DOI:
10.1039/C6SC00542J
(Edge Article)
Chem. Sci., 2016,
7, 4264-4275
“Rate-limited effect” of reverse intersystem crossing process: the key for tuning thermally activated delayed fluorescence lifetime and efficiency roll-off of organic light emitting diodes†
Received
3rd February 2016
, Accepted 4th March 2016
First published on 15th March 2016
Abstract
Issues concerning excited state lifetime (τTADF) tuning of thermally activated delayed fluorescence (TADF) materials are critical for organic light emitting diode (OLED) applications and other specific fields. For TADF-OLEDs, employing emitters with a short τTADF gives rise to suppressed singlet–triplet annihilation (STA) and triplet–triplet annihilation (TTA), leading to reduced efficiency roll-off at practical relevant brightness (100 and 1000 cd m−2 for display and illumination applications, respectively). Through molecular design, exciton dynamic process rate constants including fluorescence (kF), intersystem crossing (kISC), internal conversion (kIC) and reverse intersystem crossing (kRISC) are selectively altered, affording four representative TADF emitters. Based on lifetime and quantum yield measurements, kF, kISC, kIC and kRISC are calculated for four emitters and their interrelationship matches corrected time-dependent density functional theory simulation. Among them, even with a small kF, low photoluminescence quantum efficiency (Φ) and large kISC, molecules with a small singlet–triplet splitting energy (ΔEST) and lowest charge transfer triplet excited state (3CT) eventuate in shortening the τTADF. Herein, kRISC, which is inversely proportional to ΔEST, turns out to be the rate-limited factor in tuning the τTADF (“rate limited effect” of the RISC process). As revealed by flexible potential surface scanning, PyCN–ACR exhibited a moderate kF, reduced kIC and enlarged kRISC, resulting in a short τTADF and a moderate Φ with orange-red emission. OLEDs containing PyCN–ACR as the emitting guest achieved orange-red TADF-OLEDs with an emission peak at 590 nm and the best external quantum efficiencies (EQEs) of 12.4%/9.9%/5.1% at practical luminances of 100/1000/10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2.
000 cd m−2.
Introduction
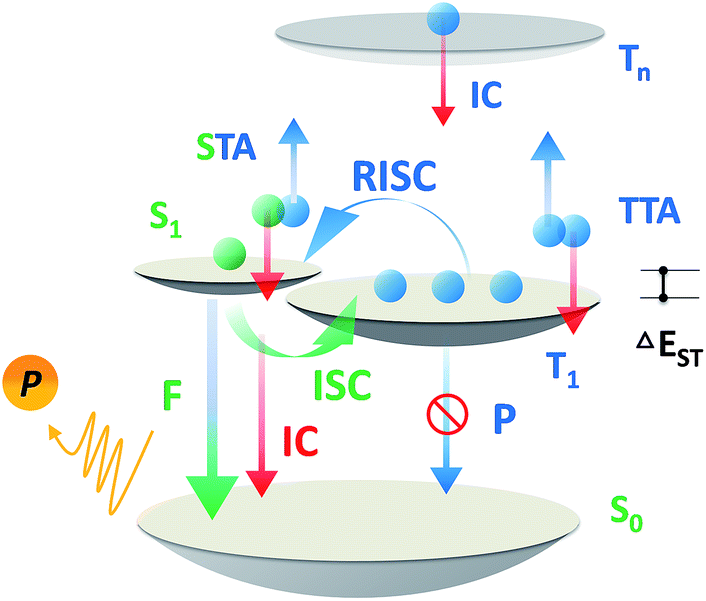
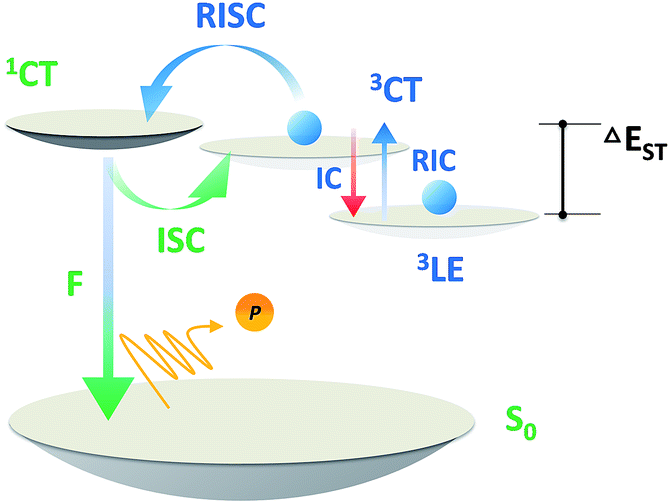
Over the past two decades, the usage of phosphorescent materials has exploited the non-radiative triplet state by introducing heavy metal atoms, improving the overall electroluminescence (EL) internal quantum efficiency (IQE) of organic light emitting diodes (OLEDs) to 100%.1,2 However, more than the usage of noble metals like iridium (Ir) and platinum (Pt), which are expensive and non-renewable,3 the problem facing phosphorescent OLEDs (PH-OLEDs) is severe triplet–triplet annihilation (TTA) and triplet–polaron annihilation (TPA) at high brightness,4 which are fatal to practical PH-OLED application, not least because the EQE at luminances of 100 cd m−2 (display relevant) and 1000 cd m−2 (illumination relevant) is of great importance.5 An alternative approach for 100% IQE in OLEDs is utilizing thermally activated delayed fluorescence (TADF) materials.6 75% of the electric-generated triplet excitons can be up-converted to the radiative singlet state to yield delayed fluorescence thanks to a very small ΔEST.7–9 Analogous to PH-OLEDs, however, the EQEs of TADF-OLEDs at high brightness are also subjected to reduction by a large margin.10,11 Except for TTA, a higher singlet exciton density than Ir-based PH-OLEDs from an efficient RISC process produces a non-negligible STA.12,13 These superimposed factors make the reduced efficiency roll-off of TADF-OLEDs challenging on the march towards future high brightness applications (Scheme 1).
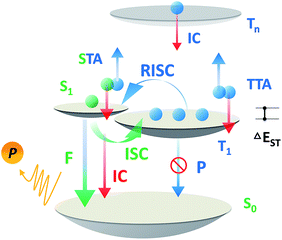 |
| | Scheme 1 Exciton dynamic processes for a typical TADF emitter in OLEDs. Singlet and triplet excitons are generated in a 1:3 ratio depending on the spin degeneracy. F, P, ISC, IC, STA, TTA and RISC are abbreviations of fluorescence, phosphorescence, intersystem crossing, internal conversion, singlet–triplet annihilation, triplet–triplet annihilation, and reverse intersystem crossing. | |
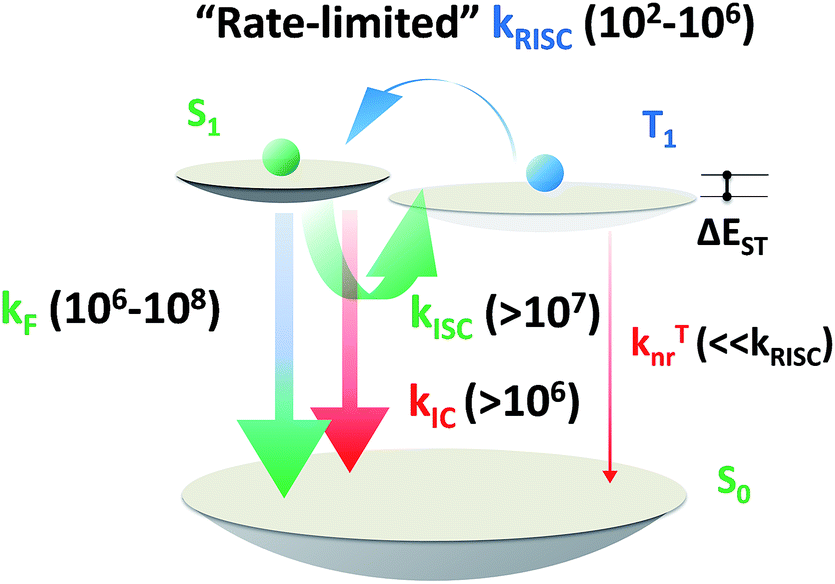
The short excited state lifetime of TADF materials (τTADF) is important to reduce the efficiency roll-off of TADF-OLEDs by suppression of the TTA and STA.4 For TADF emitters, the rate constants of fluorescence (kF), intersystem crossing (kISC), reverse intersystem crossing (kRISC), internal conversion (kIC), and non-radiative decay from the triplet state (kTnr) play roles in exciton dynamic processes and could govern τTADF. Given that some of these factors are also involved in the EQE annihilation model of TADF-OLEDs (2c, ESI†),13 it is reasonable to deduce that tuning these factors via chemical modification could eventuate in various EQE roll-off curves even in exactly the same device architecture. Notably, it can be complicated to comprehend and design materials taking all factors into account, and the critical point for tuning τTADF remains unclarified. Therefore, selectively tuning these factors does help to find out the key to govern τTADF, and ultimately not only assists in promoting efficiencies of TADF-OLEDs at practically relevant brightness, but can be used for reference towards material design in other specific fields where excited lifetime tuning is required.
To handle this, the governing of these factors should be linked to molecular design. To our knowledge, kRISC is proportional to exp(−ΔEST/kBT),8 where kB denotes the Boltzmann constant and T denotes temperature. ΔEST is proportional to the orbital overlap (Sif) of the initial (Φi) and final (Φf) state wavefunctions of the S1 transition. Meanwhile, kF is related to the transition integral (Hif) and is roughly proportional to Sif.14,15 Therefore, kF and kRISC cannot be cut apart for TADF emitters, because a large kF originating from a large frontier orbital overlap could enlarge ΔEST and reduce kRISC. A convenient approach to tune kF and kRISC is introducing twisted intramolecular charge transfer (TICT) systems with different dihedral angles between electron donating and withdrawing moieties.7,9 Enlarging the dihedral angles between D–A systems minimizes the transition integral, resulting in reduced kF and ΔEST and vice versa. For emitters without heavy atoms, metals or halogens, kISC can be tuned using emitters with π–π* or n–π* transition character, for the latter ones facilitate the forbidden transition from the singlet to triplet excited state that violates spin conservation to enhance kISC.16–18 For kIC, utilizing rigid chemical moieties and compact molecular structure design can remarkably suppress non-radiative channels. Notably, non-radiative processes in the triplet state are much slower than others given that T1 is relatively stable for pure aromatic compounds, and the influence of kTnr can be temporarily ignored for simplicity.19 By the aforementioned criteria, molecules exhibiting specific characters can be designed.
In this work, instead of starting from host engineering or device architecture management, we design four typical TADF emitters to elaborate the connection among τTADF governing, device efficiency roll-off control and molecular design. Among the diverse chemical moieties, carbonyl-type compounds are chosen for their special n–π* transition features, which inevitably renders a small kF and a significant ISC process.17,20–22 Along this line of thinking, the benzil (DC) moiety is selected. On one hand, its orthogonal n and π* orbitals diminishes the exchange integral of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), enabling a fairly small ΔEST around 0.1–0.2 eV.14,23 Additionally, alternation of the electronic configuration can be realized via simple modification of the DC core, affording another representative radiative conducive π–π*-favoured constituent. The replacement of benzil by dicyano-pyrazine (PyCN) offers typical fluorophores with a large kF, reduced kIC and kISC.24,25 Aside from the acceptor cores, the donors are fixed on TC (3,6-di-tert-butylcarbazole) and ACR (9,10-dihydroacridine) moieties, respectively. The six-membered ACR ring induces enlarged D–A twisting compared with the five-membered TC heterocycle,9 leading to a reduced ΔEST and enhanced molecular rigidity for DC–ACR and PyCN–ACR. The aforementioned design thinking embodies four distinct and representative types of molecules as part of a large class of TADF emitters with analogous characters. The influence of molecular design on governing τTADF by tuning kF, kISC, kIC and kRISC are investigated thoroughly by examining the photophysical properties and quantum chemical simulations. Lastly, OLEDs are fabricated to ascertain the affiliation between molecular design and device performance concerning efficiency roll-off (Fig. 1).
