
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methane activation by gold-doped titanium oxide cluster anions with closed-shell electronic structures†
Yan-Xia
Zhao
a,
Xiao-Na
Li
a,
Zhen
Yuan
ab,
Qing-Yu
Liu
ab,
Qiang
Shi
*a and
Sheng-Gui
He
*a
aBeijing National Laboratory for Molecular Sciences, State Key Laboratory for Structural Chemistry of Unstable and Stable Species, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: shengguihe@iccas.ac.cn; qshi@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 29th March 2016
Abstract
The reactivity of closed-shell gas phase cluster anions AuTi3O7− and AuTi3O8− with methane under thermal collision conditions was studied by mass spectrometric experiments and quantum chemical calculations. Methane activation was observed with the formation of AuCH3 in both cases, while the formation of formaldehyde was also identified in the reaction system of AuTi3O8−. The cooperative effect of the separated Au+ and O2− ions on the clusters induces the cleavage of the first C–H bond of methane. Further activation of the second C–H bond by a peroxide ion O22− leads to the formation of formaldehyde. This study shows that closed-shell species on metal oxides can be reactive enough to facilitate thermal H–CH3 bond cleavage and the subsequent conversion.
Introduction
Methane, the major component of natural gas and shale gas, represents an important feedstock for the production of value-added chemicals.1–6 However, the direct conversion of methane poses a serious challenge in contemporary catalysis owing to the significant energy required for C–H bond cleavage.1 Many heterogeneous and homogeneous catalytic systems have been studied to transform methane, while it is challenging to uncover the elementary reactions and molecular level (ML) mechanisms associated with methane activation and conversion.3–7 In the last decades, model investigations of the elementary reactions between methane and gas phase atomic clusters with state-of-the-art mass spectrometric experiments and quantum chemistry calculations have been serving as an important approach to discovering the ML mechanisms of methane activation and transformation.8–16 The identified mechanisms can be very useful for catalyst design and optimization.17–20Many atomic clusters including oxides,8–11 carbides,21 noble metals,13–16 and so on have been identified to be able to activate methane under thermal collision conditions. Investigations of these cluster systems have revealed three types of mechanism to activate the C–H bond of methane:
| [MO˙−] + CH4 → [M(O–H)−] + CH3˙ | (1) |
| [M] + CH4 → [H–M–CH3] | (2) |
| [ML] + CH4 → [M–CH3] + H–L | (3) |
| [M+⋯O2−] + CH4 → [CH3–M⋯(O–H)−] | (4) |
The ability of two atoms with different polarity to promote chemical activity of atomic clusters in reactions with small molecules has been previously identified in the literature. Castleman, Khanna, and their co-workers have reported that a complementary active site composed of a pair of adjacent Al atoms that respectively act as a Lewis acid and Lewis base can activate a variety of polar molecules such as water, alcohols, aldehydes, and thiols.30–33 Recently, we have found that a pair of non-adjacent ions Au+⋯O2− on AuCeO2+ and AuCe2O4+ cations can activate the non-polar dihydrogen.34 However, these clusters are still not reactive enough to activate methane. This study reports that the cooperation of the separated Au+ and O2− ions on gold-doped titanium oxide clusters AuTi3O7− and AuTi3O8− can bring about methane activation at thermal energies. Subsequent conversion of methane to a stable organic compound, formaldehyde, has also been identified. It is noteworthy that for the thermal activation of methane, most of the reactive clusters reported have open-shell electronic structures and the very few reactive species with closed-shell electronic structures are all mononuclear cations.26,29 The cluster anions were generally found to be much less reactive than the corresponding cations in the reactions with methane.10,13,16 For the first time, we report thermal methane activation by cluster anions with closed-shell electronic structures.
