
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence resonance energy transfer-based hybridization chain reaction for in situ visualization of tumor-related mRNA†
Jin
Huang
,
He
Wang
,
Xiaohai
Yang
,
Ke
Quan
,
Yanjing
Yang
,
Le
Ying
,
Nuli
Xie
,
Min
Ou
and
Kemin
Wang
*
State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Hunan University, Changsha, P. R. China. E-mail: kmwang@hnu.edu.cn
First published on 25th February 2016
Abstract
The ability to visualize tumor-related mRNA in situ in single cells would distinguish whether they are cancer cells or normal cells, which holds great promise for cancer diagnosis at an early stage. Fluorescence resonance energy transfer (FRET) and hybridization chain reactions (HCRs) were combined with amplified sense tumor-related mRNA (TK1 mRNA) in situ with high sensitivity in single cells and tissue sections. Using this strategy, each copy of the target mRNA can propagate a chain reaction of hybridization events between two alternating hairpins to form a nicked duplex that contains repeated FRET units, amplifying the fluorescent signal. The detection limit of 18 pM is about three orders of magnitude lower than that of a non-HCR method (such as the binary-probe-system). Meanwhile, due to the FRET strategy, complicated washing steps are not necessary and experimental time is sharply reduced. As far as we know, this is the first report of a fluorescence in situ hybridization (FISH) strategy that can simultaneously fulfil signal amplification and is wash-free. We believe that this FRET-based HCR strategy has great potential as a powerful tool in basic research and clinical diagnosis.
Introduction
Cancer, also known as a malignant tumor, is a group of diseases involving abnormal cell growth with the potential to invade other parts of the body, with several million deaths every year.1–3 The survival of cancer patients is strongly associated with the stage of the tumor at the time of diagnosis. Up to now, there is increasing evidence to suggest that cancer is a genetic disease, in which gene changes control the way cancer cells function, especially how they grow and divide.4–6 Thus, tumor-related mRNA has been widely used as a specific marker to assess whether it is a cancer cell or a normal cell.7–10 Therefore, the detection of tumor-related mRNA can provide new tools for identifying cancer cells at an early stage and holds great promise for increasing the survival rate of cancer patients.Recently, a series of bulk measurements, including quantitative polymerase chain reaction (qPCR),11,12 northern blotting,13,14 and DNA microarray,15,16 has been developed to measure specific mRNA levels following its isolation from cells. However, the detection and identification of foreign or mutated mRNA are often difficult in a clinical setting due to the low abundance of diseased cells in blood or sputum samples. Furthermore, in population-based assays that analyze the content of many cells, molecules in rare cells escape detection. Also, such assays provide no information concerning which of the molecules detected originate from which cells. Expression in single cells can vary substantially from the mean expression detected in a heterogeneous cell population.
Apparently, the shift from population to single-cell studies can address the intrinsic heterogeneity of cell population, which is masked in bulk measurements. Fluorescence in situ hybridization (FISH) is a powerful and widely used technique which analyzes the expression pattern of a gene of interest in situ at single-cell resolution.17–21 Fixed cells retain much of their structural organization and provide a controlled setting for visualizing the subcellular distribution of mRNA or other targets using FISH. FISH images are composed of points of light with variable intensities resulting either from hybridization or from background fluorescent noise, where cells or tissue sections are fixed and permeabilized to increase the probe delivery efficiency and unbound probes are removed by washing. Nevertheless, the traditional FISH method, composed of fixation, hybridization, washing and visualization, is restricted by two problems. First, only one signal probe can be hybridized to the target mRNA molecule, thereby yielding inadequate fluorescence to quantify the mRNA expression in individual cells, especially in cells with very low amounts or small changes in the expression levels of mRNA that are required to be evaluated. Second, with in situ hybridization, complicated and time-consuming washing steps are subsequently required to remove unbound probes prior to imaging of fluorophores, that either label the probes directly or are localized in the vicinity of probes during a subsequent amplification step. Recently, PCR22 and rolling circle amplification (RCA)23 based FISH methods have been developed for in situ sensitive detection of mRNA expression. Yet, these methods require enzymes and complicated operations. Very recently, Pierce et al. developed a hybridization chain reaction (HCR)-based FISH method, where a polymerization reaction of two types of fluorescently labeled hairpin monomer was controllably catalysed, then the fluorescent signal associated with a given mRNA expression was amplified and could be imaged readily using fluorescence microscopy.24,25 However, it needs repeated washing steps and a relatively long time to accomplish the experiment. Thus, a better strategy to improve the FISH technique would be the combination of signal amplification and a wash-free strategy into one detection system. The development of simultaneous signal amplification and a wash-free strategy for in situ visualization of mRNA expression patterns has been largely unexplored.
