dT is observed for oligonucleotides containing the O4MedT, O4EtdT, DFP, and TTP modifications. The NMR structure of a duplex containing an O4MedT·dA pair indicated that the O4–Me group is syn and that the bases adopt a wobble alignment with one hydrogen bond formed between the imino nitrogen of O4MedT and the amino group of dA.15 The restricted anti-orientation of the methylene group for DFP or TPP modifications does not significantly impact duplex stability compared to O4MedT or O4EtdT.
Steady-state kinetics of individual nucleotide incorporation opposite the DFP and TTP modifications by hPol η demonstrated preferred insertion of purine nucleotides relative to the pyrimidines, similar to O4MedT and O4EtdT. The efficiency of nucleotide insertion (kcat/Km) for the correct nucleotide (dAMP) across the lesions followed the order DFP > TPP > O4MedT > O4EtdT. For dGMP, a similar efficiency of nucleotide insertion occurred for DFP, TPP, and O4MedT whereas a drop was observed for O4EtdT. In agreement with studies involving Saccharomyces cerevisiae DNA polymerase η (yPol η), a reduction in incorporation efficiency due to the presence of an O4MedT insert was observed.17 However, whereas the yeast homolog revealed a significant preference for dGMP, which was incorporated approximately 80 times more efficiently than dAMP,17 hPol η displayed almost equal selectivity at incorporating dAMP (f = 0.94) as dGMP opposite O4MedT. A comparable 80-fold preference for dGMP over dAMP was exhibited by yPol η for the bulkier O4-carboxymethylthymidine lesion.25 The rationale for the preferred incorporation of dGMP opposite O4MedT by yPol η was attributed to a dG·O4MedT wobble base pairing. Differences observed for nucleotide incorporation opposite O4MedT by the yeast and human homologs of Pol η may be influenced in part by different sequence contexts, as previously observed.5,26 In addition, homologs of Pol η have exhibited differences in nucleotide incorporation across some types of DNA damage. For example, yPol η accurately inserts dCMP across 8-oxodG whereas hPol η is less accurate, inserting some dAMP across this lesion as well.5,27 Interestingly, similar misinsertion profiles have been observed in bypass experiments of hPol η and yPol η with O6MedG, a lesion which also protrudes in the major groove of the DNA duplex.28
For the bulkier O4EtdT and conformationally restricted analogs DFP and TTP, a preference for nucleotide incorporation of dAMP over dGMP was observed. For O4EtdT, hPol η was more proficient at incorporating dAMP over dGMP with catalytic efficiencies of 0.10 and 0.06 μM−1 s−1, respectively. These values are approximately two-fold lower compared to those observed for the O4MedT-containing template, but can be rationalized by the increased bulk of the ethyl group, which may influence dNTP incorporation in the hPol η active site. Ethylation of the O4-position of dT has been shown to stall the human Y-family DNA polymerases hPol κ and hPol ι but not hPol η (although steady-state analysis was not reported for oligonucleotides containing O4EtdT).18 Bypass of O4EtdT by hPol η revealed dGMP misincorporation at 55% compared to 19% for dAMP insertion, in agreement with our data despite different sequence contexts.
For the conformationally restricted DFP and TPP modifications, incorporation efficiency was observed to be ∼1.5-fold higher for dAMP (0.27 and 0.24 μM−1 s−1, respectively) and comparable for dGMP relative to O4MedT. These results demonstrate that hPol η is more proficient at incorporating both the correct (dAMP) and incorrect (dGMP) nucleotides across from these more conformationally restricted lesions. In addition, the increase in steric bulk from the DFP to TPP slightly decreases incorporation efficiency. Exposure of the hydrogen bonding face of the DFP or TPP modifications may have a greater influence on stabilizing the wobble alignment geometry that has been suggested for the O4MedT·dA pairing. The conformational restriction of the alkyl group to an anti-orientation around the C4–O4 bond, as in the DFP and TPP modifications, would direct the O4-methylene group away from the amino group of dA, which could account for the enhancement of incorporation of the correct nucleotide (dAMP) compared to O4MedT. Incorporation of dGMP may not be as influenced by orientation of the alkyl group around the C4–O4 bond as the proposed hydrogen bonding interaction, based on the NMR structure of the duplex containing the O4MedT·dG, which occurs between the amino group of dG and O2-atom of O4MedT.24 In the case of O4EtdT, the combination of the syn-orientation and the size of the ethyl group may both contribute to the reduced efficiency of nucleotide insertion of dAMP and dGMP in this series.