Results
Reactivity of AuTi3O7− and AuTi3O8− with methane
The AuTi3O7− and AuTi3O8− cluster anions were prepared by a reaction of O2 with metal plasmas generated by laser vaporization of a solid disk compressed with Au and 48Ti powders. The clusters of interest were mass-selected by a quadrupole mass filter and entered into a linear ion trap reactor, where they were thermalized by collisions with a pulse of He gas (maximal instantaneous pressure around 2–4 Pa) and then interacted with a pulse of CH4, CD4, or CH2D2 for a period of time.35 Upon the interaction of AuTi3O7− with 50 mPa CH4 for about 1.07 ms (Fig. 1b), a strong product peak that can be assigned to Ti3O7H− was observed, suggesting the following reaction channel:| AuTi3O7− + CH4 → Ti3O7H− + AuCH3 | (5) |
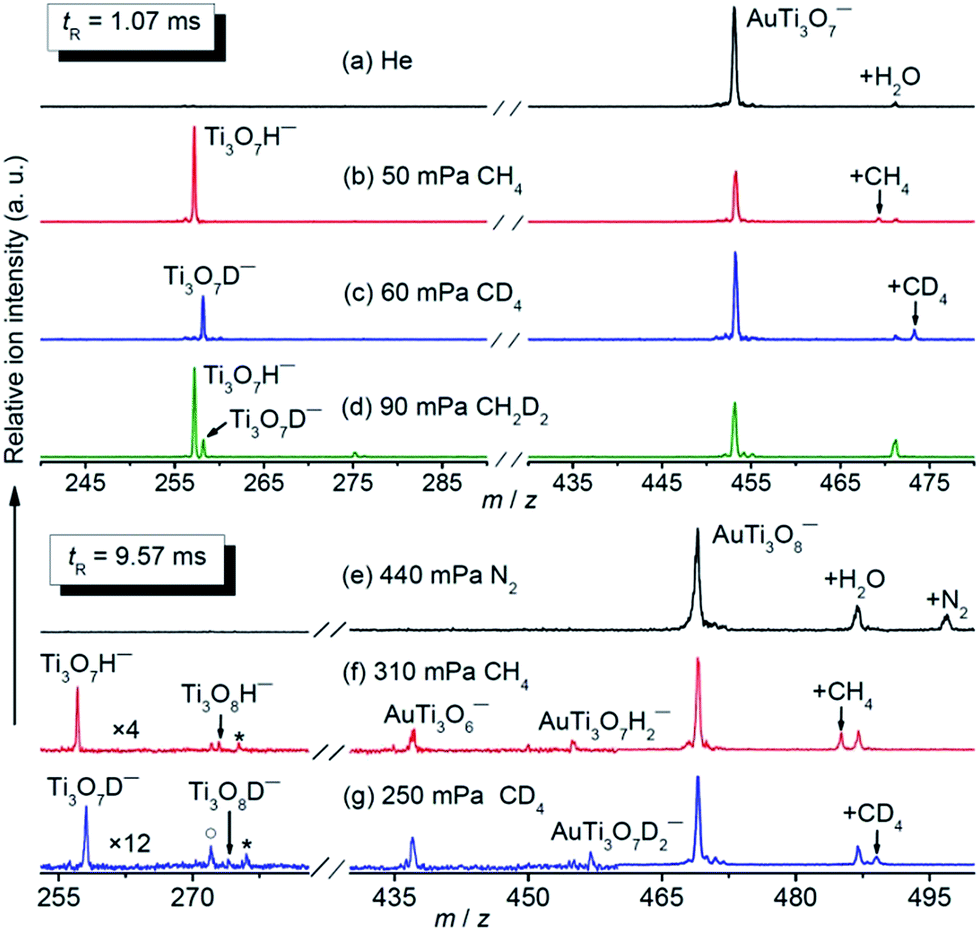 | ||
| Fig. 1 Time-of-flight mass spectra for the reactions of mass selected AuTi3O7− and AuTi3O8− with CH4 (b and f), CD4 (c and g), CH2D2 (d), and N2 (e). The peaks marked with asterisks and hollow circles in panels (f) and (g) represent water adsorption products (Ti3O7HH2O− or Ti3O7DH2O−) and Ti3O8− from collision induced dissociation, respectively. The peaks marked with +X (X = H2O, CH4, CD4, and N2) in panels (a–c) and (e–g) denote the association products with AuTi3O7− and AuTi3O8−, respectively. The reactant gas pressures are shown. The reaction times (tR) are 1.07 ms for (a–d) and 9.57 ms for (e–g). The signal magnitudes below m/z 460 are amplified by 4 and 12 for (f) and (g), respectively. | ||
The isotopic labelling experiments with CD4 (Fig. 1c) and CH2D2 (Fig. 1d) confirmed the above reaction. The generation of Ti3O7D− was observed from AuTi3O7− + CD4 while both Ti3O7H− and Ti3O7D− were produced from AuTi3O7− + CH2D2. The inter- and intra-molecule isotopic effects were apparently observed. In addition to Reaction (5), a minor association reaction channel generating AuTi3O7CH4− (4% of the total product ions) was also observed.