Results and discussion
Herein, we present a fluorescence resonance energy transfer (FRET)-based HCR for in situ visualization of tumor-related mRNA, where the in situ target mRNA fragment acts as a reaction initiator and activates a HCR, constructing multiple DNA repeating units, each of which generates a FRET signal. As illustrated in Scheme 1, this strategy is based on the use of three programmable DNA hairpins (H1, H2 and H3, see Table S1 in the ESI†). H1 is designed to contain a sequence complementary to the target TK1 mRNA. H2 and H3 are labeled with a fluorescence acceptor (TAMRA) and donor (FAM) at the appropriate positions, respectively. In the presence of target, H1 is opened by the target (step 1). The opened H1 pairs with the sticky end of H2, which undergoes an unbiased strand-displacement interaction to open the hairpin (step 2). The newly exposed H2 nucleates at the sticky end of H3 and opens the hairpin to expose a sticky end on H3, which can open H2 again (step 3). In this way, each copy of the target can propagate a chain reaction of hybridization events between alternating H2 and H3 hairpins to form a nicked duplex (repeat steps 2 and 3). Each H2–H3 pair hybridization event leads to FRET signal generation, amplifying the fluorescence signal. Upon irradiation of the sample at the donor excitation wavelength, the energy transfer can be detected as a decrease in the fluorescence emission of the donor and an enhancement in the emission intensity of the acceptor fluorophore. The ratio of the last to the first emission intensities gives a relative FRET measure, where higher ratios indicate higher energy transfer efficiency.26,27 However, the hairpins stay metastable when no target is present, due to the closed formation of the hairpin stems. A FRET-based approach has been implemented in our system to avoid multiple washing steps and prevent false positive signals that can be observed in quencher/dye systems as a result of probe accumulation or degradation. As far as we know, this is the first report of such a FISH strategy that can simultaneously satisfy signal amplification and is wash-free.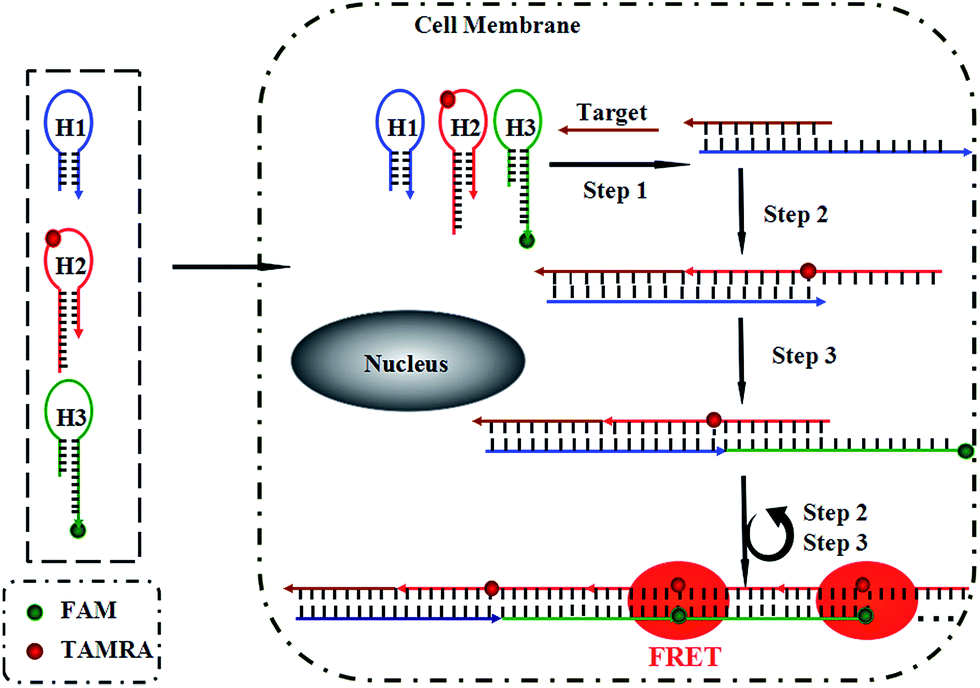 | ||
| Scheme 1 Working principle for the in situ detection of TK1 mRNA using the FRET-based HCR method. | ||
To demonstrate the feasibility of this principle using in-tube experiments, we designed three DNA hairpins (H1, H2 and H3) to make sure that the hairpins exhibit metastability in the absence of targets but the introduction of targets can trigger HCR (for details see Table S1 in the ESI†). The resulting electrophoresis illustrates the three hairpins meet the above conditions that only the presence of target and the three hairpins can trigger the polymerization reaction (see Fig. S1 in the ESI†). As expected, various lengths of large molecular weight products appear as a bright band in this case, which indicates that it is feasible to use the three-hairpin-system as an amplification technique. To further confirm the switching of the FRET signaling of the detection system with the addition of targets, we detected the fluorescence profile before and after adding target. The results showed that the acceptor (TAMRA) fluorescence increased and the donor (FAM) fluorescence decreased (see Fig. S2 in the ESI†), which suggests that the adding of targets can switch the FRET signal. To test the in vitro target sensing behavior of the FRET-based HCR detection system, we monitored the kinetics toward 10 nM target DNA. The result showed that the FRET signals gradually increase with time until 200 min (see Fig. S3 in the ESI†). We subsequently examined the response to varying concentrations of synthetic DNA targets, instead of mRNA, at 37 °C. The results in Fig. 1 illustrate excellent FRET signal change according to different concentrations of the targets. It suggests that the acceptor-to-donor ratio is dependent upon the target concentrations. Fluorescence intensities of the FRET-based HCR assay increases with increasing concentration of targets and an estimated detection limit is about 18 pM (three times the standard deviation in the blank solution).
