
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand enhanced upconversion of near-infrared photons with nanocrystal light absorbers†
Zhiyuan
Huang‡
a,
Duane E.
Simpson‡
a,
Melika
Mahboub
b,
Xin
Li
a and
Ming L.
Tang
*a
aDepartment of Chemistry, University of California, Riverside, 900 University Ave., Riverside, CA 92521, USA. E-mail: mltang@ucr.edu
bDepartment of Materials Science & Engineering, University of California, Riverside, 900 University Ave., Riverside, CA 92521, USA
First published on 4th March 2016
Abstract
We designed and synthesized a tetracene derivative 4-(tetracen-5-yl)benzoic acid (CPT) as a transmitter ligand used in PbS/PbSe nanocrystal (NC) sensitized upconversion of near infrared (NIR) photons. Under optimal conditions, comparing CPT functionalized NCs with unfunctionalized NCs as sensitizers, the upconversion quantum yield (QY) was enhanced 81 times for 2.9 nm PbS NCs from 0.021% to 1.7%, and 11 times for 2.5 nm PbSe NCs from 0.20% to 2.1%. The surface density of CPT controls the solubility of functionalized NCs and the upconversion QY. By increasing the concentration of CPT in the ligand exchange solution, the number of CPT ligand per NC increases. The upconversion QY is maximized at a transmitter density of 1.2 nm−2 for 2.9 nm PbS, and 0.32 nm−2 for 2.5 nm PbSe. Additional transmitter ligands inhibit photon upconversion due to triplet–triplet annihilation (TTA) between two neighboring CPT molecules on the NC surface. 2.1% is the highest reported QY for TTA-based photon upconversion in the NIR with the use of earth-abundant materials.
Introduction
Multi-excitonic processes can be harnessed to reorganize the energy contained in light in order to improve the performance of photovoltaic devices or photocatalysts.1 Reshaping the solar spectrum to match the optical properties of common semiconductors will allow the efficient use of all incident light. While many efforts e.g. hot carrier devices,2 intermediate band3 or multi-exciton generation solar cells,4 offer a route to manipulating incoming photons, the conversion of low energy near-infrared (NIR) photons to higher energy photons is particularly appealing, especially when considering NIR radiation comprises 53% of the solar spectrum.The upconversion of NIR photons at the solar flux has not been demonstrated. If this formidable challenge is met, sub-bandgap photons that are currently not absorbed by common semiconductors can be utilized. Photon upconversion is predicted to increase the power conversion efficiency of a single p–n junction silicon solar cell from 28% to 43%,5 beyond the Shockley–Queisser limit.6 Currently, the upconversion of incident photons at power densities commensurate with the solar flux has only been demonstrated for the conversion of green to violet light, via a triplet–triplet annihilation (TTA) based mechanism.7 This is because other upconverting platforms, like the lanthanides8 or the chromophores for multi-photon absorption9 (used in bioimaging) require high excitation densities for appreciable efficiency. TTA-based photon upconversion can be efficient when molecular10 or nanocrystal11 (NC) light absorbers are used to sensitize triplet states on molecules. For molecular sensitized upconversion, triplets are created via intersystem crossing. In NC sensitized upconversion, triplet energy transfer was observed from NC to molecular triplet states11 and later confirmed with transient absorption spectroscopy.12 Two triplets can encounter each other and undergo TTA to emit a high-energy photon. Internal upconversion quantum yields (QYs) as high as 35–36%13 and 10%11 have been reported for the upconversion of green to violet light with palladium porphyrins and CdSe NCs as sensitizers respectively. However, in terms of harvesting NIR photons, molecular sensitizers that absorb strongly in the NIR generally have low fluorescence QYs due to strong internal conversion,14 as predicted by the energy gap law. In contrast, the size, shape and material dependent optical properties of NCs make them ideal as light absorbers for photon upconversion, and it has been recently demonstrated that PbS or PbSe NCs are able to serve as NIR sensitizers in solution11 and thin film.15
Here, we report that specially designed tetracene-based transmitter ligands can vastly increase the upconversion QY of NIR photons in a hybrid NC-molecular platform. Using the tetracene derivative 4-(tetracen-5-yl)benzoic acid or CPT (Fig. 1a), the upconversion QY increases from 0.021% (as synthesized NCs) to 1.7% (for NCs functionalized with CPT) using PbS NCs as the light absorbers, or sensitizers, and rubrene, as the light emitter, or annihilator. The corresponding increase in upconversion QY for PbSe NCs is 0.20% to 2.13%. For the first time, we demonstrate the potential of transmitter ligands to enhance the upconversion of NIR photons, with an 81-fold and 11-fold enhancement for PbS and PbSe respectively. This rationally designed transmitter ligand addresses the problem of poor energy transfer in the original NC-rubrene platform,11 and shows a clear path towards obtaining high QYs for the upconversion of NIR light, unhindered by the limitations of molecular light absorbers. The upconversion QY obtained here is the highest reported in the literature for TTA-based photon upconversion achieved without using precious metals.