The AuTi3O8− cluster is much less reactive than AuTi3O7− and a longer reaction time and higher methane pressures were used for AuTi3O8− + CH4 (Fig. 1f), in which the generation of Ti3O7H−, Ti3O8H−, AuTi3O6−, and AuTi3O7H2− was observed and the assignments were confirmed by the experiments with CD4 and CH2D2 (Fig. 1g and S1 ESI†). In a reference experiment with N2 (Fig. 1e), the AuTi3O6− product was not generated, indicating that this product cluster in Fig. 1f and g was due to a chemical reaction rather than collision induced dissociation (CID, such as AuTi3O8− + CH4 → AuTi3O6− + O2 + CH4). In addition to molecular association, the following four reaction channels are suggested by the experiments:
| AuTi3O8− + CH4 → Ti3O8H− + AuCH3 | (6) |
| AuTi3O8− + CH4 → AuTi3O7H2− + CH2O | (7) |
| AuTi3O8− + CH4 → Ti3O7H− + AuCH3O (CH2O + AuH) | (8) |
| AuTi3O8− + CH4 → AuTi3O6− + CH4O2 (CH2O + H2O) | (9) |
The branching ratios of generating AuTi3O8CH4− (41%), Ti3O7H− (35%), and AuTi3O6− (15%) are much larger than those of AuTi3O7H2− (6%) and Ti3O8H− (3%) (Fig. 1f).
An atomic cluster often has different structural isomers with very different reactivities.29 The analysis of the methane-pressure dependent reactivity indicated that (87 ± 1)% of the experimentally generated AuTi3O7− ions (Fig. S2†) and only (37 ± 4)% of the AuTi3O8− ions (Fig. S3†) were reactive with CH4. For the reactive component of AuTi3O7−, the pseudo first-order rate constant (k1) of Reaction (5) is (7.7 ± 2.3) × 10−11 cm3 per molecule per second, corresponding to a reaction efficiency (Φ)36 of (7.7 ± 2.3)%. The intra and inter-molecular kinetic isotope effects (KIEs) amount to 5.0 ± 1.1 and 4.5 ± 1.3, respectively. For the reactive component of AuTi3O8−, the summed k1 value of Reactions (6)–(9) is (1.0 ± 0.3) × 10−12 cm3 per molecule per second [Φ = (0.1 ± 0.03)%]. The inter-molecular KIE amounts to 4.0 ± 1.3.
Reaction mechanisms of AuTi3O7− and AuTi3O8− with CH4
Density functional theory (DFT) calculations at the TPSS level, which shows the overall best performance in calculating several critical bond energies among the 18 tested methods (Table S1, ESI†), have been conducted to explore the detailed reaction mechanisms. A Fortran code based on the genetic algorithm was used to search the global minimum structures of AuTi3O7− as well as AuTi3O8− clusters with different spin multiplicities.37 To determine the lowest-energy isomers of AuTi3O7− and AuTi3O8−, further single-point energy calculations with a high-level quantum chemistry method of a restricted coupled-cluster method with single, double, and perturbative triple excitations [RCCSD(T)] have also been performed for the TPSS optimized structures. In the reaction pathway of AuTi3O8− + CH4, concomitant cleavage and formation of several chemical bonds are involved in important transition states. It is known that commonly used density functionals do not correctly describe the long- and mid-range dispersion interactions, which can influence the chemical reaction energies.38 Thus, TPSS functional calculated energies with dispersion corrections38 are given throughout the reaction pathways.The lowest-lying isomer of AuTi3O7− (Fig. 2 and S5†) has two terminally-bonded oxygen anions (Ot2−, −0.62e) and a one-fold coordinated gold cation (Au1f+, +0.42e). It is noteworthy that the superscripts “2−” of O2− and “+” of Au+ denote the formal oxidation states rather than the net charges on the atoms. As marked in Fig. 2, the Au1f+ cation is separated from the Ot2− anions. The Au-side of AuTi3O7− is the least negatively charged, so it is expected that the Au1f+ atom traps CH4 to form the encounter complex I1 with a significant binding energy (54 kJ mol−1). Then the CH4 is delivered to be close to a Ot2− ion (TS1) so that the cleavage of one C–H bond and the concomitant formation of one Au–CH3 bond and one O–H bond can take place. Such a process (I1 → TS1 → I2) is subject to an energy barrier of 13 kJ mol−1 which is surmountable by the binding energy of I1. The formation of I2 can release a high energy of 198 kJ mol−1, which is enough to evaporate the AuCH3 species rather than Au + CH3 (endothermic by 244 kJ mol−1) from the reaction complex to form the experimentally observed Ti3O7H− cluster (Fig. 1b).