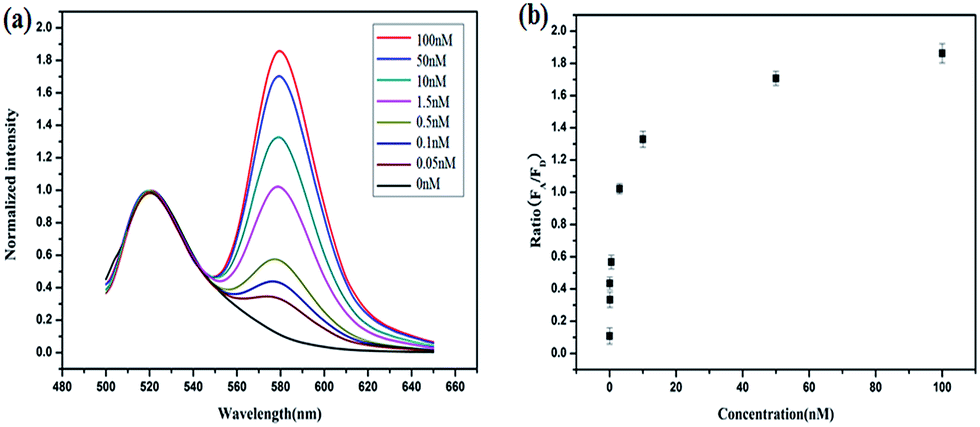 | ||
| Fig. 1 (a) Fluorescence profile of the FRET-based HCR method responding to different synthetic DNA targets in vitro at 37 °C. (b) A plot of acceptor-to-donor ratio as a function of target concentrations. | ||
To demonstrate the feasibility of the strategy in the cell system, we chose TK1 mRNA as a tumor-related target mRNA, which is associated with cell division and is proposed to be a marker for tumor growth.28,29 Our previous studies revealed that TK1 mRNA was commonly overexpressed in tumor cell lines (HepG2 and MCF-7) compared with the normal cell control (L02).30,31 In this work, HepG2 (a human liver hepatocellular carcinoma cell line) and MCF-7 (a breast cancer cell line) were selected as the target cells. And L02 (a normal human hepatocyte cell line) was selected as the control cell. Herein, the FRET-based HCR strategy was used to image TK1 mRNA in the three cell lines. Fig. 2 shows that strong FRET signals (TAMRA fluorescence) for TK1 mRNA in HepG2 and MCF-7 are observed, and almost no FRET signal in L02 is detected. The results are also consistent with the results of another conventional technique qPCR (see Fig. S4 in the ESI†) and further indicate that the signals of the FRET-based HCR strategy correlate very well with the levels of mRNA expression. Moreover, the results suggest that the FRET-based HCR strategy possesses high specificity.
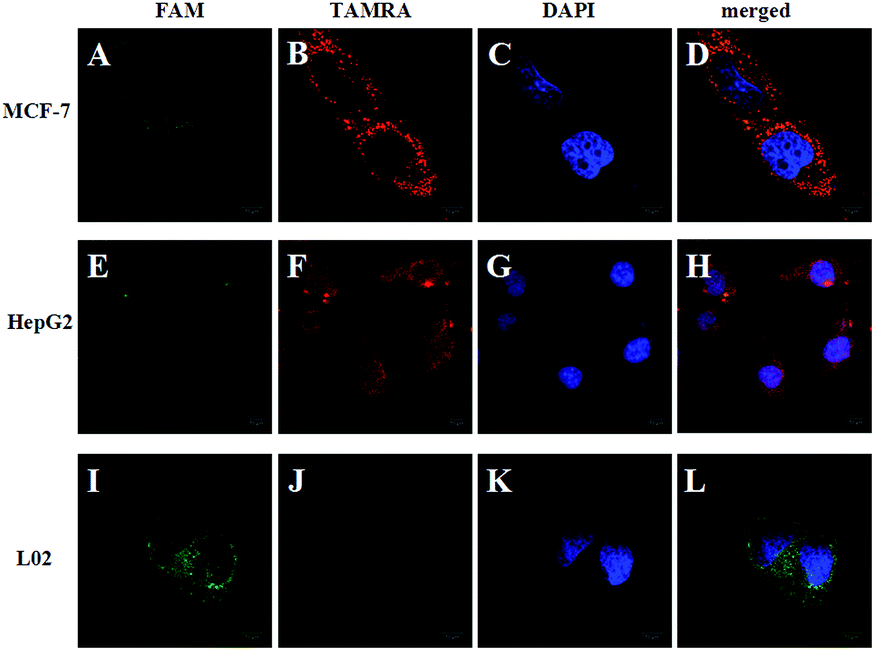 | ||
| Fig. 2 Fluorescence images of TK1 mRNA in MCF-7, HepG2 and L02 cells using the FRET-based HCR method. The green fluorescence represents FAM (A, E and I), the red fluorescence represents TAMRA (B, F and J), and the blue fluorescence represents DAPI stained cell nuclei (C, G and K). The merged image represents an overlay of the FAM, TAMRA and DAPI signals (D, H and L). The excitation wavelength is 488 nm, and the images were collected in the ranges of 505–560 nm (FAM) and >560 nm (TAMRA). Scale bar: 10 μm. | ||
To further prove the mechanism of the FRET-based HCR strategy, we designed four control experiments. Firstly, only two hairpins (H1 and H2, H1 and H3, H2 and H3) were incubated with the fixed MCF-7 cells under the optimized conditions. As shown in Fig. 3, we notice that no FRET signal (TAMRA fluorescence) is detected in this case (A–C in Fig. 3), whereas the sample with three hairpin probes shows a highly intense FRET signal (E in Fig. 3), verifying each of the three hairpins is strongly necessary to anchor these signal probes on the position of target TK1 mRNA. Secondly, a superfluous inhibitor, a single strand specific DNA, which could block the target TK1 mRNA binding location (see Table S1 in the ESI†), was used to pretreat the fixed cells before FRET-based HCR detection. As expected, we did not observe any distinct TAMRA fluorescence signals in this case (D in Fig. 3), suggesting that the FRET signals were specific target mRNA-dependent, rather than induced by nonspecific binding. From the above four control experiments, we conclude the fact that the target RNA recognizes H1 and the newly exposed sequence of H1 triggers HCR between alternating H2 and H3 to form a long nicked double-helix. In this state, numerous FAMs and TAMRAs are brought into close proximity to produce bright FRET signals.