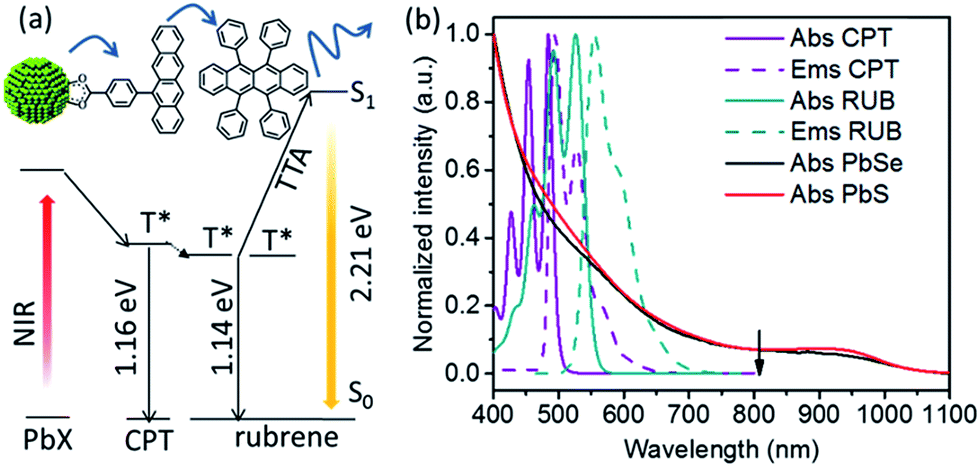 | ||
| Fig. 1 (a) Schematic of energy transfer during upconversion in this hybrid system with PbX (X = S, Se) as sensitizer, CPT as transmitter and rubrene as annihilator. (b) Absorption and emission spectra of CPT (purple), rubrene (dark cyan), 2.9 nm PbS (red) and 2.5 nm PbSe (black) in toluene at room temperature, with excitation wavelength (808 nm) indicated by the black arrow. | ||
Results and discussion
The components of this hybrid photon upconversion system and their optical properties are shown in Fig. 1. The first step in this upconversion scheme occurs when PbX NCs absorb a NIR photon (red arrow). Triplet energy transfer (TET) is enhanced in the presence of CPT directly anchored on the NC surface. TET subsequently occurs between CPT and rubrene in solution. Two rubrene molecules then undergo TTA to emit a visible photon at 570 nm (yellow arrow). Here, a CPT scaffold is chosen as the transmitter because its T1 energy level is a little larger (1.16 eV, estimated from triplet energy of 5-phenyltetracene16) than that of rubrene,17 thus forming a cascade for directional energy transfer. The absorption and emission spectra of the PbX NCs, CPT, and the rubrene annihilator are shown in Fig. 1b. CPT has a fluorescence QY of 0.53 and an extinction coefficient of 9340 M−1 cm−1 at its absorption maxima of 484 nm (Table S1†). The synthesis of CPT is shown in Scheme 1. 5-Bromotetracene18 was used in a palladium catalyzed Suzuki cross-coupling reaction to give the esterified analog of CPT. Deprotection in KOH and repeated recrystallization in toluene/THF afforded CPT. Ligand exchange was performed at room temperature in solution as outlined in the ESI.†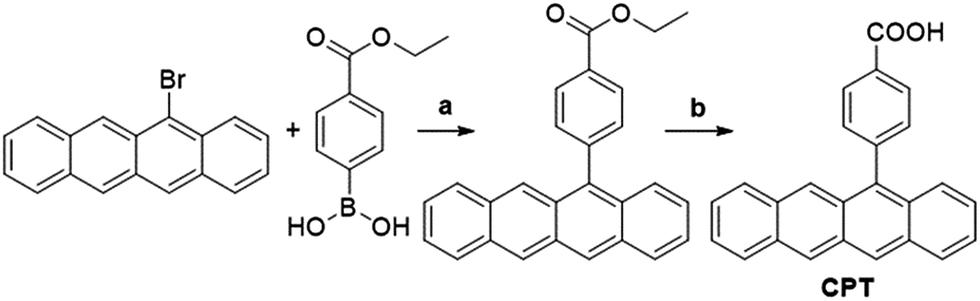 | ||
Scheme 1 Synthesis of transmitter tetracene derivative 4-(tetracen-5-yl)benzoic acid (CPT). Reagents and conditions: (a) Cs2CO3, Pd(dppf)Cl2·CH2Cl2, toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 3:1, 60 °C, overnight. (b) 2 M KOH aqueous solution, THF:MeOH = 1:1, reflux, 3 h. :H2O = 3:1, 60 °C, overnight. (b) 2 M KOH aqueous solution, THF:MeOH = 1:1, reflux, 3 h. | ||