 | ||
| Fig. 2 TPSS functional calculated potential energy profile for AuTi3O7− + CH4 → Ti3O7H− + AuCH3. All of the species are in a singlet spin state. The dispersion corrected energies of the reaction intermediates (I1 and I2), transition state (TS1), and products (P1) with respect to the separated reactants (R1) are given in kJ mol−1. Bond lengths are given in pm. | ||
The gold atom in the lowest-lying isomer of AuTi3O7− is one-fold coordinated (Fig. 2). In a low-lying isomer of AuTi3O7− (Fig. S5†), the Au+ ion can be two-fold coordinated (Au2f+). Such a cluster anion with Au2f+ can hardly trap (binding energy is only 8 kJ mol−1) the reactant molecule CH4. The activation of CH4 by AuTi3O7− with the Au2f+ is subject to an additional transformation so that the Au2f+ ion becomes Au1f+ (as in I1 of Fig. 2). The Au2f+ → Au1f+ transformation has an overall positive barrier (3 kJ mol−1) which hinders subsequent methane activation, suggesting that the AuTi3O7− isomer with Au2f+ corresponds to the un-reactive component (13%) of AuTi3O7− in the experiments.
When one of the Ot2− ions in the lowest-lying isomer of AuTi3O7− is replaced by a peroxide unit (O22−), a low-lying isomer of AuTi3O8− with Au1f+ and Ot2− ions can be formed (see I3 of Fig. 3). However, this isomer with Au1f+ is less stable by 24 kJ mol−1 than the lowest-lying isomer of AuTi3O8− that contains an Au2f+ ion (Fig. S6†). The activation of CH4 by the lowest-lying isomer of AuTi3O8− also involves the Au2f+ → Au1f+ conversion which is hindered by an overall positive reaction barrier (1 kJ mol−1). As a result, this lowest-lying isomer with Au2f+ accounts for the 63% un-reactive component of the experimentally generated AuTi3O8− ions. The 37% reactive ions can then be assigned to the low-lying isomer with Au1f+ and the reaction mechanism is shown in Fig. 3.