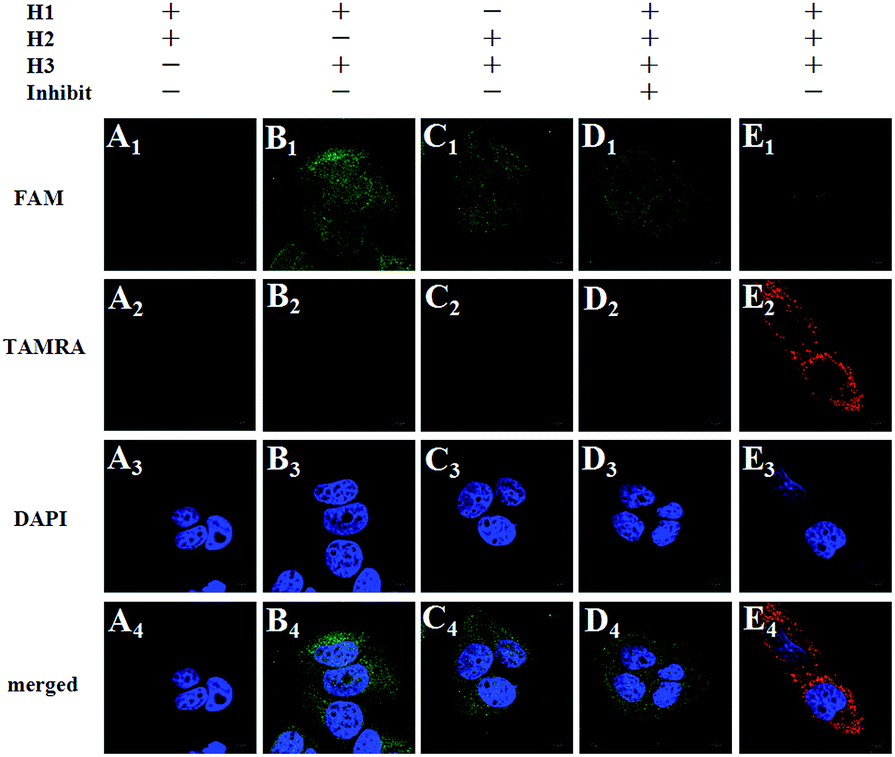 | ||
| Fig. 3 Control experiments: fluorescence images of TK1 mRNA in MCF-7 cells under different conditions. Cells are incubated with H1 + H2 (A1–A4), H1 + H3 (B1–B4), H2 + H3 (C1–C4), H1 + H2 + H3 + inhibitor (D1–D4), and H1 + H2 + H3 (E1–E4), respectively. Scale bar: 10 μm. | ||
To demonstrate the signal amplification effect of the FRET-based HCR method, we arranged the FRET-based binary probes for in situ mRNA detection as a control (see Table S1 in the ESI†). In the FRET-based binary probes, one part of the probe is conjugated with a donor while another contains an acceptor fluorophore.32,33 When hybridized to the target, the oligonucleotides bring the two dyes in close proximity, thus enabling FRET. Because one target molecule induces only one FRET signal, this method does not involve signal amplification and is used as a reference method to compare the sensitivity with the FRET-based HCR method. For in-tube testing, fluorescence intensities of the FRET-based binary probe assay also increased with increasing concentration of target, a result obviously produced by the hybridization of the target with the probes (see Fig. S5 in the ESI†). An estimated detection limit is about 9.8 nM (three times the standard deviation in the blank solution), the sensitivity of which is about 3 orders of magnitude lower than that of the FRET-based HCR method. The reason for the signal amplification is changing the “one target–one FRET” signal to “one target–multiple FRET” signals.
When comparing the in-cell models, as shown in Fig. 4, the FRET-based HCR method exhibits highly intense TAMRA fluorescence signals in MCF-7 cell lines, whereas relatively weak signals could be observed by the FRET-based binary probe method under the same conditions. Furthermore, flow cytometry reveals that the MCF-7 cell population treated with the FRET-based HCR probes is more fluorescent than the population treated with the FRET-based binary probes (see Fig. S6 in the ESI†). These flow cytometry experiments are in excellent agreement with confocal imaging and demonstrate the signal amplification effect in the cell system. These in-tube and in-cell experiments clearly demonstrate that the FRET-based HCR technique can indeed enhance the sensitivity of mRNA detection and provide high imaging contrast, which is of significant importance in the detection of trace levels of a specific mRNA as well as the determination of small changes in the expression levels of mRNA.