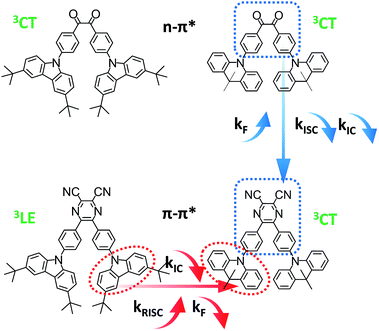 |
| | Fig. 1 Molecular structures of the investigated compounds and molecular design for tuning kF, kIC, kISC and kRISC. | |
Results and discussion
Computation simulation
In order to demonstrate the influence of molecular design on governing τTADF by tuning kF, kISC, kIC and kRISC, comprehensive pictures of the molecules should be provided. Through adopting the appropriate methodology, density functional theory (DFT) and time-dependent density functional theory (TD-DFT) simulations can provide relatively reasonable descriptions of the investigated molecules.26–28 Initially, the ground state (S0) geometries of the four emitters were optimized at the B3LYP/6-31G* level.28,29 Different from the S0 geometry, S1 geometries for ICT molecules strongly depend on the exchange–exchange (XC) functional.30 Besides, selection of a proper functional to depict the vertical absorption transition energy (EVA) and emission energy (EVE) is required, because non-hybrid functionals neglect long-range coulombic attraction and underrate the CT transition energy while pure Hartree–Fock (HF) functionals suffer from electron correlation problems and overrate the excited energy.31 Lastly, the configuration interaction (CI) description of the S1 transition on coupling between the charge transfer excited state (CT) or locally excited state (LE) also relies on the XC functional.9 To simplify the simulation, a semi-empirical approach using experimental EVA(S1) to find the optimal percentage of the HF fraction (OHF) into functionals was adopted. Different HF% functionals were employed to compute EVA(S1) based on the optimized S0 geometry. HF% was plotted against EVA(S1) and the relationship was established (Fig. S7†). The optimized HF% (OHF) was determined to be close to 42% (BMK) by fitting the simulated EVA(S1) with well-defined CT absorption peaks from the ultraviolet-visible (UV-vis) spectroscopy measurement of all molecules.32 As S1 is sensitive towards XC functionals and solvent effects, it is necessary to optimize the S1 geometries, calculate EVE and analyze the configuration interaction (CI) description at the BMK/6-31G* level utilizing the polarized continuum model (PCM) and so potential deviation can be minimized.33,34
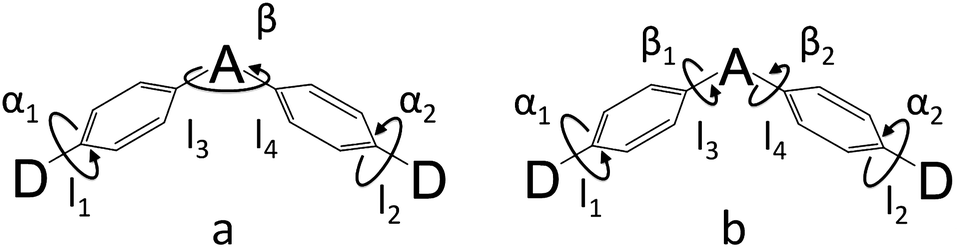
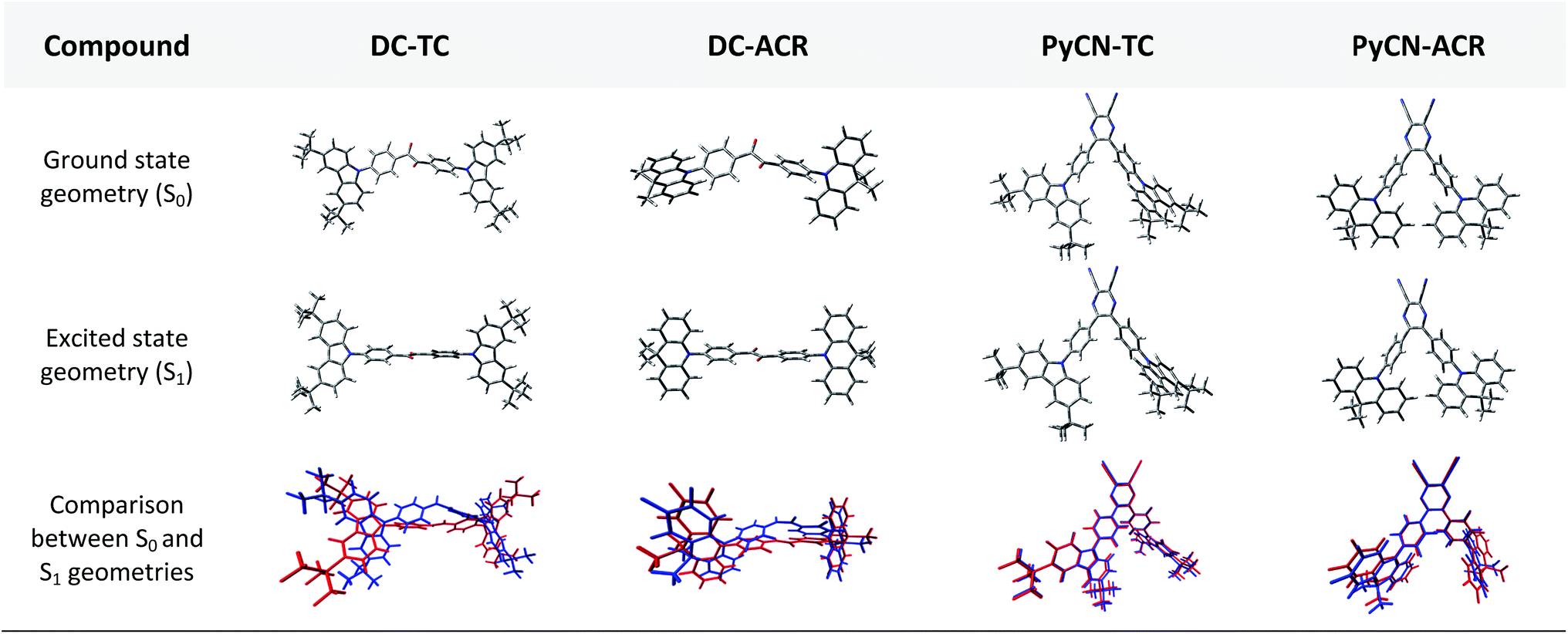
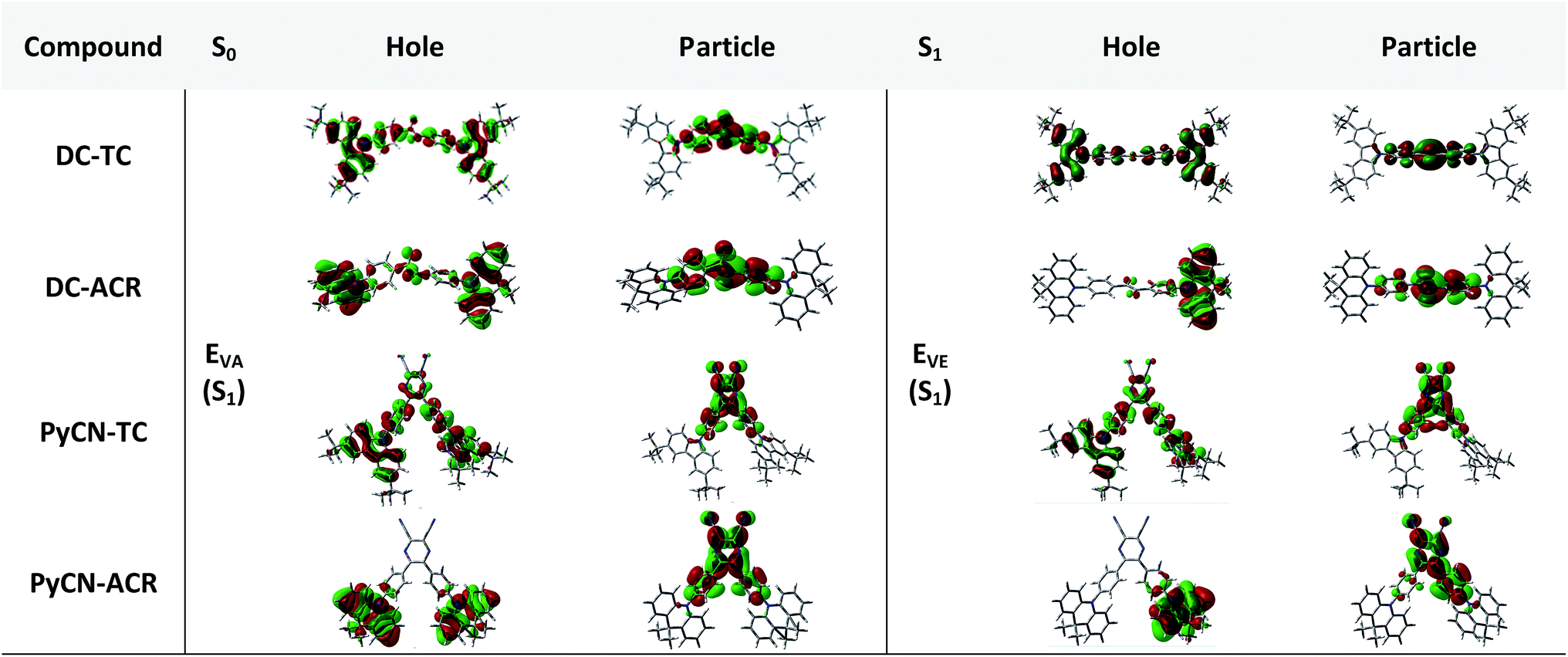
The simulated results of the four D–π–A–π–D compounds can be divided into two categories (Scheme 2). For the S0 and S1 geometries, detailed parameters including the bond length (l) and dihedral angle (α or β) are listed in Table 1. To visualize the frontier orbital transition of all compounds, natural transition orbital (NTO) analyses in view of singular value decomposition of the one-particle transition density matrix were carried out to comprehend the electron transitions, as NTO analyses provide fewer orbital pairs with maximum eigenvalues and simplify our interpretation.35,36 The geometries and NTOs of the four molecules are presented (Fig. 2 and 3), respectively.