 | ||
| Fig. 3 TPSS functional calculated potential energy profile for AuTi3O8− + CH4 to generate the products Ti3O8H− + AuCH3 (P2), AuTi3O7H2− + CH2O (P3), Ti3O7H− + CH2O + AuH (P4), and AuTi3O6− + CH2O + H2O (P5). All of the species are in a singlet spin state. The dispersion corrected energies of the reaction intermediates (I3–I8), transition states (TS2–TS6), and products (P2–P5) with respect to the separated reactants (R2) are given in kJ mol−1. Bond lengths are given in pm. | ||
Similarly to AuTi3O7− + CH4 in Fig. 2, the Au1f+ on AuTi3O8− traps CH4 and delivers CH4 to be close to the Ot2− ion for C–H bond cleavage (I4 → TS3 → I5, Fig. 3). The large amount of exothermic energy released (191 kJ mol−1) can evaporate the AuCH3 unit to produce Ti3O8H− (I5 → P2, Reaction (6)). Alternatively, I5 can overcome the energy barrier (122 kJ mol−1, I5 → TS4) involving the activation of a second C–H bond by the O22− unit (I5 → TS4 → I6) to produce a neutral CH2O molecule and AuTi3O7H2− ions (I6 → TS5 → I7 → TS6 → I8 → P3, Reaction (7)). The formation of AuTi3O7H2− and CH2O is highly exothermic (ΔH0 = −261 kJ mol−1), so the resulting AuTi3O7H2− has enough internal energy to evaporate AuH and H2O (Fig. S7†) to form the product ions Ti3O7H− (Reaction (8)) and AuTi3O6− (Reaction (9)), respectively. It can be seen that Reactions (7)–(9) all involve the generation of formaldehyde (CH2O) and the DFT calculations correctly predict that Reactions (6)–(9) are all kinetically and thermodynamically favorable. Furthermore, the lower energy of TS4 (−69 kJ mol−1) than that of P2 (−46 kJ mol−1) can well rationalize the experimental branching ratios that show Reaction (6) as a minor channel.
In the reaction of AuTi3O8− with CH4, the Au1f+ cation can also deliver CH4 to be close to the O22− anion to activate the first C–H bond of methane (Fig. S9†). Subsequent transformation to form the intermediate I5 is kinetically less favorable than the reaction path of Fig. 3. A reaction path to form I7 (Fig. S10†) is slightly more favorable than the path of Fig. 3 kinetically (−45 kJ mol−1versus −38 kJ mol−1 for the critical transition states). This alternative path has a very deep potential well (209 kJ mol−1) which can hinder the further transformation of the reaction complex into separate products and leads to the formation of the association species AuTi3O8CH4−. This result is consistent with the experimental observation that molecular association is a major reaction channel (41%) for AuTi3O8− + CH4 (Fig. 1f).
Discussion
Many metal oxide clusters including homo-nuclear (MxOy±) and hetero-nuclear oxide clusters (M1x1M2x2Oy±) have been found to react with methane under thermal collision conditions.8–11,22–25 All of the reactive oxide clusters were open-shell systems with oxygen radical centers and methane activation primarily followed Reaction (1). Recently, it has been demonstrated that the PtAl2O4− cluster can activate CH4 through Reaction (2) and then the oxygen radical accepts the transferred H atom.39 Herein, the AuTi3O7− and AuTi3O8− clusters are closed-shell systems (without radical oxygen species) and they activate methane through the new mechanism, Reaction (4). Previously, a few positively charged mononuclear species26,29 with closed-shell electronic structures were shown to activate methane through Reactions (2) and (3).The observed reactivity of AuTi3O7− and AuTi3O8− with methane (Reactions (5)–(9)) can be closely related to the extraordinary properties of gold, and results from the strong relativistic effect on this element.40 The high electro-negativity of gold leads to a rather weak Au–O chemical bond (bond energy of 219 kJ mol−1)41 so that the O–Au1f+ bond can be flexible for the delivery of CH4 to be close to O2− (Fig. 2 and 3) or O22− (Fig. S9†) for C–H activation. Moreover, the analogous Au/H42 results in relatively strong Au–CH3 (232 kJ mol−1 by the TPSS functional) and Au–H (292 kJ mol−1 by the TPSS functional) bonds so that the evaporation of AuCH3 (Reactions (5) and (6)) and AuH (Reaction (8)) from the reaction complex is possible. Upon generation of the CH2O moiety in AuTi3O8− + CH4, the Au atom is bonded with the metal atom Ti (I8 of Fig. 3) and the gold atom becomes negatively charged (−0.20e), which is also a result of the relativistic effect.40,43,44
In addition to the extraordinary properties of gold, the co-participation of both Au1f+ and Ot2− ions is very important for the activation of methane (Fig. 2 and 3). The reaction paths for the C–H bond cleavage of CH4 by the single Au1f+ cation and a single Ot2− anion of AuTi3O7− have been also followed. These processes are subject to very high energy barriers (Fig. S11†). In addition, the reaction of AuTi3O7− with CH4 on the triplet potential energy surface has also been calculated. It turned out that all of the triplet reactants, intermediates, and transition states are much higher (>100 kJ mol−1) in energy than the corresponding singlet counterparts. As a result, the cooperative activation by the Au1f+ and Ot2− ions, as shown in Fig. 2, is the only mechanism of methane activation by AuTi3O7−.