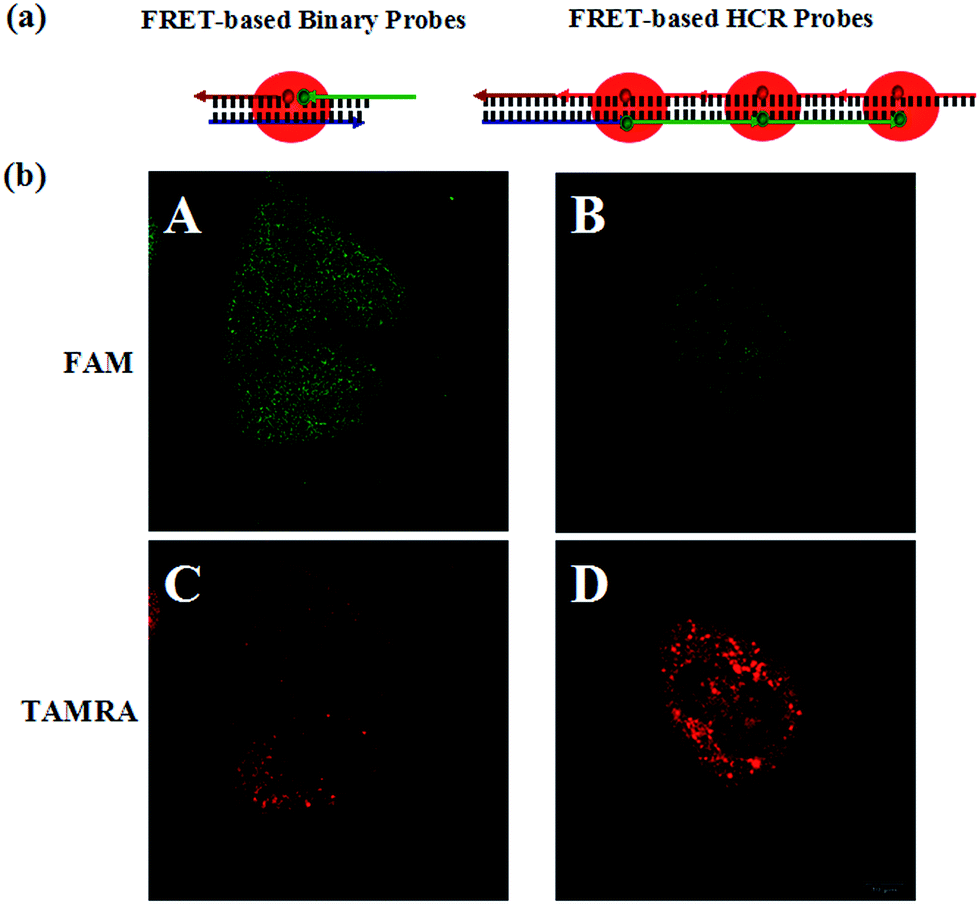 | ||
| Fig. 4 (a) Schematic of target triggered FRET-based binary probes and FRET-based HCR probes, respectively. (b) Fluorescence images of TK1 mRNA in MCF-7 cells (A and C) using the FRET-based binary probes and the FRET-based HCR probes (B and D), respectively. The excitation wavelength is 488 nm, and the images are collected in the ranges of 505–560 nm (FAM) and >560 nm (TAMRA). Scale bar: 10 μm. | ||
The ability of the FRET-based HCR method to identify the changes in the TK1 mRNA expression level in MCF-7 cells was then studied. It was reported that tamoxifen induced the down-regulation of TK1 mRNA expression and β-estradiol induced the up-regulation of TK1 mRNA expression.13 The MCF-7 cells were separated into three groups in parallel. One group was treated with tamoxifen to decrease the TK1 mRNA expression, and another was treated with β-estradiol to increase the TK1 mRNA expression. An untreated group served as a control. The FRET-based HCR method was subsequently applied to image the TK1 mRNA in cells of the three groups. As shown in Fig. 5, the TAMRA fluorescence intensity is lower in the tamoxifen-treated cells and higher in the β-estradiol-treated cells compared to that in the untreated cells. These results indicate that the FRET-based HCR method is capable of sensing changes in gene expression levels in cancer cells.
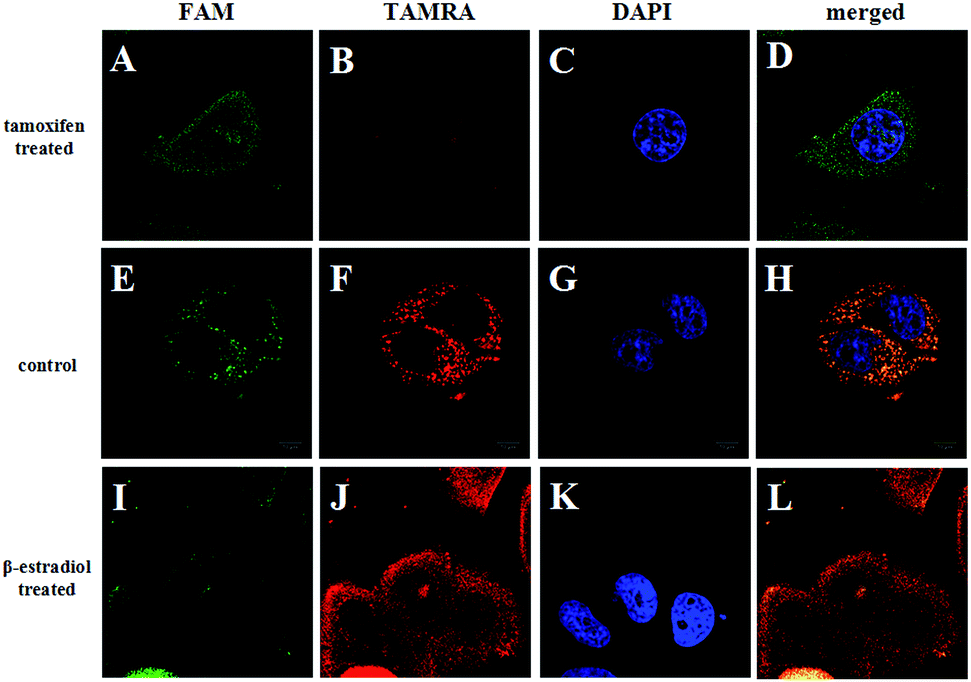 | ||
| Fig. 5 Fluorescence images of TK1 mRNA in MCF-7 cells before (E–H) and after treating with tamoxifen (A–D) and β-estradiol (I–L), respectively. Scale bar: 10 μm. | ||
Cancer histopathology is currently the preferred method for detecting microscopic anatomical changes in tissue sections, making the discovery of cancer biomarkers critical for early diagnosis and treatment.34 Tissue immunostaining is critically important in clinical applications. Alternatively, tumor-related mRNA is also an ideal biomarker for clinical applications in cancer diagnosis and imaging. In order to demonstrate the ability of the FRET-based HCR technique applied at the tissue section level, we chose tumor-related TK1 mRNA as a target. As shown in Fig. 6, for liver cancer tissue sections that highly expressed TK1 mRNA, the results exhibit highly intense cytoplasmic fluorescent signals, and for liver normal tissue sections that expressed TK1 mRNA in a very low amount, almost no signal could be detected under the same conditions. These results were consistent with the results of the cell experiments, which validated the ability of the FRET-based HCR technique for specific in situ detection of mRNA at the tissue section level.