Primer extension reactions in the presence of all four dNTPs for templates containing the O4MedT, O4EtdT, DFP, and TTP modifications demonstrated that hPol η was proficient at incorporating nucleotides across and past the adducted site. However, both O4MedT and O4EtdT exhibited a greater accumulation of non-full length oligonucleotide products at reduced reaction times (30 and 60 min), which was not observed for bicyclic DFP or TPP analogs. The LC-MS/MS analyses of the extension products from the in vitro primer bypass studies revealed that dGMP incorporation across the lesion was preferred over dAMP in all cases except the control (dT). The ratio of dGMP:dAMP incorporation by hPol η, assessed from the extension products, was found to decrease in the series TPP (4.6:1) > DFP (3.3:1) ≈ O4EtdT (3.2:1) > O4MedT (2.1:1). The presence of the larger alkyl group for O4EtdT or the analogs with the O4-methylene group in an anti-conformation (TPP and DFP) clearly promotes dGMP misincorporation in the presence of all four nucleotides. In addition, the DFP and TPP modification were not found to induce a significant amount of frameshifts in the products compared to O4MedT. Differences in fidelity observed between the steady-state kinetic and LC-MS/MS full-length experiments have been observed previously.17,19 The variance may be attributed to accommodation of the incoming dGTP relative to dATP for these modifications, highlighting that adduct size and the conformation of the O4-methylene group can influence interactions in the active site of hPol η. It should be noted, however, that other steric and/or stereoelectronic effects may have an impact on hPol η bypass processivity of the conformationally locked analogues relative to O4MedT or O4EtdT, respectively. In the case of the analogs investigated, hPol η continued extension of the primer in an error-free manner after incorporation of dATP or dGTP across from the damaged site on the template.
Several observations based on nucleotide incorporation profiles attest to the distinct effects on hPol η bypass synthesis exerted by the O4MedT and O4EtdT lesions. These concern (i) the more error-prone bypass caused by O4MedT, i.e. dGTP is favored relative to dATP (Fig. 2a), (ii) increased accumulation of the +1 product in the full-length extension reaction for O4MedT (Fig. 1), and (iii) significantly more frameshift products caused by the O4MedT lesion (Fig. 2b). Interestingly, the structural data for insertion-stage hPol η complexes with either O4MedT or O4EtdT in the template strand reveal starkly different orientations of the two adducted nucleotides at the active site. O4MedT is trapped in an orientation that keeps it at a considerable distance from the incoming purine nucleotide triphosphates. Conversely, O4EtdT pairs opposite both dATP and dGTP with formation of bifurcated H-bonds (whereby the latter pair features a sheared orientation of the two partners, with G being pushed toward the minor groove). The increased proclivity for insertion of dG opposite O4MedT compared to O4EtdT is not surprising if one considers the strict preference by the O4-methyl substituent for a syn conformation. The syn conformation precludes adoption of an O4MedT:dA pair with standard Watson–Crick geometry, but the sheared pairing mode seen in the case of O4EtdT:dG(MPNPP) (Fig. 4c and d), also presumably adopted by the O4MedT:dG pair, is compatible with a syn conformation of the substituent. This conclusion is borne out by the observations that the ethyl moiety in the O4EtdT:dA(MPNPP) pairs assumes an anti conformation (Fig. 4a, b, e and f), whereas its conformation is syn in the O4EtdT:dGMPNPP pair. Furthermore, the TPP adduct opposite dGMPNPP was modeled from the O4EtdT:dG(MPNPP) ternary crystal structure coordinates. The configuration of the adduct seen in the model is consistent with the enhanced incorporation of dG observed in the primer extension experiments since the constrained anti conformation of the bicyclic system does not hinder the guanine nucleobase from shifting towards the major groove and potentially form two H-bonds with TPP (Fig. S51†).
On one hand, one could argue that the higher fraction of frameshifts for O4MedT relative to O4EtdT is consistent with the structural data that show the former is not engaged opposite the incoming nucleotide but trapped adjacent to the ‘entrance’ of the active site. Perhaps the more pronounced accumulation of the +1 product in the case of the full-length extension reactions opposite O4MedT compared to the other O4 adducts tested here are the result of non-templated insertion. Thus, purine nucleoside triphosphates would be favored and their incorporation would not be affected by the particular conformation of the O4-methyl group, syn or anti. This scenario is certainly not inconsistent with the structural data that reveal no interaction between the O4MedT lesion and the incoming dATP or dGMPNPP. Clearly, it is intriguing that both activity and structural data show distinct consequences of the O4MedT and O4EtdT lesions for bypass by hPol η. However, it is important to note that the position of the O4MedT lesion at the active site, unique among all crystal structures of hPol η complexes analyzed to date, represents one state during bypass. Perhaps other orientations and interactions of the adducted nucleotide occur during bypass, which precludes all steps involved in the mechanism of O4-alkyl bypass synthesis by hPol η.