To explore the excellent ability of the cooperative Au1f+ and Ot2− ions to promote methane activation by the closed-shell cluster anions of AuTi3O7− and AuTi3O8− under thermal collision conditions, variation of the geometrical structures of the reaction intermediates and the change of natural charge (Table S2†) as well as the Wiberg bond order (Table S3†) of critical atoms and chemical bonds have been analyzed for the reaction system of AuTi3O7− + CH4 (Fig. 2). During the course of C–H bond cleavage (I1 → TS1 → I2), the electron population on the CH3 group and Au atom increases (−0.11e → −0.41e for CH3 and +0.44e → +0.28e for Au) and that on the transferring H decreases (+0.18e → +0.47e), indicating that the C–H bond may be cleaved in a heterolytic manner and the reaction may follow a Lewis acid–base pair mechanism taking into account that the separated Au1f+ and Ot2− ions in AuTi3O7− can be considered as a Lewis acid and Lewis base, respectively. However, the change in the natural charges of the CH3 moiety and H is not very large (around 0.3e), additionally, the charge increase on the H is normal for a bond conversion of C–H → O–H. In contrast to the Lewis acid–base mechanism, another mechanism of the flexible switch of the roles of the two Ot atoms in [Ti3O7]2− which enables the favorable C–H activation is proposed from a bonding point of view. The reactant of AuTi3O7− can be viewed as Au+[Ti3O7]2−, in which Au+ (+0.42e) is attached to one of the Ot in [Ti3O7]2− (−1.42e) with a bond strength of 1058 kJ mol−1 and the Ot becomes a bridging-bonded oxygen (Ob, as marked in Fig. 2). During the C–H activation, such Ob releases gold to bond with the CH3 moiety and the Ob itself switches to Ot after AuCH3 evaporation (Fig. 2). At the same time, a different Ot in [Ti3O7]2− switches to Ob after attaching to H+ (+0.46e) with a much stronger bond strength of 1710 kJ mol−1 (Fig. 2). Thus, the overall increased bond strengths in the products drive the C–H activation thermodynamically. The switch of the roles of the two Ot in [Ti3O7]2− can also be evidenced by the change of the Wiberg bond order of Ti–Ot (Table S3†). The Ti1–Ot and Ti2–Ot bonds (Fig. 2) gradually switch from single/double bonds (187/167 pm) in the reactant to double/single bonds (166/187 pm) in the product. This mechanism suggests that the C–H bond may be cleaved in a homolytic manner, namely, hydrogen atom transfer (HAT).
The identification of the Lewis acid–base mechanism or HAT for AuTi3O7− + CH4 relies on the transfer mode of one electron (e−) and one proton (H+) of a H atom. HAT is characterized by the transfer of the electron and proton to a single site. In contrast, a Lewis acid–base mechanism corresponds to the transfer of the electron and proton to different acceptor sites. Such a transfer mode can be called an electron–proton transfer (EPT).45 It was proposed that a key element in the theoretical characterization of the mechanisms of proton and electron transfer is the formulation of their localized diabatic states.46 However, the electron and proton described by standard quantum mechanical methods tend to be delocalized, and the analysis of the electron or proton acceptors such as a molecular orbital or a chemical bond depends on the adopted computational level of theory.45 Consequently, it is hard to distinguish exactly the two proposed mechanisms of AuTi3O7− + CH4 at the present level of theory. More advanced methods such as multistate DFT, in which the electron and proton localized diabatic configurations can be constructed through block-localization of Kohn–Sham orbitals, should be employed to study the potential energy surfaces of the HAT and EPT, which may provide clues to recognize the mechanistic details of our reaction systems.