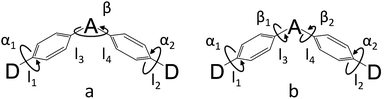 |
| | Scheme 2 Two models for donor–π–acceptor–π–donor (D–π–A–π–D) molecules ((a) for DC–TC and DC–ACR, (b) for PyCN–TC and PyCN–ACR). α and β denote dihedral angles. l1 and l2 denote the bond lengths between the donor and phenyl bridge, while l3 and l4 denote the bond lengths between the acceptor and phenyl bridge. | |
Table 1 Calculated twisting angles (α1, α2 and β1, β2), bond lengths (l1, l2, and l3, l4), transition energies (EVA and EVE based on OHF functionals), and oscillator strengths (fVA and fVE based on OHF functionals) of all investigated molecules in the gas phase in S0-optimized geometries and in solvents in S1-optimized geometries
| |
S0 geometry |
S1 geometry |
| Compound |
α
1/α2 (°) |
β or β1/β2 (°) |
l
1/l2 (Å) |
l
3/l4 (Å) |
E
VA(S1) (eV) |
f
VA
|
α
1/α2 (°) |
β or β1/β2 (°) |
l
1/l2 (Å) |
l
3/l4 (Å) |
E
VE(S1) (eV) |
f
VE
|
| DC–TC |
47.4/47.7 |
44.8 |
1.41/1.41 |
1.48/1.48 |
3.23 |
0.7712 |
50.0/50.3 |
3.0 |
1.41/1.41 |
1.50/1.50 |
2.80 |
0.5561 |
| PyCN–TC |
47.2/48.5 |
32.9/34.4 |
1.41/1.41 |
1.48/1.48 |
2.85 |
0.3632 |
50.0/47.1 |
21.9/28.8 |
1.41/1.41 |
1.46/1.47 |
2.55 |
0.4677 |
| DC–ACR |
89.1/89.3 |
49.5 |
1.43/1.43 |
1.49/1.49 |
2.77 |
0.0002 |
86.8/86.5 |
5.8 |
1.43/1.43 |
1.49/1.49 |
2.06 |
0.0011 |
| PyCN–ACR |
89.3/89.3 |
35.1/35.1 |
1.43/1.43 |
1.48/1.48 |
2.59 |
0.0001 |
89.9/89.4 |
40.7/15.8 |
1.43/1.44 |
1.49/1.43 |
2.11 |
0.0022 |
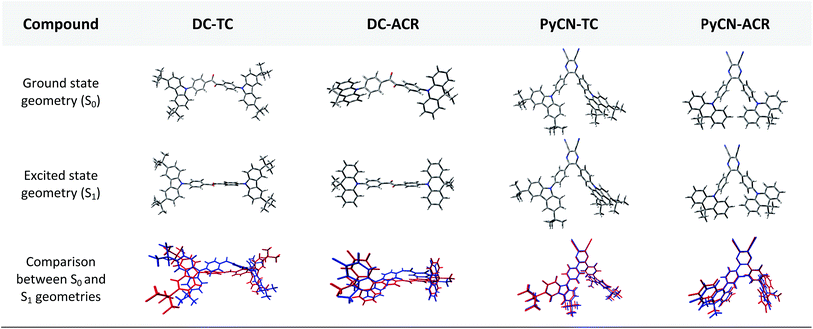 |
| | Fig. 2 Front views of the DFT-optimized ground state geometries from B3LYP/6-31G* in the gas phase and excited state geometries from BMK/6-31G* in toluene using the PCM model for the four compounds. The bottom geometries are intuitive pictures comparing the S0 (blue) and S1 (red) geometries with minimum root-mean-square-deviation (RMSD). | |
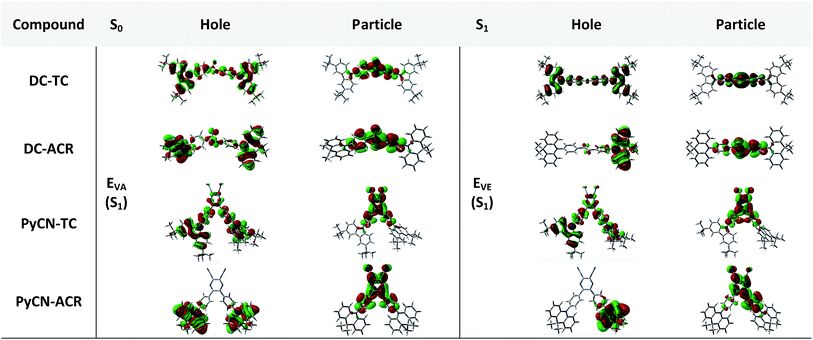 |
| | Fig. 3 Natural transition orbital (NTO) analyses of the four investigated molecules. Hole and particle distributions of the HONTO and LUNTO of the S0 and S1 geometries are presented for intuitive comparison. | |
In the S0 geometry, the six-membered ACR offers a large steric hindrance effect from repulsion between the adjacent hydrogen atoms and α = 89° was estimated, much larger than that between TC and Ph (around 47°). The benzil moiety twists and avoids forming a flat surface in S0. β between PyCN and the Ph bridge lies around 34° (Fig. S6†) and twists to avoid repulsion with the ortho-substituted Ph ring. For vertical electron excitation, the S0 to S1 transition of DC–TC locates at the benzil moiety and has a much smaller oscillator strength (f ≈ 0) than S2; the latter is regarded as S1 in the following discussion. The lowest unoccupied natural transition orbitals (LUNTOs) of DC–TC and PyCN–TC predominantly localize on the DC and PyCN cores and slightly delocalize over the adjacent Ph ring. The highest occupied natural transition orbitals (HONTOs) distribute mainly on both TC–Ph arms and a small amount on the acceptor plane. Owing to the enlarged α, DC–ACR and PyCN–ACR have small orbital overlap, with the HONTO localizing mostly on ACR and the LUNTO localizing on the DC and PyCN–2Ph centers. The decreased coplanarity between the donor and acceptor prohibits nitrogen lone pair electrons from delocalizing over the whole molecule. In view of the more limited HONTO and LUNTO overlap for DC–ACR and PyCN–ACR, a much smaller f can be anticipated than DC–TC and PyCN–TC.
In S1, the bond lengths (l1, l2, l3, l4) show rather small variations in comparison with the S0 geometry. However, α and β change significantly, suggesting molecular geometry relaxation happens in S1. β decreases from 44.8° to 3.0° for DC–TC and from 49.5° to 5.8° for DC–ACR. A similar trend is also observed for PyCN–TC, as both β1 and β2 show a slight decrease to induce a more conjugated acceptor plane. However, for PyCN–ACR, β2 decreases from 35.1° to 15.8° while β1 increases from 35.1° to 40.7°, which can be explained by a more compact geometry compared with PyCN–TC. To twist a rigid ACR–Ph arm inevitably enhances the steric hindrance of the other, which is forced to avert close interaction between the adjacent ACR–Ph arms. The images comparing the S0 and S1 geometries (Fig. 2) clearly illustrate a degree of deformation and qualitatively depict more enhanced rigidity with smaller discrepancy. The coplanarization of acceptors and the internal twist of donors can be interpreted by the minimum overlap rule, which drives more complete charge separation of the donor and acceptor so as to lower the CT state energy.37 The flexible benzil acceptor tends to elongate conjugation, and this and the slightly increased α both contribute to separate further the HONTO and LUNTO and weakens electronic coupling between the donor and acceptor.38–40 Compared with DC–TC and DC–ACR, more suppressed non-radiative deactivation processes can be expected for PyCN–TC and PyCN–ACR showing lower geometry deformation in S1.
In the optimal S1 geometries, NTO transitions analogous to those in S0 are observed for DC–TC and PyCN–TC but broken-symmetry effects are noted for DC–ACR and PyCN–ACR. Followed by geometry relaxation, the degrees of HONTO and LUNTO overlap for DC–TC and PyCN–TC still far exceed those of DC–ACR and PyCN–ACR, indicating a larger kF for the former ones. It is noteworthy that despite partially overlapping centres on the benzil moiety for the HONTO and LUNTO of DC–ACR, the n–π* transition nature of the benzil moiety hardly contributes to the overall f value.21,22 A f value roughly equal to zero is observed for such a typical n–π* transition in both the S0 and S1 geometries of the DC center, explaining the negligible fVA and fVE for DC–ACR (Fig. S5 and Table S1†).
To interpret the energy level coupling relationship, calculation of zero–zero energy levels (E0–0) is critical. A systematic comparison of EVA(S1), EVE(S1), E0–0(1CT), E0–0(3CT), E0–0(3LE) and ΔEST is listed (Table S2†). According to the Franck–Condon principle, the average values of EVA(S1) and EVE(S1) approximate the E0–0 energy level by eqn (1) and reproduce the experimental data with small deviation (Fig. S7†).41
| | | E0–0(1CT) = (EVA(S1) + EVE(S1))/2 | (1) |
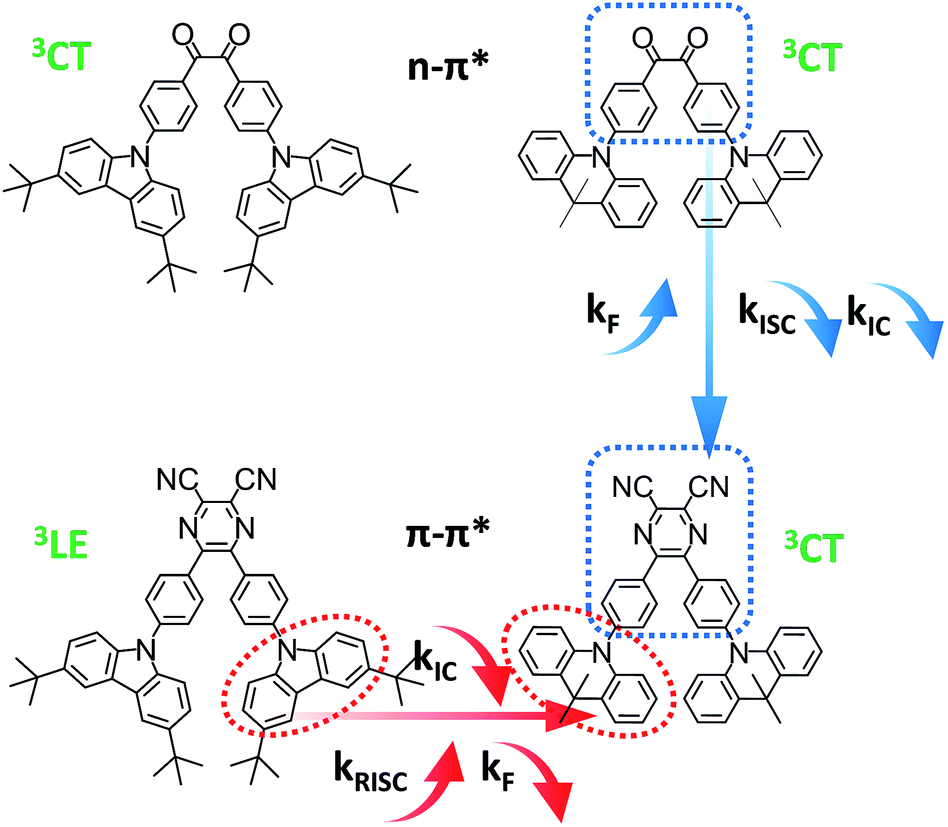
For analyzing the RISC process, coupling between the CT and LE triplet states should be carefully defined. The lower-lying 3LE state than the 3CT state ought to decrease the rate constants of the RISC process because of the slow reverse internal conversion (RIC) rate from the 3LE state to the 3CT state.9 Both E0–0(3CT) and E0–0(3LE) were computed independently based on eqn (2) and (3) (Scheme 3).42
| | | E0–0(3CT) = E0–0(S1) − (EVA(S1, OHF) − EVA(S1, OHF)/EVA(S1, BLYP) × EVA(T1, BLYP)) | (2) |
| | | E0–0(3LE) = EVA(T1)/(EVA(S1, OHF)/EVA(S1, BLYP)) − 0.09 | (3) |
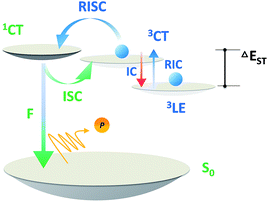 |
| | Scheme 3 Exciton dynamic process for TADF emitters with and without a local triplet excited state (3LE). RIC denotes reverse internal conversion. | |
Base on the calculated E0–0(3CT) and E0–0(3LE), the lower energy level turns out to be the lowest T1 state and ΔEST can be obtained. The reverse internal conversion (IC) rate is determined by the energy gap and the thermal equilibrium process between 3LE and the higher 3CT.9 A lower-lying 3LE state than the 3CT state impedes an efficient RISC process and enlarges ΔEST.43 Triplet exciton generation at the 3LE state is hardly up-converted into the radiative 1CT state, which leads to a high triplet exciton density. Except for PyCN–TC, all the evaluated E0–0(3LE) are higher than E0–0(3CT). The E0–0(3LE) of PyCN–TC is lower than E0–0(3CT) by 0.31 eV and could be the predominant energy level forming triplet excitons under optical excitation or in EL devices. The difference between E0–0(1CT) and lowest T1 defines ΔEST, which is proportional to the degree of orbital overlap (Sif). With qualitative analyses of the overlap extent of HONTO and LUNTO, larger ΔEST values are calculated for DC–TC (0.21) and PyCN–TC (0.46) than DC–ACR (0.01) and PyCN–ACR (0.01) because the almost orthogonal dihedral angles between ACR and the acceptor cores induce a more separated HONTO and LUNTO. Due to the existence of the low-lying 3LE state, the calculated ΔEST of PyCN–TC reaches 0.46 eV, which is over twice as much as its chemical precursor DC–TC. Since similar α values were observed for DC–TC and PyCN–TC, the distinction for ΔEST can be explained by the different electronic configuration of the acceptor moiety. When the n–π* transition DC center is chemically modified into PyCN, the increased π–π* transition nature enhances the HONTO and LUNTO overlap, promoting a radiative transition process with a higher kF and enlarged ΔEST. As mentioned, additional attention should be stressed on the CT and LE coupling of the triplet state in molecular design because 3LE could intrinsically disable an efficient RISC process. The lowest 3CT and small 1CT/3CT energy splitting enables an unhindered RISC process, which leads to reduction of the triplet exciton population under optical or electrical excitation.