As shown in Fig. 2, for both PbS and PbSe, the number of CPT transmitter ligands bound per particle, n, correlates positively with the concentration of CPT in the ligand exchange solution (denoted as [CPT]LX). For 2.9 nm diameter PbS NCs, n varies from 14 to 45 as [CPT]LX is increased from 50 to 250 μM. The corresponding transmitter ligand density increases from 0.53 to 1.7 nm−2. In comparison, there are 3 to 22 CPT ligands per 2.5 nm diameter PbSe NCs when [CPT]LX increases from 100 to 1000 μM, with ligand density ranging from 0.22 to 1.1 nm−2. The correlation between [CPT]LX and n was also confirmed by the photoluminescence (PL) quenching of 2.9 nm PbS. As shown in Fig. S1a and b,† with increasing [CPT]LX (ranging from 100 to 1500 μM), the PL of PbS was increasingly quenched, indicating very efficient energy transfer from PbS NCs to CPT. This estimate for n is obtained from the UV-vis absorption spectrum of the PbX/CPT complex that does not contain free ligand, taking into account the extinction coefficients of both the molecule and NC, assuming no charge transfer occurs (see ESI† for details). The number of CPT transmitter ligands bound affects the solubility of the PbX/CPT complex. Experimentally, it was impossible to redisperse the PbX–CPT pellet in toluene after centrifugation if [CPT]LX exceeded 1500 μM, and the PbX NCs would even spontaneously crash out of solution if [CPT]LX was over 2000 μM. Since CPT can effectively displace the native oleic acid ligands on the PbX NCs, the functionalized NCs no longer remain soluble if complete ligand exchange occurs. This is expected when the solubilizing long-chain hydrocarbons on the NC surface are completely replaced with the relatively insoluble CPT. The original oleic acid capped PbS and PbSe NCs have their surface saturated with carboxylic acid ligands with ligand densities of 3.019 and 4.220 nm−2 respectively. The surface densities of CPT on PbS and PbSe NCs that lead to aggregating structures are 0.7519 and 1.120 nm−2 respectively, consistent with the fact that the CPT molecule is around 4 times wider than oleic acid. Since the goal of this work is to establish ligand enhanced upconversion of NIR photons in solution, we used [CPT]LX below 1500 μM, where the PbS/CPT complex remains soluble.