Conclusions
The reactions of methane with negatively charged titanium oxide clusters doped with single gold atoms, AuTi3O7− and AuTi3O8−, have been identified by mass spectrometry and quantum chemistry calculations. To the best of our knowledge, this is the first example of the thermal activation and transformation of methane by atomic cluster anions with closed-shell electronic structures. Unlike the previously reported three general mechanisms including hydrogen atom abstraction, oxidative addition, and σ-bond metathesis for methane activation by atomic clusters, the cooperation of the separated Au+ and O2− induces the cleavage of the first C–H bond of methane for AuTi3O7− and AuTi3O8− clusters. Further activation of the second C–H bond of methane on the oxygen-rich system of AuTi3O8− leads to the formation of the stable organic compound formaldehyde (CH2O). The observed unique reactivity of AuTi3O7− and AuTi3O8− toward methane results from the strong relativistic effect on the gold element. This study not only serves as an important step in understanding that closed-shell species on metal oxides can be reactive enough to facilitate thermal H–CH3 bond cleavage, but also provides molecular-level insights into the design of active sites on metal oxide supported gold catalysts to activate and transform methane.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21273247, 21325314, and 21573247), the Major Research Plan of China (No. 2013CB834603), and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA09030101).Notes and references
- R. H. Crabtree, Chem. Rev., 1995, 95, 987–1007 CrossRef CAS.
- J. H. Lunsford, Catal. Today, 2000, 63, 165–174 CrossRef CAS.
- R. A. Periana, O. Mironov, D. Taube, G. Bhalla and C. Jones, Science, 2003, 301, 814–818 CrossRef CAS PubMed.
- Q. Zhu, S. L. Wegener, C. Xie, O. Uche, M. Neurock and T. J. Marks, Nat. Chem., 2013, 5, 104–109 CrossRef CAS PubMed.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- J. Gao, Y. Zheng, J.-M. Jehng, Y. Tang, I. E. Wachs and S. G. Podkolzin, Science, 2015, 348, 686–690 CrossRef CAS PubMed.
- S. Arndt, G. Laugel, S. Levchenko, R. Horn, M. Baerns, M. Scheffler, R. Schlögl and R. Schomäcker, Catal. Rev.: Sci. Eng., 2011, 53, 424–514 CAS.
- N. Dietl, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2012, 51, 5544–5555 CrossRef CAS PubMed.
- J. Roithová and D. Schröder, Chem. Rev., 2010, 110, 1170–1211 CrossRef PubMed.
- X.-L. Ding, X.-N. Wu, Y.-X. Zhao and S.-G. He, Acc. Chem. Res., 2012, 45, 382–390 CrossRef CAS PubMed.
- G. de Petris, A. Troiani, M. Rosi, G. Angelini and O. Ursini, Chem.–Eur. J., 2009, 15, 4248–4252 CrossRef CAS PubMed.
- R. Liyanage, X.-G. Zhang and P. B. Armentrout, J. Chem. Phys., 2001, 115, 9747–9763 CrossRef CAS.
- U. Achatz, C. Berg, S. Joos, B. S. Fox, M. K. Beyer, G. Niedner-Schatteburg and V. E. Bondybey, Chem. Phys. Lett., 2000, 320, 53–58 CrossRef CAS.
- S. M. Lang, T. M. Bernhardt, R. Barnett and N. U. Landman, Angew. Chem., Int. Ed., 2010, 49, 980–983 CrossRef CAS PubMed.
- D. J. Harding, C. Kerpal, G. Meijer and A. Fielicke, Angew. Chem., Int. Ed., 2012, 51, 817–819 CrossRef CAS PubMed.
- C. Adlhart and E. Uggerud, Chem. Commun., 2006, 2581–2582 RSC.
- A. W. Castleman Jr, Catal. Lett., 2011, 141, 1243–1253 CrossRef.
- S. Yin and E. R. Bernstein, Int. J. Mass Spectrom., 2012, 321–322, 49–65 CrossRef CAS.
- R. A. J. O'Hair, Int. J. Mass Spectrom., 2015, 377, 121–129 CrossRef.
- M. Diefenbach, M. Brönstrup, M. Aschi, D. Schröder and H. Schwarz, J. Am. Chem. Soc., 1999, 121, 10614–10625 CrossRef CAS.