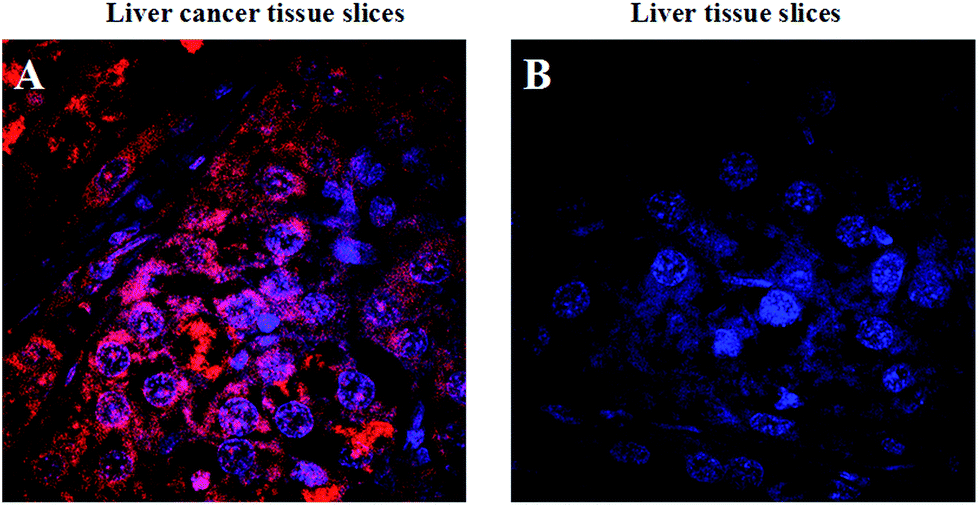 | ||
| Fig. 6 Fluorescence images of TK1 mRNA in liver cancer tissue slices and liver tissue slices, respectively. The red fluorescence represents TAMRA and the blue fluorescence represents DAPI stained cell nuclei. | ||
Conclusions
In summary, we have developed a FRET-based HCR strategy for highly sensitive and selective in situ visualization of tumor-related mRNA expression in single cells and tissue sections. It has many advantages: (1) by taking advantage of the HCR technique, highly sensitive and selective detection of mRNA is achieved; (2) HCR is an excellent isothermal signal-amplification technique which does not require an enzyme; (3) by using the FRET strategy, complicated washing steps are not necessary and experimental time is sharply shortened; (4) the signal generation based on FRET can minimize the effect of system fluctuations and avoid false positive signals that can be observed in quencher/dye systems as a result of probe accumulation or degradation. We believe that the FRET-based HCR strategy has great potential as a powerful tool for basic research and clinical diagnosis.Experimental section
Chemicals and materials
The oligonucleotides used in this work (Table S1†) were synthesized by Sangon Biological Co. Ltd (Shanghai, China). Formamide was purchased from Sinopharm Chemical Reagent Co. Ltd (Beijing, China). 4% paraformaldehyde (PFA) and sodium chloride/sodium citrate (SSC, 20×) buffer were from Dingguo Biotechnology Co. Ltd (Beijing, China). 4′,6-Diamidino-2-phenylindole (DAPI) was from Gen-view Scientific Inc. (USA). Tamoxifen and β-estradiol were from Sigma Aldrich Chemical Co. Ltd (St. Louis, MO). Tissue slices were from Huashun Biotechnology Co. Ltd (Wuhan, China). All other chemicals were of analytical grade. All solutions were prepared using ultrapure water, which was obtained through a Millipore Milli-Q water purification system (Billerica, MA, USA) with an electric resistance > 18.3 MΩ.Fluorescence experiments
In order to study the relationship between FRET-based HCR probes reaction time and target concentration, the 100 nM signal probes (H1, H2 and H3) were reacted with increasing concentrations of the DNA targets (0, 0.05, 0.1, 0.5, 1.5, 10, 50, 100 nM) for 4 h at 37 °C. In the process of reacting, the fluorescence was monitored at appropriate excitation wavelengths. The fluorescence was recorded on a F-7000 fluorescence spectrometer exciting at 488 nm and measuring emission from 510 nm to 650 nm in 1 nm increments. For FRET fluorescence detection, the excitation and emission wavelength are 485 nm, and 510–650 nm, respectively. For TAMRA fluorescence detection, the excitation and emission wavelengths are 543 nm, and 560–630 nm, respectively (the average and standard deviation were obtained by 3 parallel experiments in each trial). Selective experiments conducted via this reaction were performed with three mismatched targets. In order to study the relationship between the FRET-based binary probe reaction time and target concentration, the 100 nM signal probes (BP1 and BP2) reacted with increasing concentrations of the DNA targets (0, 10, 25, 35, 45, 50, 65, 75, 100 nM) for 4 h at 37 °C. In the process of reacting, the fluorescence was monitored at appropriate excitation wavelengths. The fluorescence was recorded on a F-7000 fluorescence spectrometer exciting at 488 nm and measuring emission from 510 nm to 650 nm in 1 nm increments. For FRET fluorescence detection, the excitation and emission wavelengths are 485 nm, and 510–650 nm, respectively. The average and standard deviation were obtained by 3 parallel experiments in each trial.Cell culture and fixation
MCF-7 cells (a breast cancer cell line), L02 cells (human hepatocyte cell line) and HepG2 cells (human liver hepatocellular carcinoma cell line) were cultured in a RPMI 1640 medium supplemented with 15% fetal calf serum, 100 μg mL−1 of streptomycin, and 100 units per mL of penicillin. Cells were all cultured at 37 °C in a humidified incubator containing 5% CO2. MCF-7, L02 and HepG2 cells were seeded on confocal laser culture slides and cultured in the culture medium for 24 h. The cells were first washed twice with phosphate buffered saline (PBS, pH 7.4, calcium and magnesium free) and then fixed on the slides with PBS containing 4% paraformaldehyde (PFA) for 15 min at room temperature followed by two PBS washes.In situ detection in fixed cells and tissue slices