 | ||
| Fig. 2 The relative upconversion QY is plotted with the upconversion fluorescence intensity normalized by the absorption at the excitation wavelength of 808 nm (black squares), and the number of bound CPT transmitter ligands per NC (hollow blue triangles) for (a) PbS, and (b) PbSe NC versus [CPT] in the ligand exchange solution. Both of the samples contain 1 mM rubrene and were measured in 200 μm thick capillary tubes sealed in air-free cuvettes. Ligand exchange condition: PbS: stirring 10 μM PbS with CPT in designated concentration for 40 min; PbSe: stirring 29 μM PbSe with CPT in designated concentration for 20 min. | ||
We found the upconversion QY reaches a maximum and then decreases as the number of bound transmitter ligands is increased (Fig. 2). Here, the relative upconversion QY is the upconversion fluorescence intensity of the rubrene emitter at 560 nm normalized by the absorption of the PbX NC at 808 nm. In Fig. 2a and b, both PbS and PbSe sensitized upconversion show the highest relative QY at the optimal [CPT]LX of 150 μM. Since more CPT ligand is bound when [CPT]LX is higher, the diminished upconversion at higher ligand loadings suggests that TET from CPT to free rubrene in solution is compromised. This suggests that the TTA process may be occurring between two neighboring CPT molecules where newly introduced ligands may be aggregating together on the NC surface, as opposed to being randomly distributed. Comparing the TTA process between bound CPT and rubrene, TTA between bound CPT is undesirable because its fluorescence QY of 53% is significantly lower than the 98% of rubrene. In addition, emission from the singlet state of surface bound CPT may be quickly quenched due to rapid Förster energy transfer to the NC acceptor. For isolated CPT, energy transfer to free rubrene avoids quenching by the NCs.11 For other reasons that are not clear, if too many transmitter ligands are installed, the upconversion QY decreases. Could it be that subsequent ligands bind to sites that result in a poor orbital overlap between the NC and the tetracene conjugated backbone? Or do these ligands bind to sites that behave as trap states for the triplet exciton? Unfortunately it is extremely challenging to interrogate the sites of bound ligands and their relative positions on the NCs to obtain a better understanding of the surface.21
To realize the maximum upconversion QY, other parameters such as the duration of ligand exchange, concentration of rubrene, and measurement setup was optimized. Other than [CPT]LX, n can also be controlled with the time allowed for ligand exchange. Fig. S2† shows the correlation between 2.5 nm PbSe sensitized upconversion QY and ligand exchange time. With 29 μM PbSe and a fixed 150 μM of [5-CPT]LX in the ligand exchange solution, the highest upconversion efficiency was obtained after 5 min of stirring. A shorter or longer ligand exchange time leads to insufficient or too many CPT ligands per PbSe NC respectively. The upconversion quantum efficiency plateaus when the ligand exchange time exceeds 15 min, indicating that equilibrium is achieved. The upconversion QY increases with the concentration of rubrene, as shown in Fig. S3,† in accordance with reports in molecular visible upconversion systems.22 Note that 20 mM is the solubility limit of rubrene in toluene. As shown in Fig. 1a, a high upconversion QY relates to efficient triplet energy transfer from CPT to rubrene, and the TTA between two rubrene molecules. The higher the concentration of rubrene, the more triplet rubrene formed, the higher the upconversion QY. Finally, to minimize the parasitic reabsorption of the upconversion signal by the NCs, the sample was put in a capillary tube with a thickness of 100 μm and sealed in an air free 1 cm by 1 cm path length cuvette. The upconversion signal was measured in a front face geometry (see Fig. S4†). Excitation power density dependence measurements were performed to confirm that all measurements occurred in the linear regime (Fig. S5†).