All results from the study (kinetic evaluation, full extension assays and crystal structures) may be integrated into one potential extension model. The crystal structure supports the notion that the O4MedT nucleobase is indeed nestled at the ceiling for an undefined period of time. The purine nucleoside triphosphates could then be imported into the active site, and would be subject to a template gap, analogous to being opposite an abasic site. According to full extension assays, there are approximately twice as many dG inserted by hPol η relative to dA. The lack of O4MedT – incoming dNTP clash in the active site may explain higher kp values observed for dATP and dGTP for O4MedT compared to O4EtdT (by 8.8 or 9.3 fold respectively). This may also aid in explaining the preference for purine insertion, in an approximate 2:1 dG:dA ratio across O4MedT, whereas a larger ratio is observed for all the other modifications as discussed above. Perhaps the other modifications prevent the modified nucleobases from accessing the conformation at the top of the active site. The ethyl group, albeit bulkier, can populate the other conformations (syn versus anti) as observed in the crystallographic data, which may contribute to the higher correct dA insertion relative to the bicyclic analogues which are locked in an anti conformation. Increase in bulk (from O4MedT to O4EtdT and DFP to TPP) leads to an increase of incorrect dG insertion. Eventually the O4MedT is required to move from the top of the active site back to the 0 or −1 (post-replicative) position(s). It is possible that this mobility causes the frameshift adduct formation observed in the full extension assays. As a result, we have provided intriguing insights on a potentially different bypass mechanism of O4MedT in comparison to the larger O4EtdT adduct.
Conclusions
Oligonucleotides containing DFP and TPP, designed as analogs of O4-alkylated thymidine, were synthesized to explore the influence of limiting the O4-alkyl lesion to an anti-orientation on nucleotide incorporation by hPol η. These modifications were shown to destabilize the DNA duplex, based on UV thermal denaturation studies, regardless of the base-pairing partner (A, G, T, or C), similar to O4MedT and O4EtdT. Primer extension assays demonstrated that these pyrimidyl modifications hindered nucleotide incorporation by hPol η. Single nucleotide incorporation studies revealed increased selectivity towards dAMP over dGMP that followed the order O4EtdT > DFP ≈ TPP. A slight preference for dGMP over dAMP incorporation was observed for O4MedT. LC-MS/MS analysis of primer extension studies (in the presence of all four dNTPs) revealed that hPol η incorporated dGMP over dAMP across the lesions in the order TPP > DFP ≈ O4EtdT > O4MedT. These trends suggest that limiting the orientation of the O4-alkylene group enhances the proficiency of dNTP incorporation by hPol η across O4-alkylated dT damage. In the presence of all four dNTPs, error-prone nucleotide incorporation by hPol η is enhanced by restricting the O4-lesion to an anti-orientation. This study exemplifies how restricting a lesion's conformational freedom impacts bypass profile by hPol η. Moreover, our results provide mechanistic insights into the mutagenicity of the biologically relevant O4MedT and O4EtdT DNA adducts.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada (Grant No. 299384-2011 to C. J. W.), the Canada Research Chair Program (Grant No. 950-213807 to C. J. W.) and the US NIH for financial support (Grants No. ES010375 to F. P. G. and M. E., Grant No. CA160032 to M. E., and ES010546 to F. P. G.). D. K. O. thanks NSERC for a Canada Graduate Scholarship (CGS) and Michael Smith Foreign Study Supplement Scholarship, and the NSERC CREATE Program in Bionanomachines for support of an exchange with Vanderbilt University. Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team at sector 21 of the Advanced Photon Source, Argonne, IL. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the United States Department of Energy, Office of Basic Energy Sciences (grant DE-AC02-06CH11357).Notes and references
- N. Shrivastav, D. Li and J. M. Essigmann, Carcinogenesis, 2010, 31, 59–70 CrossRef CAS PubMed
.
- M. P. Stone, H. Huang, K. L. Brown and G. Shanmugam, Chem. Biodiversity, 2011, 8, 1571–1615 CAS
- J. E. Sale, A. R. Lehmann and R. Woodgate, Nat. Rev. Mol. Cell Biol., 2012, 13, 141–152 CrossRef CAS PubMed
- S. D. McCulloch, R. J. Kokoska, C. Masutani, S. Iwai, F. Hanaoka and T. A. Kunkel, Nature, 2004, 428, 97–100 CrossRef CAS PubMed
- A. Patra, L. D. Nagy, Q. Zhang, Y. Su, L. Muller, F. P. Guengerich and M. Egli, J. Biol. Chem., 2014, 289, 16867–16882 CrossRef CAS PubMed
- M. R. Albertella, C. M. Green, A. R. Lehmann and M. J. O'Connor, Cancer Res., 2005, 65, 9799–9806 CrossRef CAS PubMed
- B. Singer, Nature, 1976, 264, 333–339 CrossRef CAS PubMed
- S. R. Tannenbaum, Lancet, 1983, 1, 629–632 CrossRef CAS
- S. M. Bronstein, T. R. Skopek and J. A. Swenberg, Cancer Res., 1992, 52, 2008–2011 CAS
- K. B. Altshuler, C. S. Hodes and J. M. Essigmann, Chem. Res. Toxicol., 1996, 9, 980–987 CrossRef CAS PubMed
- B. Singer, S. J. Spengler, H. Fraenkel-Conrat and J. T. Kusmierek, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 28–32 CrossRef CAS
- B. D. Preston, B. Singer and L. A. Loeb, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8501–8505 CrossRef CAS
- B. Singer, Cancer Res., 1986, 46, 4879–4885 CAS
- J. A. Swenberg, M. C. Dyroff, M. A. Bedell, J. A. Popp, N. Huh, U. Kirstein and M. F. Rajewsky, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 1692–1695 CrossRef CAS
- M. W. Kalnik, M. Kouchakdjian, B. F. Li, P. F. Swann and D. J. Patel, Biochemistry, 1988, 27, 100–108 CrossRef CAS PubMed
- R. G. Brennan, D. Pyzalska, W. J. Blonski, F. E. Hruska and M. Sundaralingam, Biochemistry, 1986, 25, 1181–1185 CrossRef CAS PubMed
- N. Andersen, J. Wang, P. Wang, Y. Jiang and Y. Wang, Chem. Res. Toxicol., 2012, 25, 2523–2531 CrossRef CAS PubMed
- N. Andersen, P. Wang and Y. Wang, Chem. Res. Toxicol., 2013, 26, 1730–1738 CrossRef CAS PubMed
- J. Y. Choi, G. Chowdhury, H. Zang, K. C. Angel, C. C. Vu, L. A. Peterson and F. P. Guengerich, J. Biol. Chem., 2006, 281, 38244–38256 CrossRef CAS PubMed
- A. Patra, Q. Zhang, L. Lei, Y. Su, M. Egli and F. P. Guengerich, J. Biol. Chem., 2015, 290, 8028–8038 CrossRef CAS PubMed
- A. C. Madril, R. E. Johnson, M. T. Washington, L. Prakash and S. Prakash, J. Biol. Chem., 2001, 276, 42857–42862 CrossRef CAS PubMed
- D. S. Daniels, T. T. Woo, K. X. Luu, D. M. Noll, N. D. Clarke, A. E. Pegg and J. A. Tainer, Nat. Struct. Mol. Biol., 2004, 11, 714–720 CAS
- K. Abdu, M. K. Aiertza, O. J. Wilkinson, J. A. Grasby, P. Senthong, A. C. Povey, G. P. Margison and D. M. Williams, Chem. Commun., 2012, 48, 11214–11216 RSC
- M. W. Kalnik, M. Kouchakdjian, B. F. Li, P. F. Swann and D. J. Patel, Biochemistry, 1988, 27, 108–115 CrossRef CAS PubMed
- A. L. Swanson, J. Wang and Y. Wang, Biochemistry, 2011, 50, 7666–7673 CrossRef CAS PubMed
- S. Rao, A. Chenna, M. Slupska and B. Singer, Mutat. Res., 1996, 356, 179–185 CrossRef PubMed
- L. Haracska, S. L. Yu, R. E. Johnson, L. Prakash and S. Prakash, Nat. Genet., 2000, 25, 458–461 CrossRef CAS PubMed
- L. Haracska, S. Prakash and L. Prakash, Mol. Cell. Biol., 2000, 20, 8001–8007 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental procedures, compound characterization, and additional discussion. The atomic coordinates and structure factors (codes 5DLF, 5DLG, 5DQG, 5DQH and 5DQI) have been deposited in the Protein Data Bank (http://www.wwpdb.org/). See DOI: 10.1039/c6sc00666c |
| This journal is © The Royal Society of Chemistry 2016 |