The slides with fixed cells were first incubated in a humidified 37 °C incubator for 4 h with 190 μL of hybridization solution containing 30 μL of 5 μM H1, 42 μL of 3 μM TAMRA-labeled H2, 42 μL of 3 μM FAM-labeled H3, 20% formamide and 2× SSC. Before imaging, the slides were stained with DAPI. If using the FRET-based binary probe procedure, the slides with fixed cells were first incubated in a humidified 37 °C incubator for 4 h with 140 μL of hybridization solution containing 3 μM FAM-labeled BP1 and 3 μM TAMRA-labeled BP2, 2× SSC, and 20% formamide. Before imaging, the slides were stained with DAPI. The tissue slices were first incubated in a humidified 37 °C incubator for 4 h with 1140 μL of hybridization solution containing 180 μL of 5 μM H1, 252 μL of 3 μM TAMRA-labeled H2, 252 μL of 3 μM FAM-labeled H3, 20% formamide and 2× SSC. Before imaging, the tissue slices were stained with DAPI.Inhibited experiment and drug treatment
The slides with fixed cells were first incubated in a humidified 37 °C incubator for 1.5 h with 50 μL hybridization solution containing 30 μL of 5 μM inhibitor, 10 μL of formamide, and 10 μL of 10× SSC. After pouring out the liquid, the slides with fixed cells were incubated in a humidified 37 °C incubator for 4 h with 190 μL of hybridization solution containing 30 μL of 5 μM H1, 42 μL of 3 μM TAMRA-labeled H2, 42 μL of 3 μM FAM-labeled H3, 20% formamide and 2× SSC. Before imaging, the slides were stained with DAPI solution. For the drug treatment, MCF-7 cells were seeded on glass slides overnight. Then the slides were first incubated with 5 μM tamoxifen or 10−2 μM β-estradiol in a humidified 37 °C and 5% CO2 incubator for 24 h and followed the above experimental steps.Confocal fluorescence imaging system
Fluorescence imaging was performed using a confocal laser scanning fluorescence microscope setup consisting of an Olympus IX-70 inverted microscope with an Olympus FluoView 500 confocal scanning system. The cellular images were acquired using a 120× and 200× objective. An Ar+ laser (488 nm) was used as an excitation source for a FAM-labeled detection probe, and a 515 nm (±10 nm) bandpass filter was used for fluorescence detection. The DAPI dye was excited with a 405 nm laser line and detected with a 460 nm (±10 nm) bandpass filter. The fluorescence images were presented after processing by Image Proplus 6.0 software and Image J version 1.38x software.qRT-PCR
Total cellular RNA was extracted from MCF-7 cells, L02 cells or HepG2 cells using Trizol reagent S5 (Sangon Co. Ltd., Shanghai, China) according to the indicated protocol. The cDNA samples were prepared by using the reverse transcription (RT) reaction with an AMV First Strand cDNA Synthesis Kit (BBI, Toronto, Canada). qPCR analysis of mRNA was performed with SG Fast qPCR Master Mix (2X) (BBI), according to the indicated protocol on an LightCycler 480 Software Setup (Roche). The primers used in this experiment were shown in Table S1.† We evaluated all of the data with respect to the mRNA expression by normalizing to the expression of GAPDH and using the 2−ΔΔCt method.Flow cytometry
For flow cytometry, cells were treated as described above, and then were analyzed by flow cytometry on a Beckman Gallios (Beckman Coulter, Brea, CA, USA) instrument using fluorescent channel FL-4 (488 nm excitation, 580 nm emission). Reported fluorescence intensities represent the median of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 analyzed cells. Background fluorescence detected in cells without staining was subtracted.
000 analyzed cells. Background fluorescence detected in cells without staining was subtracted.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21190044 and 21205032), Hunan Provincial Natural Science Foundation of China (13JJ4032), the Foundation for Innovative Research Groups of NSFC (21521063), and the Fundamental Research Funds for the Central Universities.Notes and references
- B. Taback, A. D. Chan, C. T. Kuo, P. J. Bostick, H. J. Wang, A. E. Giuliao and D. S. B. Hoon, Cancer Res., 2001, 61, 8845–8850 CAS.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69–90 CrossRef PubMed.
- H. D. Schwarzenbach, S. B. Hoon and K. Pantel, Nat. Rev. Cancer, 2011, 11, 426–437 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinherg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed.
- K. L. Nathanson, R. Wooster, B. L. Weber and K. N. Nathanson, Nat. Med., 2001, 7, 552–556 CrossRef CAS PubMed.
- X. H. Peng, Z. H. Cao, J. T. Xia, G. W. Carlson, M. M. Lewis, W. C. Wood and L. Yang, Cancer Res., 2005, 65, 1909–1917 CrossRef CAS PubMed.
- P. J. Santangelo, B. Nix, A. Tsourkas and G. Bao, Nucleic Acids Res., 2004, 32, e57 CrossRef PubMed.
- D. S. Santangelo, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef PubMed.
- N. Li, C. Chang, W. Pan and B. Tang, Angew. Chem., Int. Ed., 2012, 51, 7426–7430 CrossRef CAS PubMed.
- W. Pan, T. Zhang, H. Yang, W. Diao, N. Li and B. Tang, Anal. Chem., 2013, 85, 10581–10588 CrossRef CAS PubMed.
- T. Nolan, R. E. Hands and S. A. Bustin, Nat. Protoc., 2006, 1, 1559–1582 CrossRef CAS PubMed.
- H. D. VanGuilder, K. E. Vrana and W. M. Freeman, BioTechniques, 2008, 44, 619–626 CrossRef CAS PubMed.
- J. C. Alwine, D. J. Kemp, B. A. Parker, J. Reiser, J. Renart, G. R. Stark and M. G. Wahl, Methods Enzymol., 1979, 68, 220–242 CAS.
- G. S. Pall, C. Codony-Servat, C. J. Byrne, L. Ritchie and A. Hamilton, Nucleic Acids Res., 2007, 35, e60 CrossRef PubMed.
- J. Couzin, Science, 2006, 313, 1559a–1563 CrossRef PubMed.
- J. J. Chen, Pharmacogenomics, 2007, 8, 473–482 CrossRef CAS PubMed.
- A. M. Femino, F. S. Fay, K. Fogarty and R. H. Singer, Science, 1998, 280, 585–590 CrossRef CAS PubMed.
- W. P. Kloosterman, E. Wienholds, E. de Bruijn, S. Kauppinen and R. H. Plasterk, Nat. Methods, 2006, 3, 27–29 CrossRef CAS PubMed.
- A. N. Silahtaroglu, D. Nolting, L. Dyrskjot, E. Berezikov, M. Moller, N. Tommerup and S. Kauppinen, Nat. Protoc., 2007, 2, 2520–2528 CrossRef CAS PubMed.
- R. Pinaud, C. V. Mello, T. A. Velho, R. D. Wynne and L. A. Tremere, Nat. Protoc., 2008, 3, 1370–1379 CrossRef CAS PubMed.
- A. Lyubimova, S. Itzkovita, J. P. Junker, Z. P. Fan, X. Wu and A. Oudenaarden, Nat. Protoc., 2013, 8, 1743–1758 CrossRef PubMed.
- B. K. Patterson, M. Till, P. Otto, C. Goolsby, M. R. Furtado, L. J. McBride and S. M. Wolinsky, Science, 1993, 260, 976–979 CAS.
- C. Larsson, I. Grundberg, O. Soderberg and M. Nilsson, Nat. Methods, 2010, 7, 395–397 CrossRef CAS PubMed.
- H. M. T. Choi, J. Chang, L. A. Trinh, J. Padilla, S. E. Fraser and N. A. Pierce, Nat. Biotechnol., 2010, 28, 1208–1212 CrossRef CAS PubMed.
- H. M. T. Choi, V. A. Beck and N. A. Pierce, ACS Nano, 2014, 8, 4284–4294 CrossRef CAS PubMed.
- Z. Cheglakov, T. M. Cronin, C. He and Y. Weizmann, J. Am. Chem. Soc., 2015, 137, 6116–6119 CrossRef CAS PubMed.
- Z. Wu, G. Liu, X. Yang and J. Jiang, J. Am. Chem. Soc., 2015, 137, 6829–6836 CrossRef CAS PubMed.
- P. Broet, S. Romain, A. Daver, G. Ricolleau, V. Quillien, A. Rallet, B. Asselain, P. M. Martin and F. Spyratos, J. Clin. Oncol., 2001, 19, 2778–2787 CAS.
- C. C. Chen, T. W. Chang, F. M. Chen, M. F. Hou, S. Y. Hung, I. W. Chong, S. C. Lee, T. H. Zhou and S. R. Lin, Oncology, 2006, 70, 438–446 CrossRef CAS PubMed.
- Y. Yang, J. Huang, X. Yang, K. Quan, H. Wang, L. Ying, N. Xie, M. Ou and K. Wang, J. Am. Chem. Soc., 2015, 137, 8340–8343 CrossRef CAS PubMed.
- J. Huang, H. Wang, X. Yang, Y. Yang, K. Quan, L. Ying, N. Xie, M. Ou and K. Wang, Chem. Commun., 2016, 52, 370373 Search PubMed.
- R. Cardullo, S. Agrawal, C. Flores, P. Zamecnik and D. Wolf, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 8790–8794 CrossRef CAS.
- J. Mergny, A. Boutorine, T. Garestier, F. Belloc, M. Rougee, N. Bulychev, A. Koshkin, J. Bourson, A. Lebedev, B. Valeur, N. Thuong and C. Helene, Nucleic Acids Res., 1994, 22, 920–928 CrossRef CAS PubMed.
- Y. Pu, Z. Liu, Y. Lu, P. Yuan, J. Liu, B. Yu, G. Wang, C. Yang, H. Liu and W. Tan, Anal. Chem., 2015, 87, 1919–1924 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc00377j |
| This journal is © The Royal Society of Chemistry 2016 |