 | (1) |
With optimal conditions (see ESI†), the ligand enhanced upconversion was measured with an 808 nm laser on CPT-bound 2.9 nm PbS and 2.5 nm PbSe NCs in 20 mM rubrene. The upconversion QY, ΦUC is given by eqn (1), where Φref is the single photon quantum yield of rubrene, excited by a cw 532 nm laser. Note that there is a factor of 2 in eqn (1), so 100% QY is achieved when 50 upconverted photons are emitted with every 100 photons absorbed. For 2.9 nm PbS without the transmitter ligand, a QY of 0.021% was obtained. This upconversion QY was enhanced by 81 times to 1.7% with the CPT transmitter covalently bound to the NC surface. For 2.5 nm PbSe, CPT enhances the upconversion QY 11 times from 0.20% to 2.1%. Note that the enhancement varies with rubrene concentration. While enhancements of 81 and 11 were obtained with 20 mM rubrene, over 200 times enhancement was observed when using 1 mM rubrene for 2.9 nm PbS NCs. As hypothesized, CPT binds on PbX NCs and forms the energy cascade described in Fig. 1a, so that energy is transferred to free rubrene molecules in solution more efficiently. The 2.1% QY is the highest reported value for TTA-based NIR upconversion. In accordance with Kasha’s rule,23 this upconversion QY is expected to extend to all wavelengths of light absorbed by the NC up to 1100 nm, as demonstrated previously. This allows 43.8% more of the NIR region to be harvested compared to the maximum wavelength of 790 nm upconverted by molecular sensitizers.24 Work is underway in this laboratory to improve this QY further by passivating the NC surface and investigating other molecular candidates for transmitters.
The use of transmitter ligands results in upconversion QYs enhanced by three orders of magnitude for CdSe NCs,11 two orders of magnitude for PbS NCs, and an order of magnitude for PbSe NCs, a trend which is inversely related to their excitonic Bohr radius of 5 nm, 18 nm and 44 nm respectively. As triplet energy transfer from NCs to ligand molecules are based on the Dexter mechanism, the overlap of wave function is important. The inverse correlation between the Bohr radius of the NCs and ligand-based enhancement of the upconversion QYs supports the hypothesis that the Bohr radius relates to the delocalization of the exitonic wavefunction in nanocrystals. Empirically, we observe that materials with a larger Bohr radius have higher upconversion QYs to begin with for the as-synthesized, unfunctionalized NCs, the subsequent transmitter-based enhancement of the upconversion QY is lower.
Conclusions
In conclusion, we have demonstrated that rationally designed transmitter ligands can enhance TET from PbX NCs to the triplet states of organic molecules, resulting in 1.7% and 2.1% upconversion QYs for PbS and PbSe NCs as the light absorbers. We show that the surface coverage of the CPT ligand on PbX NPs heavily influences the upconversion QY. This work lays the foundation for the molecular engineering required to improve this Dexter energy transfer process. For example, perhaps bulky structures that inhibit TTA between bound transmitter ligands may facilitate better TET between the NC light absorber and rubrene annihilator. In terms of practical applications, we expect the CPT functionalized PbS/PbSe NCs sensitizers to enhance the upconversion quantum yield in a thin film geometry as well.15 Here the triplets are used for photon upconversion, but in principle, they could be directly extracted as electron–hole pairs in a solar cell. Antenna geometries, polymeric scaffolds and other supramolecular architectures could be harnessed to direct triplets efficiently from the NC to the annihilator, for high-performing, next-generation photovoltaic cells and photocatalysts.Acknowledgements
The authors acknowledge financial support from the US Army W911NF-15-1-0040 and the National Science Foundation CHE-1351663.Notes and references
- O. E. Semonin, J. M. Luther, S. Choi, H.-Y. Chen, J. Gao, A. J. Nozik and M. C. Beard, Science, 2011, 334, 1530–1533 CrossRef CAS PubMed; D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef PubMed; J.-H. Kim and J.-H. Kim, J. Am. Chem. Soc., 2012, 134, 17478–17481 CrossRef PubMed.