Photo-physical properties
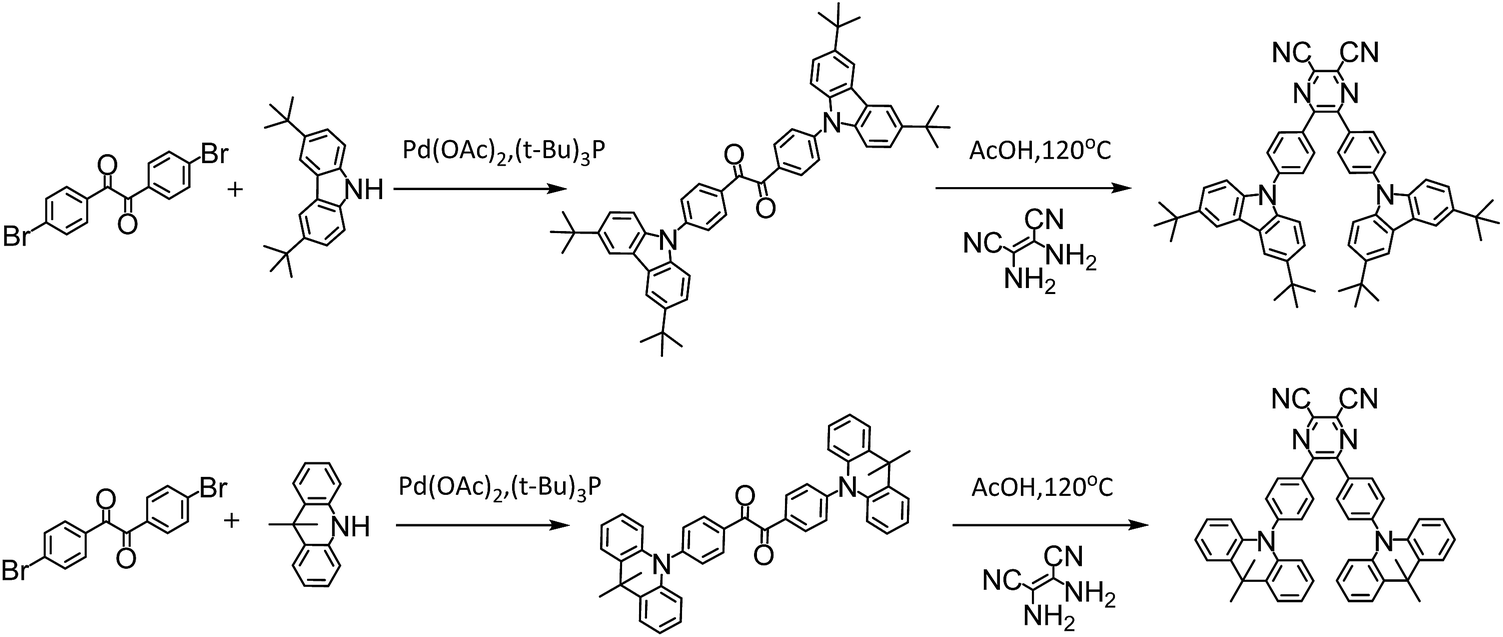
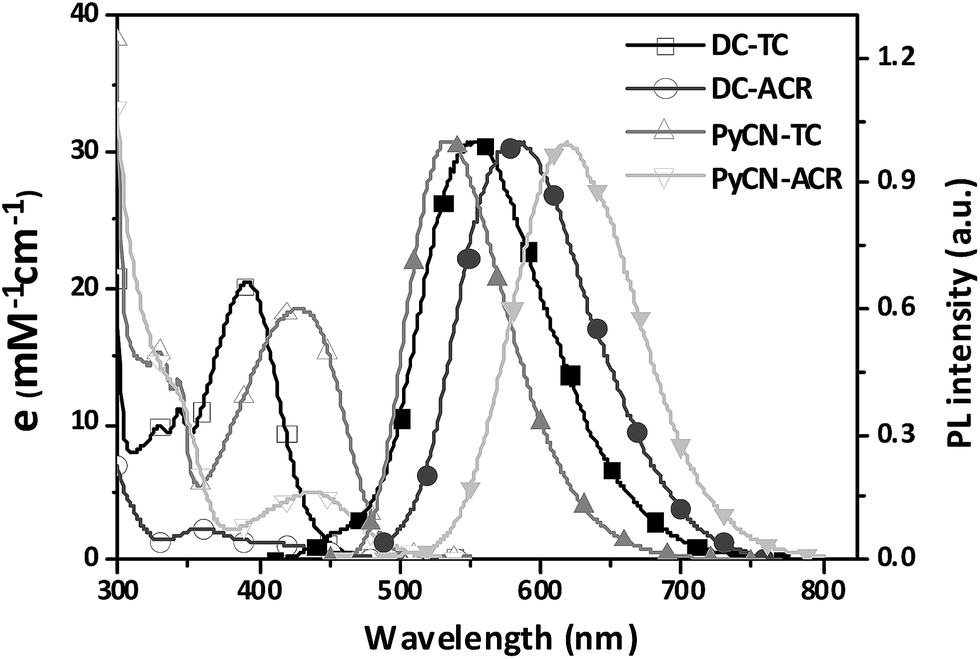
The investigated compounds DC–TC and DC–ACR were synthesized from commercially available starting materials in a one-step convenient Buchwald–Hartwig C–N coupling reaction (Scheme 4). Based on DC–TC and DC–ACR, dicyano-pyrazine ring-closing products were easily produced followed by reactions with diaminomaleonitrile (DAMN) in acetic acid in high yields (>85%), making these compounds cost effective.24,25 Their photophysical properties were studied in diluted solutions and doped films. Broad ICT absorption and photoluminescence (PL) bands were observed. For DC–TC and PyCN–TC, the vertical absorption transition exhibits a larger molar extinction coefficient (ε = 18.0–21.0 mM−1 cm−1) than those of DC–ACR and PyCN–ACR (ε = 2.5–5.0 mM−1 cm−1). The large ε of DC–TC and PyCN–TC absorption can be understood by enhancement of the transition integral (Hif) from large frontier orbital overlap. The solvatochromism effect on the absorption of all compounds was investigated (Fig. S10†). A fluctuating molar extinction coefficient intensity as well as drifting of the absorption peak position was noted. Except for a solvation effect on the Franck–Condon transition,44 diverse S0 molecular configuration distribution in various solvents can play a part. According to the DFT simulation geometry and the TD-DFT computed oscillator strength (f), the orthogonally connected ACR and Ph inevitably give rise to a forbidden HONTO to LUNTO transition (f ≈ 0). Nevertheless, a vertical transition absorption with moderate ε = 2.5–5.0 mM−1 cm−1 still can be detected, not to mention the moderate photoluminescence quantum yield (PLQY) of PyCN–ACR measured in the doped film state considering kF is closely related to the f value of ICT vertical absorption (Fig. 4).14
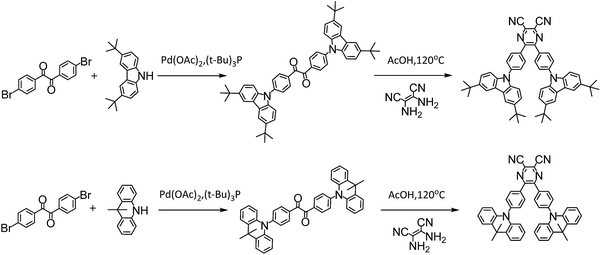 |
| | Scheme 4 Synthesis routes for all compounds and reaction conditions. | |
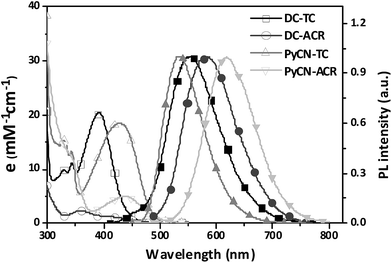 |
| | Fig. 4 UV-vis absorption and PL spectra of the investigated molecules measured in toluene (DC–ACR in n-hexane) (0.05 mM) in air at room temperature. | |
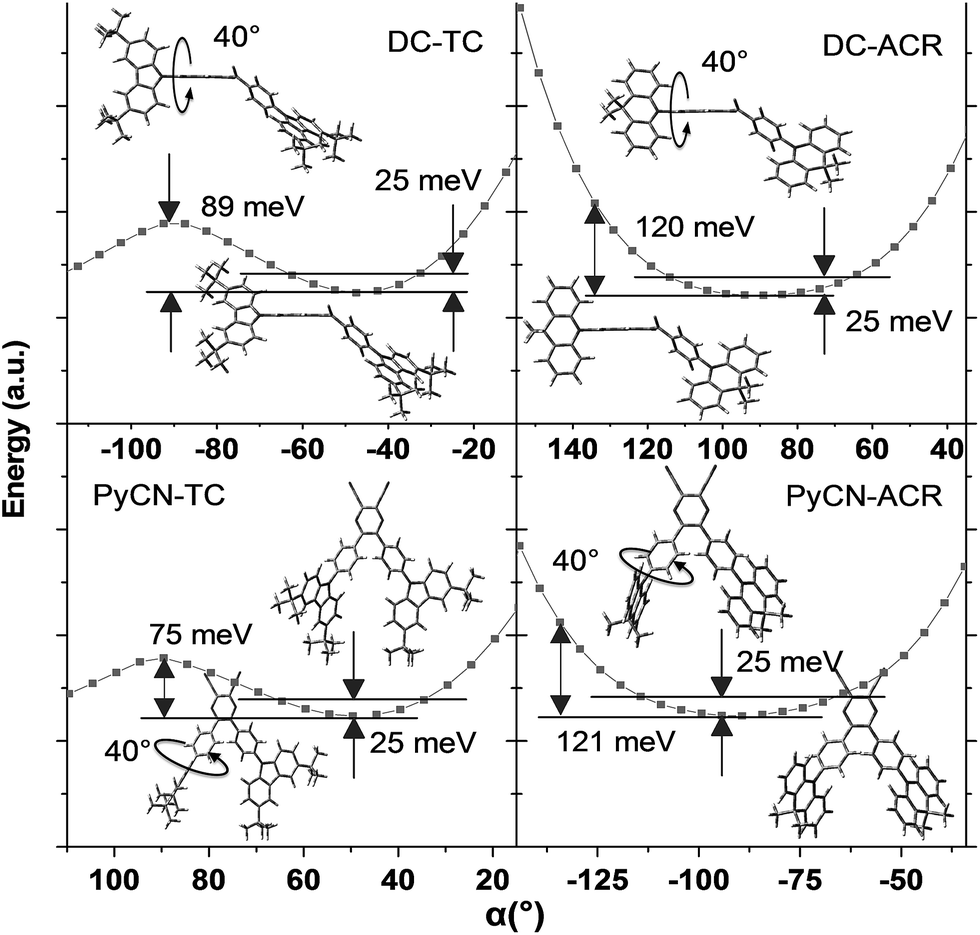
For greater understanding, flexible potential surface scans (full geometry optimization for each molecular conformation) were conducted for all molecules (Fig. 5). Due to symmetric double donor arms in the optimized S0 geometry, only one arm was selectively rotated for simplicity to explicate energy variation and molecular geometry distribution. Starting from the initial geometries, molecules were optimized (with Δα1 = ±5° a step) to relax the irrational intramolecular interaction force, affording a more accurate vertical excitation energy and rotational energy barrier. Local energy maxima of DC–TC and PyCN–TC were obtained following a rotation Δα1 = ±40°. The energy discrepancy between the minimized and local maximum energy turns out to be 89 meV for DC–TC and 75 meV for PyCN–TC. When undergoing the same twisting angle for DC–ACR and PyCN–ACR, the energy discrepancy enlarged to around 120 meV for both molecules. The more rigid ACR–Ph arm than TC–Ph accounts for this, and a steeper potential energy surface can be expected. Due to the large dihedral angle at the minimized energy geometry, the ACR–Ph arm is relatively stationary, which leads to the almost forbidden transition. However, ambient temperature (300 K) with 25 meV energy endows possible molecular rotation around Δα1 = ±25°.45,46 The transition is more permissible because the more planar D–A conformation contributing to larger orbital overlap is found around the equilibrium conformation after activation by thermal energy. Theoretical absorption spectra of DC–ACR and PyCN–ACR were simulated based on OHF% functionals according to different twisting angles (Δα1) using Multiwfn (Fig. S11†).47 When the twisted angle (Δα1) increased from 5–25° (energy in the range of 25 meV), the f of ICT absorption asymptotically increases. In light of this, a moderate kF and suppressed non-radiative process can be obtained simultaneously by the rigid but rotation-feasible ACR–Ph arms (Table 2).
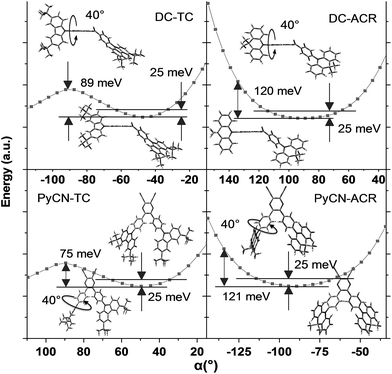 |
| | Fig. 5 Flexible potential surface scanning on the energy of the ground state geometry at different twisted angles (α) in toluene from BMK/6-31G*. The two solid lines label the thermally activated energy (25 meV) at 300 K. The molecular insets represent the stationary point energy-optimized geometry (with α = 40° rotation) and the lowest energy-optimized geometry. | |
Table 2 Thermal, electrochemical and photo-physical properties of the investigated molecules
| Compound |
T
d
/Tgb (°C) |
IP/EAc/EAd (eV) |
E
g
/Egf (eV) |
λ
abs (nm) solg |
λ
PL (nm) solg/filmh |
PLQY (%) solg/filmh |
|
Decomposition temperature (5% weight loss).
Glass transition temperature.
Ionization potential (IP) and electron affinity (EA) measured by cyclic voltammetry in 0.1 M n-Bu4NPF6 in CH2Cl2:CH3CN (4:1) solution.
EA was calculated by IP minus optical gap approximated by the absorption edge in toluene.
Energy gap estimated from cyclic voltammetry measurement.
Energy gap estimated from the absorption edge in toluene (DC–ACR in n-hexane).
The longest peak wavelength of absorption and PL measured in toluene (n-hexane for DC–ACR) and absolute PL quantum yield (PLQY) evaluated using an integrating sphere in toluene (n-hexane for DC–ACR) before and after N2 bubbling (in parenthesis).
PLQY of 7 wt% investigated molecules doped into CBP in air and N2 atmosphere (in parenthesis), respectively.
|
| DC–TC |
413/78 |
−5.56/N.A./−2.9 |
N.A./2.58 |
389 |
553/518 |
0.3 (0.6)/14.7 (19.0) |
| DC–ACR |
387/98 |
−5.35/N.A./−2.84 |
N.A./2.51 |
411 |
584/532 |
0.8 (2.0)/11.5 (13.0) |
| PyCN–TC |
403/N.A. |
−5.56/−3.32/−3.26 |
2.24/2.30 |
429 |
532/534 |
53.8 (62.9)/28.6 (38.0) |
| PyCN–ACR |
450/N.A. |
−5.33/−3.18/−3.17 |
2.15/2.16 |
438 |
618/560 |
8.6 (16.4)/39.8 (43.0) |
For the photoluminescence (PL) spectra (Fig. 6 and S12†), when increasing the polarity of the solvent, a positive solvatochromism effect was observed.48 DC–TC and DC–ACR were hardly emissive while ring-locked PyCN–TC and PyCN–ACR exhibited higher PLQY in solution. After N2 treatment, the PLQYs of the four compounds in solution increase compared with those in air equilibrium solutions, implying that the dissolved oxygen quenches the triplet state excitons.49 As revealed in TD-DFT (Fig. 3), the less rigid DC–TC and DC–ACR undergo obvious conformation deformation, leading to a quick internal conversion (IC) process in S1. When DC was chemically modified into PyCN, the increased rigidity significantly suppresses the non-radiative decay process and a higher PLQY can be obtained in liquid media. To verify such effects on restricted molecules, the PL of the four compounds in different water:THF mixtures ratio was measured (Fig. S13†). Higher water fractions increase the extent of aggregation, enabling suppression of non-radiative channels.50 Evidently, the emission of all the compounds was enhanced with increased aggregation when a high fraction of water was added.
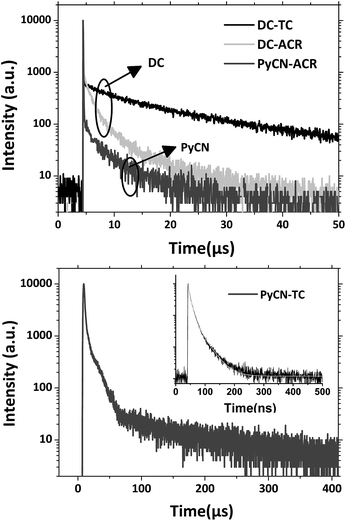 |
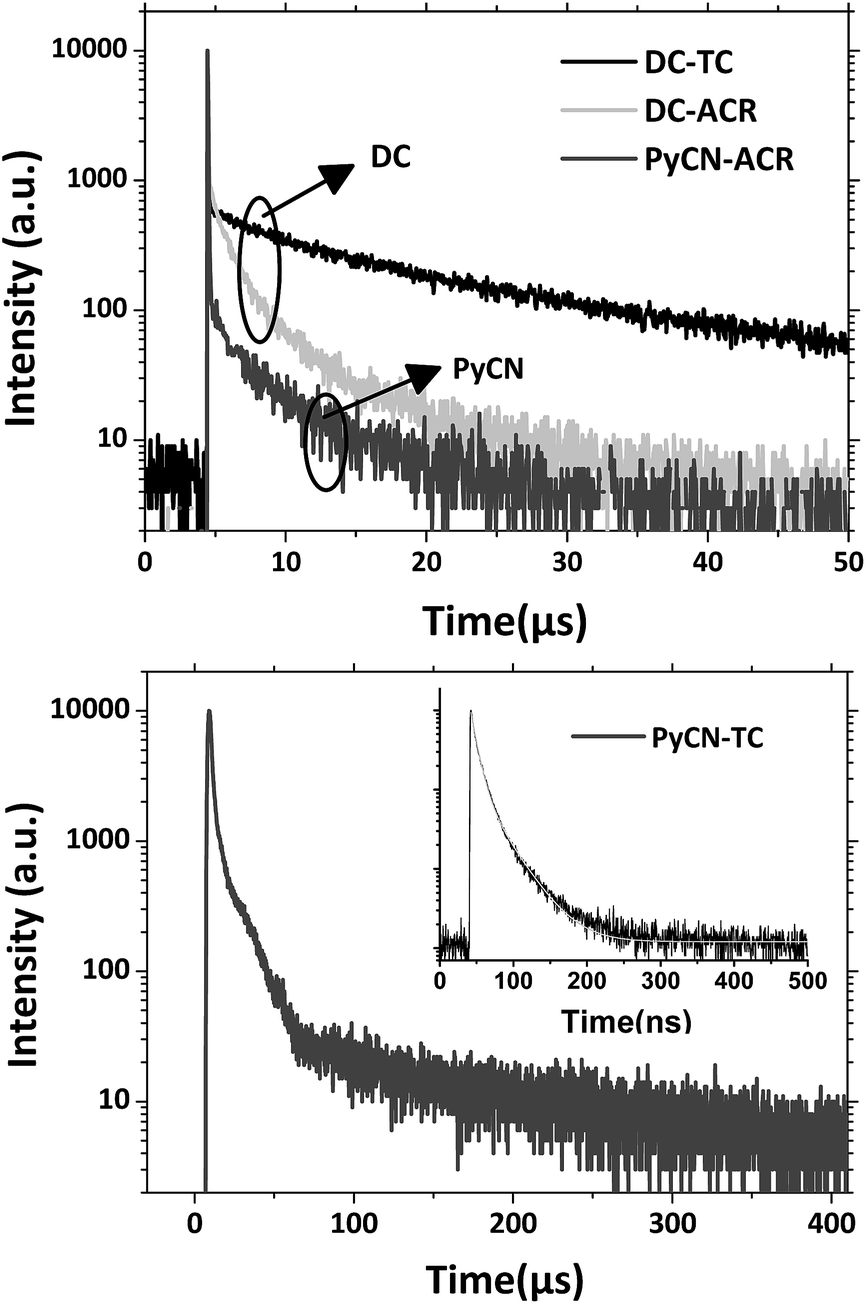
| | Fig. 6 Transient PL decay spectra of the compounds doped into CBP films (7 wt%) measured with a fluorescence lifetime spectrometer (C11367-03, Hamamatsu Photonics) at room temperature in a N2 atmosphere. | |
In addition, the character of the triplet emission was analyzed via phosphorescence spectra at 77 K. In good agreement with the TD-DFT analyses, the 3LE triplet state of PyCN–TC was experimentally confirmed. The phosphorescence spectra of PyCN–TC suggested a specific vibrational structure. For the other three, the TD-DFT-simulated lowest 3CT triplet states are verified by single ICT phosphorescence peaks. (Fig. S9†) ΔEST of all the compounds can be calculated and the order is DC–ACR (0.01 eV) > PyCN–ACR (0.08 eV) > DC–TC (0.13 eV) > PyCN–TC (0.43 eV), in good agreement with the quantum simulation results.
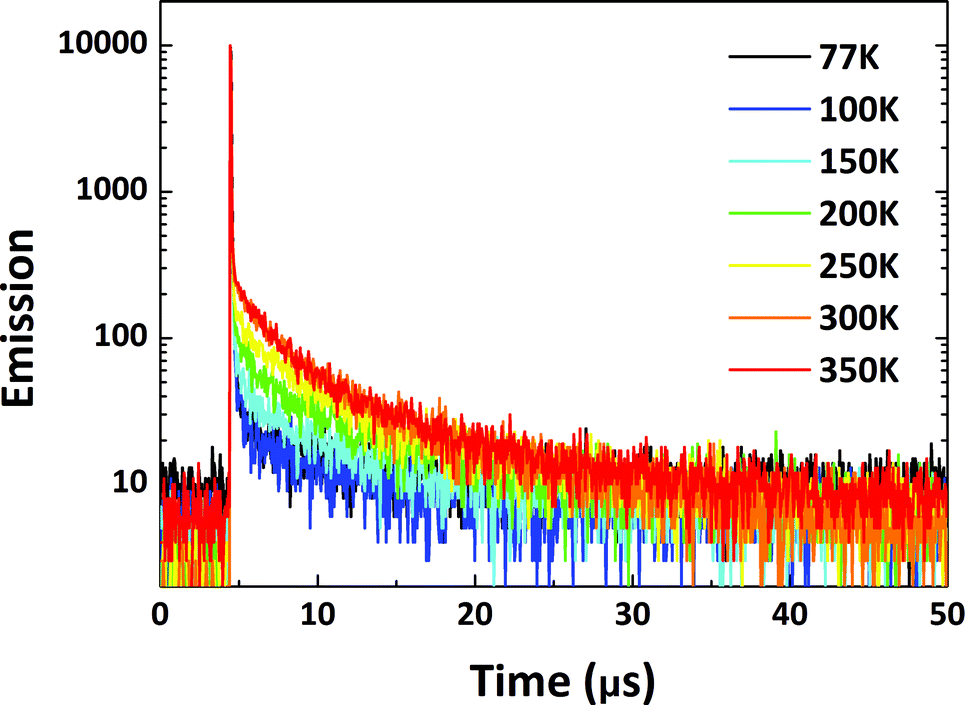
To factually assess performance of the emitting materials in devices, the solid-state photo-physical properties of all the materials were investigated by doping them into 4,4′-bis(carbazol-9-yl)biphenyl (CBP). The doping concentration was controlled at a low level to avoid aggregation-induced quenching effects. The emission in the doped films blue-shifted by around 10–60 nm in contrast to that in solution, indicating that molecular reorganization and relaxation are hindered by the surrounding solid matrix with a reduced Stokes bathochromic shift (Fig. S14†).15 To verify the TADF nature of all the materials, temperature-dependent transient PL spectra were measured from 77 to 350 K (Fig. 7 and S15–S17†). The delayed fluorescence ratio of all the materials showed enhancement when the temperature was increased, then some of the fluorescence ratios were followed by a slight decrease as the temperature was further increased to 350 K. For PyCN–ACR, for example, the monotonically enhanced ratio of the delayed fluorescence from 9.7% to 60.5% firmly depicts the enhanced contribution of RISC from 77 to 300 K. Conversely, further increasing the temperature from 300 to 350 K reduced the delayed fluorescence ratio from 60.5 to 56.2%. Other than the suppressed IC process at low temperature, it competes significantly with RISC at high temperature and results in a reduced delay fluorescence ratio, which is one of the typical TADF characters.51 An analogous phenomenon was also observed for DC–TC, DC–ACR and PyCN–TC, putting all of them into the category of TADF materials (Scheme 5).
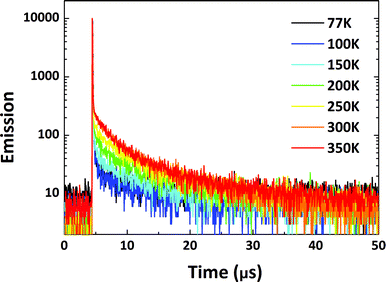 |
| | Fig. 7 Transient PL decay curves for 1 wt% PyCN–ACR doped into CBP measured from 77 to 350 K in a N2 atmosphere. | |
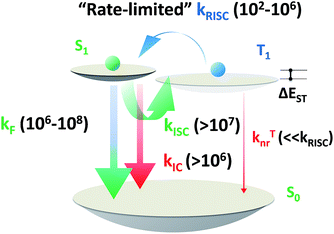 |
| | Scheme 5 Order of magnitude of rate constants involved in exciton dynamic processes for TADF emitters. As shown, kRISC becomes the rate-limited step during these processes for tuning the lifetime of the excited state (τTADF). kF, kISC, kIC and kRISC are on the orders of 106 to 108, >107, >106 and 102 to 106 respectively, as summarized by previous reports.7–9,13,15,51–54,56,58,61,64 | |
Based on the transient PL decay curve at 300 K in a N2 atmosphere (Fig. 8), the prompt (ΦF) and delayed (ΦTADF) components of the PLQY as well as the prompt decay lifetime (τp) and delayed decay lifetime (τTADF) were separated for successive calculation. kF, kISC, kRISC and kIC were estimated using the following formulas.8,15,52,53
| | | kRISC = (kpkdΦTADF)/(kISCΦF) | (7) |
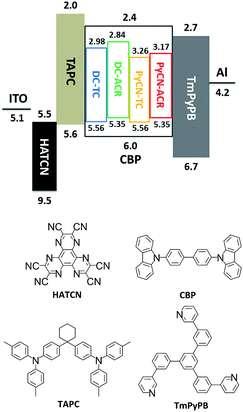 |
| | Fig. 8 Energy level diagram and molecular structures of the materials for fabricating OLED devices. The HOMO and LUMO levels of the investigated molecules were estimated by cyclic voltammetry. | |
As expected, the triplet yields (ΦISC) of DC–TC (93%) and DC–ACR (97%) are superior to those of PyCN–TC (67%) and PyCN–ACR (80%). Optically generated S1 excitons are inclined to ISC into triplet states for DC–TC and DC–ACR because the n–π* transition of carbonyl derivatives favours an ISC process.17 In contrast, when DC was chemically modified into PyCN, ΦISC and kISC were reduced and an enhanced kF was observed (PyCN–TC contrast to DC–TC, and PyCN–ACR contrast to DC–ACR, respectively). Owing to the similar α for DC–TC and PyCN–TC or DC–ACR and PyCN–ACR, the increased π–π* character induces a larger kF for the latter pair. Additionally, when comparing the rigidity of DC–TC to PyCN–TC or DC–ACR to PyCN–ACR, lower kIC values were observed for the latter owing to the more rigid PyCN core. Though PyCN–ACR shows a redder emission, the compact molecular structure results in the lowest kIC among all the emitters. The kRISC of DC–ACR and PyCN–ACR are an order of magnitude higher than that of DC–TC and PyCN–TC, indicating a more efficient RISC from T1 to S1 for the former pair. Interestingly, the delayed lifetimes of carbonyl-type DC–TC and DC–ACR differ greatly with analogous ΦISC ratios (over 90%). In this case, the shortening τTADF of DC–ACR should be ascribed to a more efficient RISC process, and an even larger kF for DC–TC (0.56 × 107) than DC–ACR (0.13 × 107) was observed. For n–π*-favored DC–ACR and π–π*-favored PyCN–ACR with different kISC and kF values, a shorter τTADF was still observed for the former, with smaller ΔEST, reduced kISC and enhanced kF contributing to the shortening of τTADF.
Based on a parallel comparison, as kF, kISC, kIC and kRISC all contribute to τTADF tuning, a large kRISC plays the leading part. This can be interpreted by the “rate-limited effect” of the RISC process. For TADF emitters, the slow dissipation of triplet excitons and an inefficient RISC process can result in a rather long excited state lifetime. As kF, kISC and kIC are larger than 106 while kRISC is from 102 to 106,7–9,54,55 the much smaller kRISC becomes the rate-limited step in exciton dynamic processes. As the triplet state can be stable and its non-radiative process is much slower than RISC, the enhanced kRISC becomes critical because the up-converted triplet excitons can be deactivated quicker radiatively or non-radiatively in the S1 state. According to the analyses, a much shorter τTADF can be expected for DC–ACR even with a large kISC and a small kF. For PyCN–TC, although the highest kF and small kISC values are calculated, τTADF of PyCN–TC is much longer than the other three emitters considering it has the smallest kRISC. Based on the molecular design, we are able to figure that increasing kRISC is the key to shortening τTADF (Table 3).
Table 3 Photo-physical data of the investigated molecules doped into CBP films (7 wt%) at room temperature
| Compound |
λ
em (nm) |
Φ
|
Φ
F
|
Φ
TADF
|
Φ
ISC
|
τ
p (ns) |
τ
TADF
(μs) |
k
F (×107 s−1) |
k
IC (×107 s−1) |
k
ISC (×107 s−1) |
k
RISC (×105 s−1) |
ΔEST (eV) |
|
Average lifetime calculated by τav = ΣAiτi2/ΣAiτi, where Ai is the pre-exponential for lifetime τi.
|
| DC–TC |
518 |
0.19 |
0.07 |
0.12 |
0.93 |
12.6 |
24.9 |
0.56 |
2.4 |
7.4 |
0.74 |
0.13 |
| DC–ACR |
532 |
0.13 |
0.03 |
0.10 |
0.97 |
22.8 |
3.0 |
0.13 |
0.9 |
4.3 |
11.4 |
0.01 |
| PyCN–TC |
534 |
0.38 |
0.33 |
0.05 |
0.67 |
25.2 |
210.5 |
1.31 |
2.1 |
2.60 |
0.011 |
0.43 |
| PyCN–ACR |
575 |
0.43 |
0.20 |
0.23 |
0.80 |
34.3 |
4.6 |
0.58 |
0.7 |
2.32 |
3.13 |
0.08 |
OLED characterization
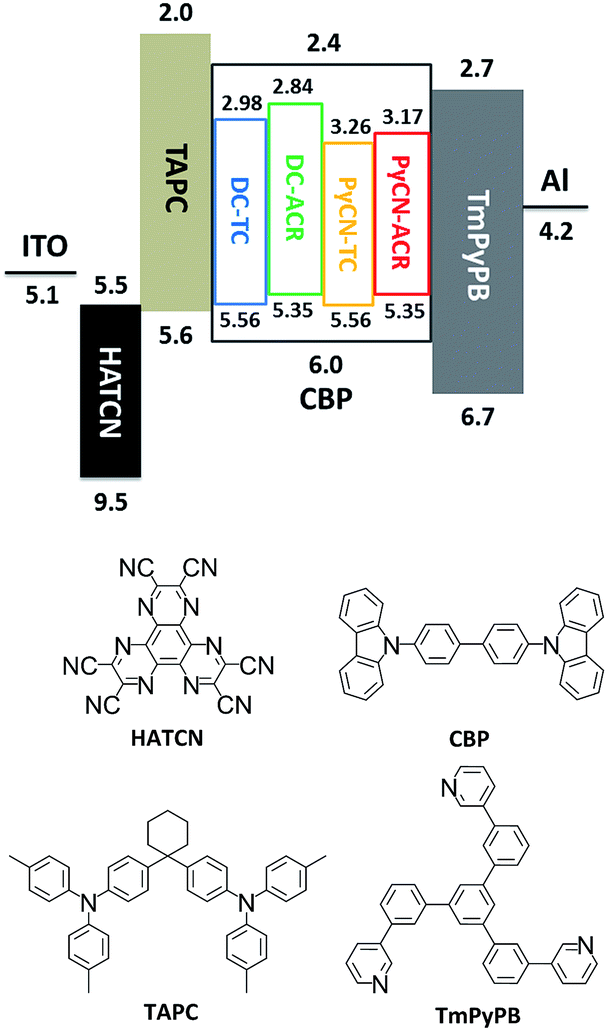
After detailed investigation on the emitting properties and energy level relationships of the four TADF molecules, differences in the electroluminescence performance is investigated for the four materials. To reduce variants, the four TADF materials were employed in the same device architecture. Given that the carrier transport abilities of the investigated guest emitters have effects on the carrier balance and could influence the roll-off character of the devices, the doping concentration of the four TADF emitters were controlled at a low level (1 wt% for DC–ACR and PyCN–ACR, 5 wt% for DC–TC and PyCN–TC) but assuring complete energy transfer from host to guest. Consequently, the device efficiency roll-off can approximately be linked to the molecular characteristics. The fabricated device architecture is indium tin oxide (ITO)/HATCN (5 nm)/TAPC (20 nm)/x wt% emitter in CBP (35 nm)/TmPyPB (55 nm)/LiF (1 nm)/Al (100 nm), in which the dipyrazino[2,3-f:20,30-h]quinoxaline-2,3,6,7,10,11-hexacarbonitrile (HATCN), (1,1-bis(4-(N,N-di(p-tolyl)-amino)-phenyl)cyclohexane) (TAPC), 1,3,5-tri(m-pyrid-3-yl-phenyl)benzene (TmPyPB) and LiF layers play the roles of hole injection, hole transport and electron blocking, electron transport and hole blocking and electron injection layers, respectively.56 An energy level diagram is displayed in accordance with the cyclic voltammetry (CV) results (Fig. 8 and S23†).4 Unlike the inefficient energy transfer from host to guest under optical excitation (Fig. S14†), suppressed CBP host emission around 400 nm was noted in the EL spectra and additional energy capture channels should be considered. As the HOMO and LUMO of all guest molecules are shallower and deeper than that of CBP, relatively equilibrium hole and electron capture abilities can be obtained.57,58 Carriers can be trapped directly into the guest instead of solely transferring the energy via long-range Förster or short-range Dexter transfer, leading to complete energy transfer from host to guest in contrast to those under optical excitation (Fig. S14†).59,60
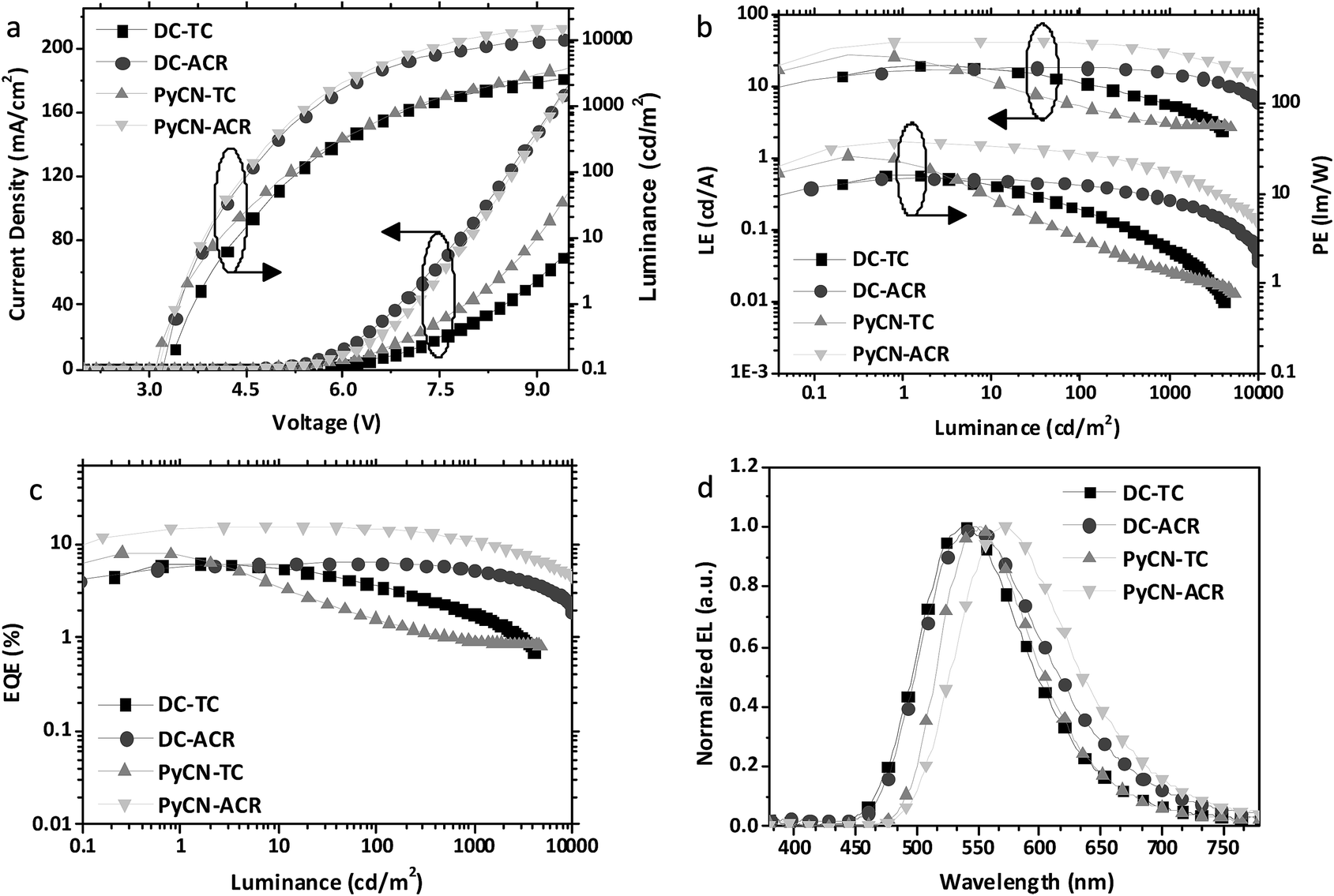
Current density–voltage–luminance (J–V–L), luminance efficiency–luminance–power efficiency (LE–L–PE), EQE–luminance (EQE–L) characteristics and EL spectra are listed in Fig. 9. From the EL spectra, emission peaks from greenish-yellow to orange locating at 538 nm for DC–TC, 546 nm for DC–ACR and PyCN–TC, and 572 nm for PyCN–ACR are observed. The turn-on voltages of the four materials are about the same at 3.4 to 3.5 V, detected at the luminance of 1 cd m−2. The maximum EQEs of the four devices are 6.2% (DC–TC), 6.3% (DC–ACR), 8.1% (PyCN–TC) and 15.6% (PyCN–ACR). Applications of benzil derivatives in OLEDs have been deemed to be impractical due to their poor PLQY. However, the achieved EQEs exceeding the 5% theoretical limitation of traditional fluorophores suggests the efficient utilization of 75% triplet excitons generated under electric excitation.
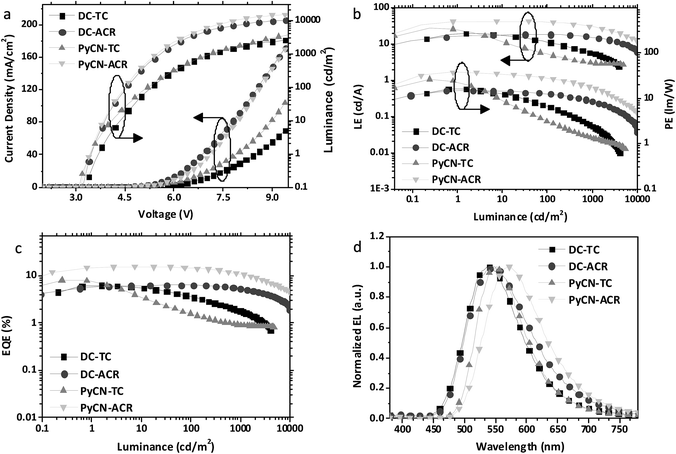 |
| | Fig. 9 (a) Current density–voltage–luminance (J–V–L), (b) luminance efficiency–luminance–power efficiency (LE–L–PE), (c) EQE–luminance characteristics, and (d) EL spectra at 10 mA cm−2 of the devices based on the investigated molecules. | |
Different degrees of efficiency roll-off at high current density were observed, eliminating the possibility of a TTA mechanism leading to the high EQE. Among the four emitters, PyCN–TC exhibits the largest ΔEST and 3LE triplet state, eventuating in τTADF exceeding 200 μs. The hardly up-converted T1 exciton contributes less to delayed fluorescence and leads to a high triplet density population even with a high kF and a small kISC. The most significant efficiency roll-off was observed with only 23% and 11% of EQEmax remaining at luminances of 100 and 1000 cd m−2 respectively.
Towards DC–TC, DC–ACR and PyCN–ACR with the lowest 3CT triplet states, efficiency roll-off can also be linked to τTADF. As mentioned, the enhanced kRISC effectively shortens τTADF, endowing reduced triplet exciton density for suppressed STA and TTA. For DC–TC, owing to the larger ΔEST than DC–ACR and PyCN–ACR, a much longer τTADF increased the ISC–RISC cycles between radiative S1 and non-radiative T1, resulting in a remarkable efficiency roll-off with only 37.1% of the EQEmax remaining at a luminance of 1000 cd m−2. In contrast, a well-controlled roll-off was achieved for DC–ACR with the shortest τTADF among four emitters, exhibiting 100% and 81% of the EQEmax at luminances of 100 and 1000 cd m−2 respectively. In spite of the close ΦISC ratio for DC–TC and DC–ACR and the even larger kF for the former, an enhanced RISC process plays the dominant role in reducing the efficiency roll-off for TADF-OLEDs. For PyCN–ACR with a larger kRISC than DC–TC, a lower device efficiency roll-off is also observed at practical brightness. Note that different doping concentrations of the guest could influence the efficiency roll-off of the device; devices with the same guest doping concentration (5 wt%) were also fabricated for comparison (Fig. S20†). For TADF-OLEDs using DC–ACR and PyCN–ACR as the emitters, the tendency is consistent with our analyses. Emitters with a large kRISC tended to exhibit low efficiency roll-off character at practical brightness (Fig. 10).
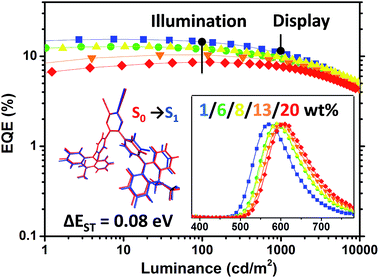 |
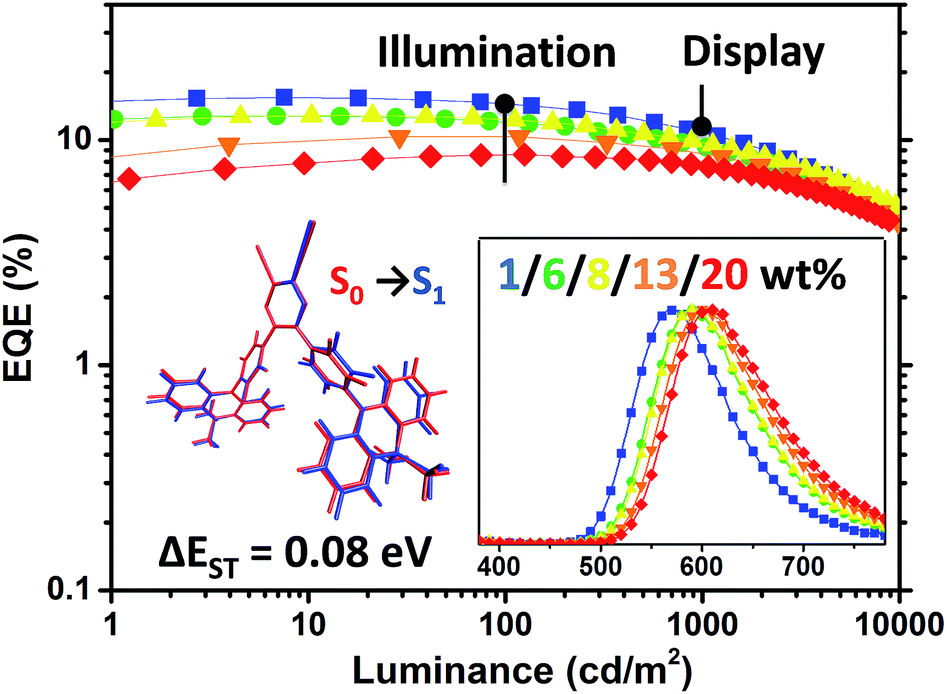
| | Fig. 10 EQE–luminance curves of the OLED devices based on PyCN–ACR at various doping concentrations in a structure of ITO/HATCN (5 nm)/TAPC (20 nm)/CBP: x wt% PyCN–ACR (35 nm)/TmPyPB (55 nm)/LiF (1 nm)/Al. Inset: (left) small geometry deformation in the excited state and ΔEST for PyCN–ACR are responsible for highly efficient and low roll-off orange-red TADF-OLEDs; (right) EL spectra of the corresponding devices at 10 mA cm−2 with different doping concentrations of PyCN–ACR in CBP. | |
Further information on the mechanism of large kRISC for reduced efficiency roll-off can be found by fitting the EQE–J curve with the EQE roll-off model of TADF-OLEDs (Fig. S22†). A high singlet exciton concentration from efficient RISC, STA and TTA processes can be the dominant source for exciton annihilation.13,61,62 For a steady-state current, the model EQEs are fitted and afford the rate constants of STA (kST) and TTA (kTT). Very small HONTO and LUNTO overlap for PyCN–ACR and DC–ACR renders a small f and the rate constant of Förster energy transfer-type STA could be low. Though the calculated kTT are two magnitudes smaller than kST, the triplet density can be over three orders higher than the singlet density and TTA could aggravate the efficiency roll-off of the device.13,63 In good agreement of our analyses, the kST and kTT of DC–ACR and PyCN–ACR are much smaller than those of the other two emitters with longer τTADF, indicating more suppressed STA and TTA for OLEDs using emitters with a large kRISC and a short τTADF.
Notably, the 1 wt% PyCN–ACR in CBP device exhibits orange EL at 572 nm. The slightly bathochromic shift (12 nm) with respect to the PL in the doped film implies geometry relaxation in the solid matrix. When the doping concentration was adjusted from 1 wt% to 20 wt%, a red-shift as large as 34 nm was observed. Under an electric field, TADF molecules with a large dipole moment tend to polarize in the host medium. Such a “solvatochromic” shift in the solid matrix can be observed when TADF emitters are doped into bipolar hosts or the doping concentration of the guest is increased.64,65 A low efficiency roll-off was noted for all the devices with different doping concentrations. The maximum efficiency roll-off turned out to be 38.8% at 1000 cd m−2 for 1 wt% PyCN–ACR doped into CBP. Especially for the devices with doping concentrations of 8 wt% and 13 wt%, EQEs of 12.4% and 10.3% at a luminance of 100 cd m−2 as well as EQEs of 9.9% and 8.8% at a luminance of 1000 cd m−2 were achieved. The roll-off was less than 25% and 15% and emission peaks were located at 590 and 600 nm, respectively. Even at a luminance of 10000 cd m−2, EQEs as high as 5.1% and 4.4% were maintained. For PyCN–ACR, the rotation-feasible ACR–Ph arms, enhanced π–π* feature and compact molecular geometry endow a moderate Φ and restricted non-radiative channel for orange-red emitters with a narrow band gap. In combination with the high kRISC, a low efficiency roll-off and highly efficient orange-red device employing PyCN–ACR can be realized, achieving the best EQE of orange-red TADF-OLEDs at application relevant brightness (100/1000/10000 cd m−2) without employing host management.
Conclusions
Via molecular design, the effects of kF, kISC, kIC and kRISC on τTADF governing were investigated based on four representative TADF emitters. τTADF can be effectively shortened by enhanced RISC (small ΔEST) even with a small kF and a large kISC. As RISC is slower than other exciton dynamic processes and T1 is relatively stable, it becomes the rate-limited step in shortening τTADF (“rate-limited effect” of the RISC process). When TADF materials with short τTADF are employed in OLEDs, more suppressed efficiency roll-off at practical relevant brightnesses (100/1000/10000 cd m−2) is observed. The shortened τTADF indicates a more efficient RISC process and eventuates in reduced triplet exciton density for suppressed STA and TTA.
Following the aforementioned guidelines, TADF materials should be carefully designed to govern τTADF. In light of the critical role of kRISC, factors concerning tuning ΔEST can effectively alter τTADF. One of the approaches is tuning the D–A strength of the emitters for enhanced ICT, which can render small frontier orbital overlap and reduce ΔEST. A more promising way is tuning the dihedral angles between electron donating and withdrawing moieties in TICT molecular systems. The enlarged dihedral angle induces separated HOMO/LUMO overlap while a flat D–A structure does the opposite. The former case reduces ΔEST and effectively shortens τTADF, giving these materials potential for fabricating low efficiency roll-off OLED devices. The latter case can elongate τTADF, making these materials promising for ultra-long lifetime delayed fluorescence applications such as bio-imaging,66 oxygen sensors67 or anti-fake,68etc. Finally, tuning 3LE and 3CT coupling also helps to tune τTADF. For TADF-OLEDs, materials with 3LE can be detrimental for shortening τTADF because the additional RIC process makes RISC tougher. OLEDs using such emitters exhibit significant roll-off, and a low EQE is expected at high brightness. Chemical moieties for fabricating TADF emitters should not have low triplet states to make sure that the locally excited state lies above the CT channel.
Acknowledgements
The authors greatly appreciate the financial support from the Ministry of Science and Technology (2015CB655003 and 2014DFA52030), the National Natural Science Foundation of China (91233116 and 51573059), and Guangdong Provincial Department of Science and Technology (2014B090901048).
Notes and references
- M. A. Baldo, D. O'brien, Y. You, A. Shoustikov, S. Sibley, M. Thompson and S. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS
 .
.
- Y. Ma, H. Zhang, J. Shen and C. Che, Synth. Met., 1998, 94, 245–248 CrossRef CAS .
-
Z. H. Kafafi, Organic electroluminescence, CRC Press, 2005 Search PubMed .
- M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10967–10976 CrossRef CAS .
- S. J. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125–2130 CrossRef CAS .
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed .
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed .
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed .
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS .
- M. Liu, Y. Seino, D. Chen, S. Inomata, S.-J. Su, H. Sasabe and J. Kido, Chem. Commun., 2015, 51, 16353–16356 RSC .
- D. Chen, G. Xie, X. Cai, M. Liu, Y. Cao and S.-J. Su, Adv. Mater., 2016, 28, 239–244 CrossRef CAS PubMed .
- K.-C. Tang, K. L. Liu and I. C. Chen, Chem. Phys. Lett., 2004, 386, 437–441 CrossRef CAS .
- K. Masui, H. Nakanotani and C. Adachi, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS .
-
N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern molecular photochemistry of organic molecules, University Science Books, Sausalito, CA, 2010 Search PubMed .
- Q. Zhang, H. Kuwabara, W. J. Potscavage Jr, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed .
- T. Itoh, Chem. Phys. Lett., 1988, 151, 166–168 CrossRef CAS .
- D. R. Kearns and W. A. Case, J. Am. Chem. Soc., 1966, 88, 5087–5097 CrossRef CAS .
- T. Förster, Discuss. Faraday Soc., 1959, 27, 7 RSC .
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed .
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef .
- M. W. Wolf, K. D. Legg, R. E. Brown, L. A. Singer and J. H. Parks, J. Am. Chem. Soc., 1975, 97, 4490–4497 CrossRef CAS .
- Y. P. Sun, D. F. Sears Jr and J. Saltiel, J. Am. Chem. Soc., 1989, 111, 706–711 CrossRef CAS .
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed .
- B. Gao, Q. Zhou, Y. Geng, Y. Cheng, D. Ma, Z. Xie, L. Wang and F. Wang, Mater. Chem. Phys., 2006, 99, 247–252 CrossRef CAS .
- S. Chen, X. Xu, Y. Liu, G. Yu, X. Sun, W. Qiu, Y. Ma and D. Zhu, Adv. Funct. Mater., 2005, 15, 1541–1546 CrossRef CAS .
- M. A. Marques and E. Gross, Annu. Rev. Phys. Chem., 2004, 55, 427–455 CrossRef CAS PubMed .
- E. Runge and E. K. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS .
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chem. Acc., 2001, 107, 48–55 CrossRef CAS .
- D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef .
- R. Magyar and S. Tretiak, J. Chem. Theory Comput., 2007, 3, 976–987 CrossRef CAS PubMed .
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed .
- A. D. Boese and J. M. L. Martin, J. Chem. Phys., 2004, 121, 3405–3416 CrossRef CAS PubMed .
- R. Improta, V. Barone, G. Scalmani and M. J. Frisch, J. Chem. Phys., 2006, 125, 054103 CrossRef PubMed .
-
P. H. Harbach and A. Dreuw, in Modeling of Molecular Properties, ed. P. Comba, Wiley-VCH, Weinheim, 2011, pp. 37–38 Search PubMed .
- D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2011, 133, 17895–17900 CrossRef CAS PubMed .
- S. Salman, D. Kim, V. Coropceanu and J.-L. Brédas, Chem. Mater., 2011, 23, 5223–5230 CrossRef CAS .
- Z. R. Grabowski, K. Rotkiewicz, A. Siemiarczuk, D. Cowley and W. Baumann, Nouv. J. Chim., 1979, 3, 443–454 CAS .
- M. Maus and W. Rettig, Chem. Phys., 1997, 218, 151–162 CrossRef CAS .
- M. Maus, W. Rettig, G. Jonusauskas, R. Lapouyade and C. Rullière, J. Phys. Chem. A, 1998, 102, 7393–7405 CrossRef CAS .
- M. Maus, W. Rettig, D. Bonafoux and R. Lapouyade, J. Phys. Chem. A, 1999, 103, 3388–3401 CrossRef CAS .
- D. Jacquemin, A. Planchat, C. Adamo and B. Mennucci, J. Chem. Theory Comput., 2012, 8, 2359–2372 CrossRef CAS PubMed .
- S. Huang, Q. Zhang, Y. Shiota, T. Nakagawa, K. Kuwabara, K. Yoshizawa and C. Adachi, J. Chem. Theory Comput., 2013, 9, 3872–3877 CrossRef CAS PubMed .
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed .
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed .
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS .
- L. Yao, Y. Pan, X. Tang, Q. Bai, F. Shen, F. Li, P. Lu, B. Yang and Y. Ma, J. Phys. Chem. C, 2015, 119, 17800–17808 CAS .
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed .
- P. Wang and S. Wu, J. Photochem. Photobiol., A, 1995, 86, 109–113 CrossRef CAS .
- T. Serevicius, T. Nakagawa, M. C. Kuo, S. H. Cheng, K. T. Wong, C. H. Chang, R. C. Kwong, S. Xia and C. Adachi, Phys. Chem. Chem. Phys., 2013, 15, 15850–15855 RSC .
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed .
- J. Li, T. Nakagawa, J. MacDonald, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Adv. Mater., 2013, 25, 3319–3323 CrossRef CAS PubMed .
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS .
- H. Tanaka, K. Shizu, J. Lee and C. Adachi, J. Phys. Chem. C, 2015, 119, 2948–2955 CAS .
- J. Li, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2014, 105, 013301 CrossRef .
- M. Numata, T. Yasuda and C. Adachi, Chem. Commun., 2015, 51, 9443–9446 RSC .
- G. Xie, X. Li, D. Chen, Z. Wang, X. Cai, D. Chen, Y. Li, K. Liu, Y. Cao and S.-J. Su, Adv. Mater., 2016, 28, 181–187 CrossRef CAS PubMed .
- S. J. Su, E. Gonmori, H. Sasabe and J. Kido, Adv. Mater., 2008, 20, 4189 CAS .
- D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050–5055 CrossRef CAS PubMed .
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CAS .
- M. Baldo, M. Thompson and S. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS PubMed .
- D. Kasemann, R. Brückner, H. Fröb and K. Leo, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 115208 CrossRef .
- Y. Zhang and S. R. Forrest, Phys. Rev. Lett., 2012, 108, 267404 CrossRef PubMed .
- T. Furukawa, H. Nakanotani, M. Inoue and C. Adachi, Sci. Rep., 2015, 5, 8429 CrossRef CAS PubMed .
- G. Méhes, K. Goushi, W. J. Potscavage and C. Adachi, Org. Electron., 2014, 15, 2027–2037 CrossRef .
- C. F. Madigan and V. Bulović, Phys. Rev. Lett., 2003, 91, 247403 CrossRef PubMed .
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. Peng, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef CAS PubMed .
- S. Nagl, C. Baleizao, S. M. Borisov, M. Schaferling, M. N. Berberan-Santos and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2007, 46, 2317–2319 CrossRef CAS PubMed .
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental tables and figures including synthesis, quantum chemical computation, roll-off model simulation, thermal properties and cyclic voltammetry of the investigated compounds. See DOI: 10.1039/c6sc00542j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a