- C. J. Cassadyt and S. W. McElvany, J. Am. Chem. Soc., 1990, 112, 4188–4191 Search PubMed.
- H. Schwarz, Isr. J. Chem., 2014, 54, 1413–1431 CrossRef CAS.
- S. Feyel, J. Döbler, D. Schröder, J. Sauer and H. Schwarz, Angew. Chem., Int. Ed., 2006, 45, 4681–4685 CrossRef CAS PubMed.
- S. Feyel, J. Döbler, R. Höckendorf, M. K. Beyer, J. Sauer and H. Schwarz, Angew. Chem., Int. Ed., 2008, 47, 1946–1950 CrossRef CAS PubMed.
- X.-N. Wu, X.-N. Li, X.-L. Ding and S.-G. He, Angew. Chem., Int. Ed., 2013, 52, 2444–2448 CrossRef CAS PubMed.
- M. Armélin, M. Schlangen and H. Schwarz, Chem.–Eur. J., 2008, 14, 5229–5236 CrossRef PubMed.
- P. B. Armentrout and J. L. Beauchamp, Acc. Chem. Res., 1989, 22, 315–321 CrossRef CAS.
- M. Schlangen and H. Schwarz, Helv. Chim. Acta, 2008, 91, 2203–2210 CrossRef CAS.
- R. Kretschmer, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2013, 52, 6097–6101 CrossRef CAS PubMed.
- P. J. Roach, W. H. Woodward, A. W. Castleman Jr, A. C. Reber and S. N. Khanna, Science, 2009, 323, 492–495 CrossRef CAS PubMed.
- A. C. Reber, P. J. Roach, W. H. Woodward, S. N. Khanna and A. W. Castleman Jr, J. Phys. Chem. A, 2012, 116, 8085–8091 CrossRef CAS PubMed.
- W. H. Woodward, A. C. Reber, J. C. Smith, S. N. Khanna and A. W. Castleman Jr, J. Phys. Chem. C, 2013, 117, 7445–7450 CAS.
- Z. Luo, J. C. Smith, C. Berkdemir and A. W. Castleman Jr, Chem. Phys. Lett., 2013, 590, 63–68 CrossRef CAS.
- J.-H. Meng and S.-G. He, J. Phys. Chem. Lett., 2014, 5, 3890–3894 CrossRef CAS PubMed.
- Z. Yuan, Z.-Y. Li, Z.-X. Zhou, Q.-Y. Liu, Y.-X. Zhao and S.-G. He, J. Phys. Chem. C, 2014, 118, 14967–14976 CAS.
- G. Kummerlöwe and M. K. Beyer, Int. J. Mass Spectrom., 2005, 244, 84–90 CrossRef.
- J.-B. Ma, B. Xu, J.-H. Meng, X.-N. Wu, X.-L. Ding, X.-N. Li and S.-G. He, J. Am. Chem. Soc., 2013, 135, 2991–2998 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Y.-X. Zhao, Z.-Y. Li, Z. Yuan, X.-N. Li and S.-G. He, Angew. Chem., Int. Ed., 2014, 53, 9482–9486 CrossRef CAS PubMed.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed.
- J. B. Pedley and E. M. Marshall, J. Phys. Chem. Ref. Data, 1983, 12, 967–1031 CrossRef CAS.
- L. S. Wang, Phys. Chem. Chem. Phys., 2010, 12, 8694–8705 RSC.
- X.-N. Li, Z. Yuan and S.-G. He, J. Am. Chem. Soc., 2014, 136, 3617–3623 CrossRef CAS PubMed.
- M. Jansen, Chem. Soc. Rev., 2008, 37, 1826–1835 RSC.
- A. K. Harshan, T. Yu, A. V. Soudackov and S. Hammes-Schiffer, J. Am. Chem. Soc., 2015, 137, 13545–13555 CrossRef CAS PubMed.
- A. Cembran, M. R. Provorse, C. Wang, W. Wu and J. Gao, J. Chem. Theory Comput., 2012, 8, 4347–4358 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The detailed experimental and computational methods as well as additional experimental and computational results. See DOI: 10.1039/c6sc00539j |
| This journal is © The Royal Society of Chemistry 2016 |