- R. T. Ross and A. J. Nozik, J. Appl. Phys., 1982, 53, 3813–3818 CrossRef CAS.
- A. Luque and A. Martí, Phys. Rev. Lett., 1997, 78, 5014–5017 CrossRef CAS.
- R. D. Schaller and V. I. Klimov, Phys. Rev. Lett., 2004, 92, 186601 CrossRef CAS PubMed.
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- P. Mahato, A. Monguzzi, N. Yanai, T. Yamada and N. Kimizuka, Nat. Mater., 2015, 14, 924–930 CrossRef CAS PubMed; Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817–20830 RSC; A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322–4332 RSC.
- F. Auzel, C. R. Acad. Sci., 1966, 262, 1016–1019 Search PubMed.
- Y. I. Park, K. T. Lee, Y. D. Suh and T. Hyeon, Chem. Soc. Rev., 2015, 44, 1302–1317 RSC.
- J. S. Lissau, D. Nauroozi, M.-P. Santoni, S. Ott, J. M. Gardner and A. Morandeira, J. Phys. Chem. C, 2013, 117, 14493–14501 CAS.
- Z. Huang, X. Li, M. Mahboub, K. M. Hanson, V. M. Nichols, H. Le, M. L. Tang and C. J. Bardeen, Nano Lett., 2015, 15, 5552–5557 CrossRef CAS PubMed.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369–372 CrossRef CAS PubMed.
- R. S. Khnayzer, J. Blumhoff, J. A. Harrington, A. Haefele, F. Deng and F. N. Castellano, Chem. Commun., 2012, 48, 209–211 RSC; B. Wang, B. Sun, X. Wang, C. Ye, P. Ding, Z. Liang, Z. Chen, X. Tao and L. Wu, J. Phys. Chem. C, 2014, 118, 1417–1425 Search PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2015, 10, 31–34 CrossRef.
- A. M. Müller, Y. S. Avlasevich, W. W. Schoeller, K. Müllen and C. J. Bardeen, J. Am. Chem. Soc., 2007, 129, 14240–14250 CrossRef PubMed.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of photochemistry, CRC Press, 1993 Search PubMed.
- R. A. Minns, K. D. Hutchinson, E. S. Kolb and D. A. Waldman, WO2004059389A3, 2004 CrossRef CAS PubMed; T. Okamoto, T. Suzuki, H. Tanaka, D. Hashizume and Y. Matsuo, Chem.–Asian J., 2012, 7, 105–111 CrossRef CAS PubMed.
- I. Moreels, Y. Justo, B. De Geyter, K. Haustraete, J. C. Martins and Z. Hens, ACS Nano, 2011, 5, 2004–2012 CrossRef CAS PubMed.
- I. Moreels, B. Fritzinger, J. C. Martins and Z. Hens, J. Am. Chem. Soc., 2008, 130, 15081–15086 CrossRef CAS PubMed.
- A. J. Morris-Cohen, M. Malicki, M. D. Peterson, J. W. J. Slavin and E. A. Weiss, Chem. Mater., 2013, 25, 1155–1165 CrossRef CAS; Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211–1221 CrossRef.
- T. N. Singh-Rachford, A. Haefele, R. Ziessel and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 16164–16165 CrossRef CAS PubMed.
- Z. Huang, X. Li, B. D. Yip, J. M. Rubalcava, C. J. Bardeen and M. L. Tang, Chem. Mater., 2015, 27, 7503–7507 CrossRef CAS.
- V. Yakutkin, M. A. Filatov, I. Z. Ilieva, K. Landfester, T. Miteva and S. Baluschev, Photochem. Photobiol. Sci., 2015 10.1039/C5PP00212E.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc00257a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |