
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic studies and radiofluorination of structurally diverse pharmaceuticals with spirocyclic iodonium(III) ylides†
Benjamin H.
Rotstein‡
 ab,
Lu
Wang‡
a,
Richard Y.
Liu
c,
Jon
Patteson
a,
Eugene E.
Kwan
c,
Neil
Vasdev
*ab and
Steven H.
Liang
*ab
ab,
Lu
Wang‡
a,
Richard Y.
Liu
c,
Jon
Patteson
a,
Eugene E.
Kwan
c,
Neil
Vasdev
*ab and
Steven H.
Liang
*ab
aDivision of Nuclear Medicine and Molecular Imaging & Gordon Center for Medical Imaging, Massachusetts General Hospital, 55 Fruit Street, Boston, Massachusetts 02114, USA
bDepartment of Radiology, Harvard Medical School, 55 Fruit Street, Boston, Massachusetts 02114, USA. E-mail: vasdev.neil@mgh.harvard.edu; liang.steven@mgh.harvard.edu
cDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA
First published on 24th March 2016
Abstract
Synthesis of non-activated electron-rich and sterically hindered 18F-arenes remains a major challenge due to limitations of existing radiofluorination methodologies. Herein, we report on our mechanistic investigations of spirocyclic iodonium(III) ylide precursors for arene radiofluorination, including their reactivity, selectivity, and stability with no-carrier-added [18F]fluoride. Benchmark calculations at the G2[ECP] level indicate that pseudorotation and reductive elimination at iodine(III) can be modeled well by appropriately selected dispersion-corrected density functional methods. Modeling of the reaction pathways show that fluoride–iodonium(III) adduct intermediates are strongly activated and highly regioselective for reductive elimination of the desired [18F]fluoroarenes (difference in barriers, ΔΔG‡ > 25 kcal mol−1). The advantage of spirocyclic auxiliaries is further supported by NMR spectroscopy studies, which bolster evidence for underlying decomposition processes which can be overcome for radiofluorination of iodonium(III) precursors. Using a novel adamantyl auxiliary, sterically hindered iodonium ylides have been developed to enable highly efficient radiofluorination of electron-rich arenes, including fragments of pharmaceutically relevant nitrogen-containing heterocycles and tertiary amines. Furthermore, this methodology has been applied for the syntheses of the radiopharmaceuticals 6-[18F]fluoro-meta-tyrosine ([18F]FMT, 11 ± 1% isolated radiochemical yield, non-decay-corrected (RCY, n.d.c.), n = 3), and meta-[18F]fluorobenzylguanidine ([18F]mFBG, 14 ± 1% isolated RCY, n.d.c., n = 3) which cannot be directly radiolabeled using conventional nucleophilic aromatic substitution with [18F]fluoride.
Introduction
Radiofluorination of arenes, particularly non-activated or sterically hindered positions, with fluorine-18 remains a major challenge and a key limitation for the development of new radiotracers for in vivo imaging with positron emission tomography (PET).1 Taking into consideration its convenient half-life (109.8 min), 18F is, with carbon-11, among the most desirable nuclides for small molecule PET radiotracers for imaging and quantification of biological processes, such as receptor expression and occupancy, metabolic activity, and cellular proliferation.2 Increased availability of cyclotron-produced (18O(p,n)18F) no-carrier-added [18F]fluoride for radiosynthesis has promoted the routine production of validated and target-selective PET radiopharmaceuticals suitable for pathology research, disease diagnosis, and drug development. Formation of 18F–Csp3 bonds using [18F]fluoride is generally much more facile than aryl radiofluorination, and can be achieved by nucleophilic displacement using primary or secondary alkyl electrophiles with [18F]fluoride. Nucleophilic aromatic substitution (SNAr) using nitroarene, aryl halide, or aryltrimethylammonium salt precursors is a pragmatic strategy for radiofluorination of activated (i.e., electron-deficient) arenes, but is of limited utility for non-activated or deactivated (i.e., electron-rich and/or sterically hindered) substrates. Similarly electrophilic radiofluorination using carrier-added [18F]F2 or the rarely utilized Balz–Schiemann and Wallach reactions using [18F]fluoride are incapable of producing structurally complex products in high specific activity. A host of more selective radiofluorination methods for non-activated arenes has been developed with [18F]fluoride,3,4 including oxidative strategies5 and transition metal-mediated reactions (Fig. 1A).6–9 While these methods have demonstrated innovative reactivity, aside from hypervalent iodonium-mediated methods, they have not been deployed in validated radiopharmaceutical syntheses for clinical imaging applications and appear to engender technical challenges that are preventing their routine use.§ Hypervalent iodonium10–14 and sulfonium15,16 precursors offer metal-free radiofluorination with varying levels of reactivity and selectivity. Of these, diaryl iodonium salt precursors have been the most well-established alternative to SNAr in the preparation of 18F-labeled compounds (Fig. 1B).17,18 In order to achieve high regioselectivity, arenes such as anisole and thiophene are often used as directing groups based on electronic discrimination, with the incoming [18F]fluoride intended for less electron-rich arenes.11 In the presence of a copper catalyst, the regioselectivity of diaryliodonium salts during radiofluorination can be controlled with high selectivity (Fig. 1B).9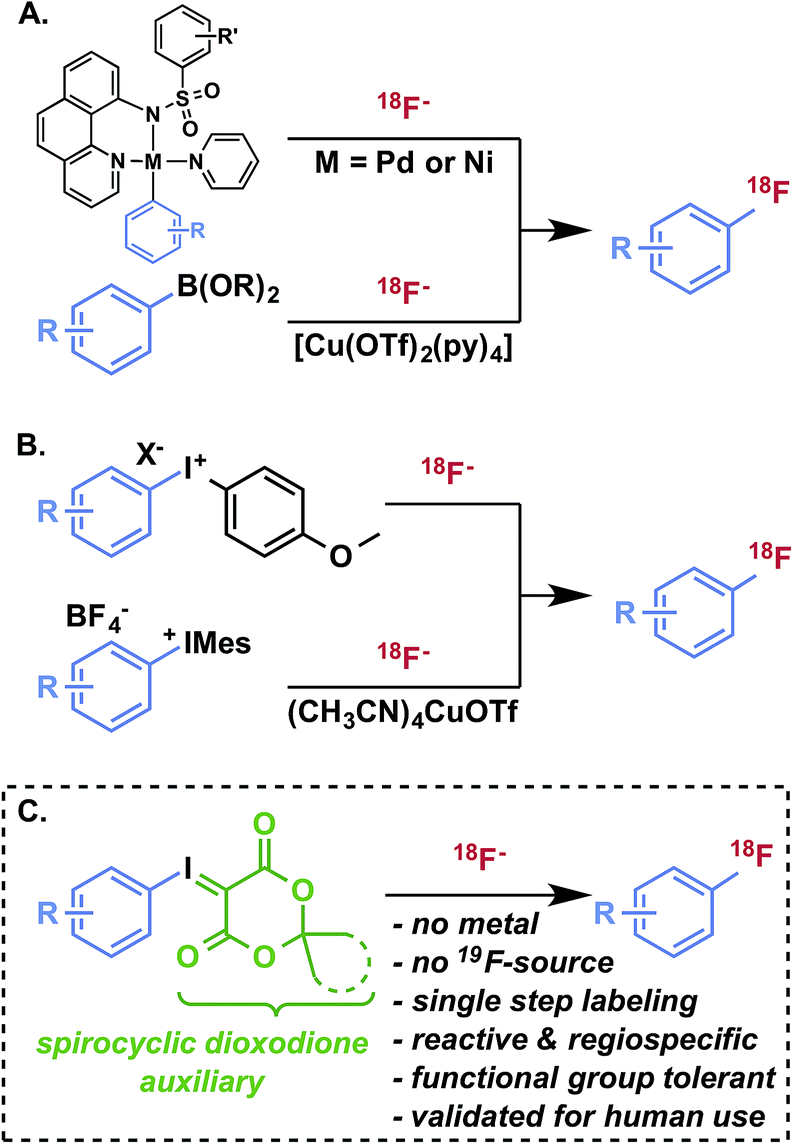 | ||
| Fig. 1 Transition metal- and iodonium-mediated arene radiofluorination. | ||
Recently, we introduced spirocyclic iodonium ylides as arene radiofluorination precursors for hindered and non-activated substrates (Fig. 1C).14 Iodonium ylides present several advantages for radiofluorination over diaryliodonium salts,12,13,19 foremost being the lack of a counterion and an auxiliary arene. As a result, iodonium ylides can be readily prepared and purified by flash chromatography and radiofluorination can be expected to proceed with high specific activity from [18F]fluoride with complete regioselectivity. While we have demonstrated the utility of these precursors for synthesis of a radiopharmaceutical20 and bioconjugation reagents,21–23 the underlying characteristics of these radiofluorination reactions, including mechanism and auxiliary substitution effects, remained uncertain and represented a major hurdle to further advance these reactions in drug labeling and radiotracer development. In this report, we detail benchmarked theoretical studies of iodonium(III) fluoride intermediates, which help to explain the high reactivity and selectivity of iodonium(III) ylides for radiofluorination, and thermostability studies and reaction monitoring by NMR, which provides insight into the advantages of a new class of adamantyl spirocyclic auxiliaries. Finally, using these precursors we demonstrate the practical radiosyntheses of drug fragments, as well as [18F]safinamide and two clinically relevant radiopharmaceuticals, 6-[18F]fluoro-meta-tyrosine and meta-[18F]fluorobenzylguanidine.
Results and discussion
Regioselectivity and reactivity of hypervalent iodonium precursors for radiofluorination
The successful labeling of [18F]FPEB20 and 18F-labeled aromatic azides21–23 with spirocyclic hypervalent iodine(III) precursors prompted us firstly to seek reasonable theoretical approaches to uncover the apparent regioselectivity and reactivity differences between iodonium ylides and diaryliodonium salts, and second to probe why these species are superior precursors for radiofluorination of deactivated arenes. DFT calculations have been shown to be useful in the study of iodine(III) compounds24 and their reactivity toward fluorination.25 Generally, B3LYP or a member of the M06 family has been chosen as the functional and a standard double-zeta basis set was employed, using LANL2DZ or SDD on the iodine atom. However, benchmarks have not been reported, especially on systems that are likely to proceed through reductive elimination at iodine.26 Furthermore, while iodine(III) compounds bearing ligands such as carboxylate,24b chloride,24e bromide, or azide24g have been studied, modeling of the aryl fluoride extrusion process hypothesized in iodonium(III) ylide mediated radiofluorination would provide an in depth understanding of hypervalent iodine-mediated fluorination reactions.As model reactions, we selected the reductive elimination and pseudorotation of diphenyliodonium fluoride. Using several DFT methods including those most commonly used for modeling hypervalent iodine molecules, the calculated barriers were compared to those obtained from an extension of the Gaussian-2 method reported by Radom (G2[ECP], a high level composite ab initio procedure developed specifically to reproduce the properties of iodine-containing molecules).27 G2[ECP] is a well-established extension of G-2 method and is itself benchmarked to ground and transition state halogen properties, including bond lengths, vibrational frequencies, ionization energies, and reaction barriers. Using geometries obtained at B3LYP/6-31G(d)/LANL2DZ(I)/PCM and single point energies obtained with the aug-cc-pVTZ/SDB-aug-cc-pVTZ(I) basis sets, we found the PBE0 functional accurately reproduced the ab initio energies, with an additional BJ-damped D3 dispersion correction further reducing error in agreement with G2[ECP] values (Table 1). Of all the functionals evaluated, PBE0 with D3 dispersion most closely approximated the G2[ECP] benchmark for both reductive elimination and pseudorotation of an I(III) species. Furthermore, while solvation modeling greatly affected barriers at the double-zeta level, it has only minimal effects with larger basis sets (Table S2, ESI†). It is notable that M06-2X had poor agreement with the benchmark values, as did B3LYP without dispersion correction.
| Functional | Error in kcal mol−1 | |||
|---|---|---|---|---|
| Reductive elimination barrier vs. G2[ECP] | Pseudorotation barrier vs. G2[ECP] | |||
| Without dispersion | With D3 dispersion | Without dispersion | With D3 dispersion | |
| B3LYP | −2.2 | −0.9 | −2.2 | −1.1 |
| B3PW91 | −1.9 | −0.6 | −1.6 | −0.5 |
| B97D | −4.1 | −2.3 | −3.9 | −2.0 |
| BH&HLYP | +2.2 | — | +1.8 | — |
| M06-2X | +3.8 | +3.8 | +1.9 | +1.8 |
| MP2 | +5.6 | — | +4.4 | — |
| PBE0 | −0.9 | −0.3 | −0.2 | +0.3 |
| TPSSh | −3.6 | −2.7 | −1.8 | −1.1 |
| ωB97XD | +0.4 | — | +0.7 | — |
We have demonstrated that hypervalent iodonium precursors are efficient for radiofluorination of non-activated aromatic rings, and these transformations are believed to operate by initial coordination of fluoride to the iodine center, followed by reductive elimination to form an 18F–Csp2 bond.26,28 In order to understand the selectivity of radiofluorination with hypervalent iodonium precursors, we modeled possible reductive elimination pathways of fluoride–iodonium complexes derived from diaryliodonium salts and iodonium ylides. Based on previously reported isolated trivalent diarylfluoroiodonium complexes,29 we hypothesized that the rate limiting step is reductive elimination of aryl fluoride from iodine(III).26 This is consistent with the observation of more facile radiofluorination of electron-deficient arenes from hypervalent iodonium precursors. All attempts to locate other multi-step pathways were unsuccessful, including solvent-assisted or dimeric pathways (see ESI† for details).30
For both diaryliodonium and iodonium ylide derived species, the barriers to pseudorotation between conformers Ia/Ib and IIa/IIb were high, though lower than the respective barriers to reductive elimination (Fig. 2), suggesting that a Curtin–Hammett scenario may be operative.31 The energies of the two ground-state conformations were nearly identical, with the phenyl substituent slightly favoring the pseudo-equatorial position (Ia). For (4-methoxyphenyl)phenyliodonium fluoride (Ia/Ib), electronic differences between the aryl substituents can induce measurable levels of regioselectivity, though the respective barriers to reductive elimination were comparable (TS-Ia and TS-Ic, ΔΔG‡ = 2.9 kcal mol−1), favoring formation of the less electron-rich aryl fluoride. In accordance with previous experimental findings,31 it may be anticipated that more electron-rich substrate arenes will demand higher activation energies for product reductive elimination, and consequently, lower levels of regioselectivity.
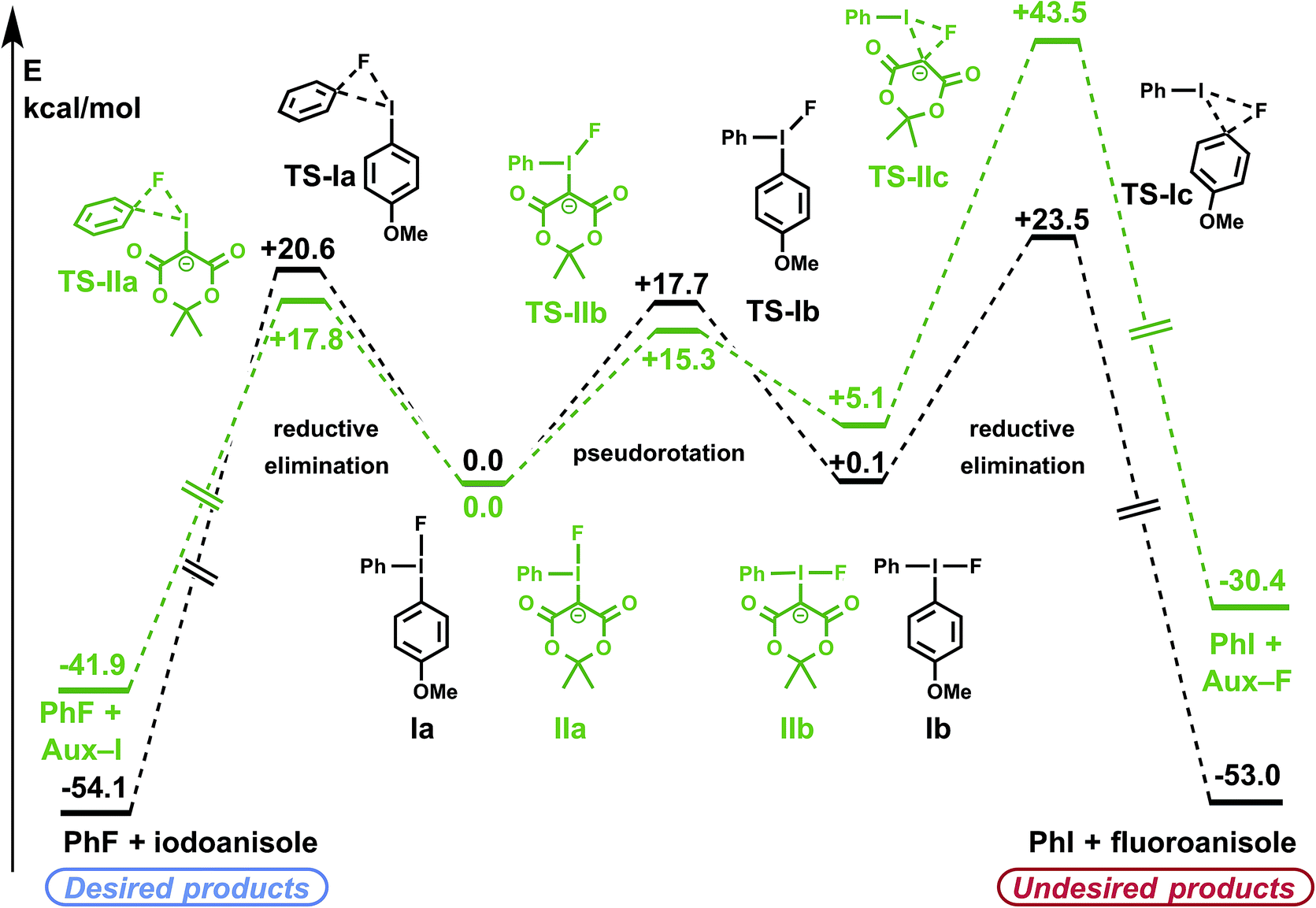 | ||
| Fig. 2 Calculated reaction profiles for iodonium fluoride reductive elimination pathways. PBE0-D3/aug-cc-pVTZ/SDB-aug-cc-pVTZ(I) single point, B3LYP/6-31G(d)/LANL2DZ(I)/PCM(DMF) geometry and vibrational correction. | ||
Regiospecificity was predicted for aryl fluoride reductive elimination from iodonium ylide-derived species. This difference originates from a combination of ground state and transition state effects. Compared with the diaryliodonium analog, pseudorotamers derived from the iodonium ylide showed a greater preference for the phenyl group in the pseudo-equatorial position (IIa). The presence of the dicarbonyl auxiliary reduces the barrier to aryl fluoride reductive elimination to 17.8 kcal mol−1 (TS-IIa). Notably, the undesired Csp3–F reductive elimination pathway has a high barrier (TS-IIc), more than 25 kcal mol−1 greater than the desired Csp2–F reductive elimination. In agreement with the calculations, fluorinated auxiliary products (Csp3–F reductive elimination) have not been experimentally observed from any of the >100 iodonium ylides we have tested in radiofluorination.
Influence of arene electronics on radiofluorination
To investigate the effect of conjugated electron-donating and electron-withdrawing groups on radiofluorination of arenes, we prepared a series of 1-iodosonaphthalenes using the adamantyl-substituted SPIAd auxiliary (vide infra) and featuring nitro-, cyano-, proto-, or methoxy-substitution at the 4-position. The fluorinated products (2) are all involatile, such that we could accurately quantify RCC by radioTLC. Under our radiofluorination conditions, each of 1a–d produced a single radioactive product, which co-eluted with independently prepared standards of 2a–d by HPLC (Fig. 3).32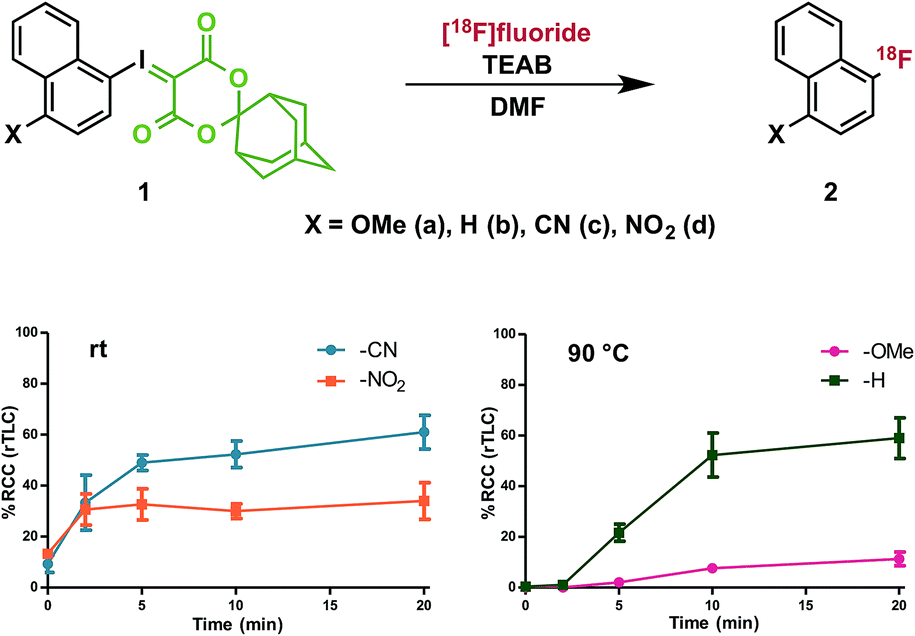 | ||
| Fig. 3 Effect of substituent electronics on radiofluorination. n = 3 for each data point; see ESI† for detailed procedure; error bars represent SD. | ||
Precursors 1a–d were evaluated under radiofluorination conditions at a series of temperatures including ambient temperature, 60, 90, and 120 °C. Aliquots of each reaction mixture were withdrawn at the following predetermined time points: 0 min (upon addition of [18F]fluoride solution), 2, 5, 10, and 20 min. Activated substrates (i.e., cyano- and nitro-substituted precursors 1c and 1d) underwent rapid radiofluorination at ambient temperature to reach RCCs of 49 ± 5% at 5 min and 30 ± 5% at 2 min respectively, after which only slight increases in conversion were measured. The methoxy- and proto-substituted ylides (1a and 1b) did not undergo radiofluorination below 90 °C. At this temperature the RCC of 1b increased to 59 ± 14%, and the electron-rich 1a was only 11 ± 5% RCC at 20 min. The greater extent of radiofluorination of electron deficient aryliodonium ylides is consistent with the hypothesis that reductive elimination is rate-determining in iodonium-mediated radiofluorination, as our calculations found that electron withdrawing groups dramatically lower the barrier to reductive elimination of the corresponding fluoroarenes (Fig. 4).
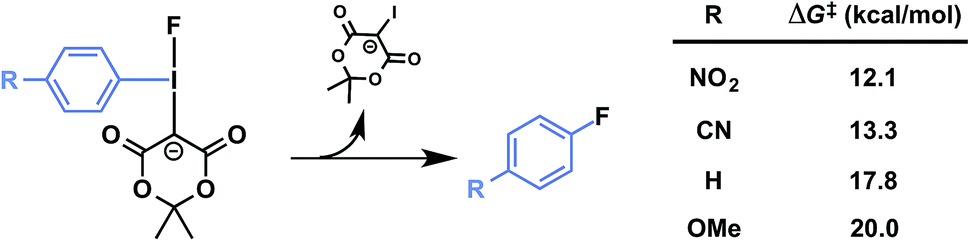 | ||
| Fig. 4 Predicted influence of arene electronics on reductive elimination. PBE0-D3/aug-cc-pVTZ/SDB-aug-cc-pVTZ(I) single point, B3LYP/6-31G(d)/LANL2DZ(I)/PCM(DMF) geometry and vibrational correction. | ||
Origins of the “ortho” effect
Though sterically hindered ligands typically participate poorly in reductive elimination processes, unsymmetrical diaryliodonium salts have shown preference for reductive elimination of ortho-substituted arenes,31 in some cases overcoming the electronic biases of a given substrate. This effect has been ascribed to steric tension in the tetragonal pyramidal precursor to reductive elimination, and can be exacerbated by larger nucleophiles.28 In the case of iodonium ylides, radiofluorination is regiospecific and greater radiochemical conversion of ortho-substituted arenes is similarly observed. Our computational model using isomeric (methoxyphenyl)iodonium fluoride complexes illustrates that this effect originates from a sterically induced ground state destabilization of the ortho-C–H⋯F interaction, which has not previously been observed (Fig. 5A). If the methoxy substituent is located at the para position, the arene resides in the F–I–C plane, perhaps in part due to a stabilizing hydrogen bond with the basic fluoride. This is the conformation typically observed for arenes lacking ortho-substituents. To the contrary, the 2-methoxyphenyl ligand is rotated out of this plane due to steric interactions of the ortho-substituent. As reductive elimination proceeds through an out-of-plane transition state conformation, the latter system needs not pay the energetic cost of arene rotation prior to reductive elimination. This difference explains the relative barriers to arylfluoride formation, which are predicted to differ by over 5 kcal mol−1, as well as the experimentally observed differences in reactivity of isomeric para- and ortho-benzyloxyphenyl substrates 3a and 4a (Fig. 5B).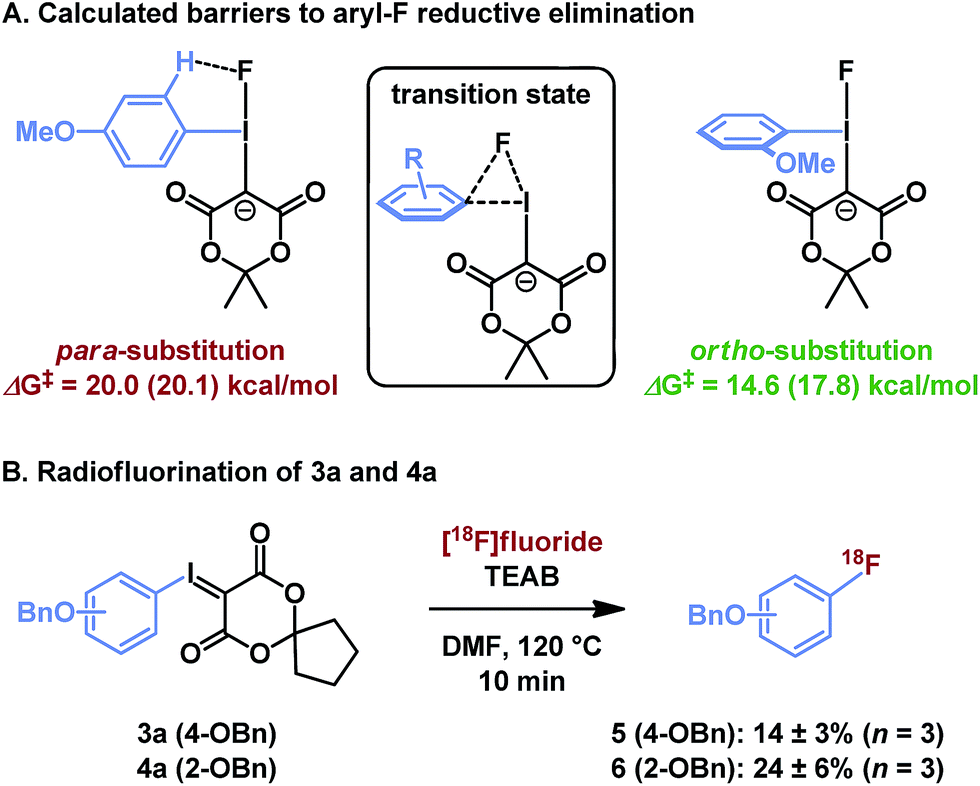 | ||
| Fig. 5 Influence of ortho-substituents on reductive elimination. (A) Calculation method: PBE0-D3/aug-cc-pVTZ/SDB-aug-cc-pVTZ(I) single point, B3LYP/6-31G(d)/LANL2DZ(I)/PCM(DMF) geometry and vibrational correction. The parenthetical energy is calculated at the same level with PCM(DMF) solvation model on the single point energy. (B) Experimental results, see ESI† for details. | ||
Thermal stability of iodonium ylides is dependent on auxiliary substitution
The preceding rationalization for the regioselectivity of iodonium ylide radiofluorination implicates the “doubly anionic” character that would develop during any reductive elimination involving the dicarbonyl auxiliary. Therefore, we would expect that aryl fluoride reductive elimination should always proceed as the major productive pathway, with little sensitivity to structural variation of the auxiliary so long as the anionic charge is preserved. Indeed, none of the auxiliaries that were experimentally surveyed show any propensity to the disfavored reductive elimination pathway leading to alkyl fluorides. To determine whether the experimentally observed effects of auxiliary substitution on radiofluorination conversion could be reproduced computationally and explained by an influence on the activation energy of reductive elimination, the barriers to reductive elimination of fluorobenzene from hypervalent iodonium complexes bearing several classes of cyclic dicarbonyl auxiliaries were calculated using the benchmarked method described above. The reaction barriers were insensitive to the presence of oxygen or methylene groups within the ring, ring size, and/or substitution at the distal position (Fig. S2†). Significantly, our computational model demonstrated that auxiliary effects on radiofluorination of arenes must originate from processes other than the reductive elimination step. The auxiliary may either affect earlier elementary processes in the productive mechanism or, more likely, influence nonproductive processes, such as the rate of precursor decomposition.After systematically surveying arrays of structurally diverse auxiliaries (Table S1†), we identified a distinctly effective auxiliary for radiofluorination, spiroadamantyl-1,3-dioxane-4,6-dione (SPIAd, Scheme 1), which was prepared from malonic acid and commercially available 2-adamantanone 7. After purification by recrystallization, SPIAd is soluble in aqueous bicarbonate and can be applied for synthesis of radiofluorination precursors under our previously described conditions.14
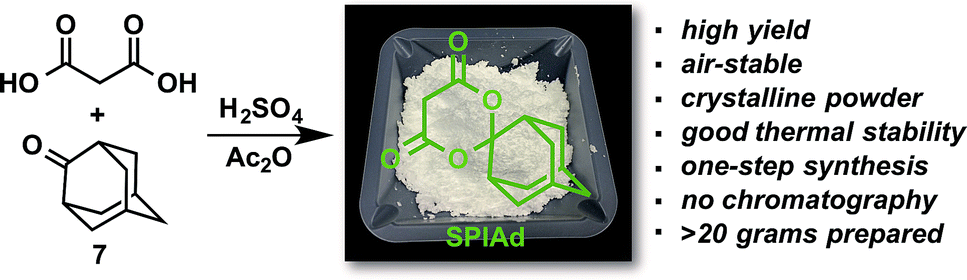 | ||
| Scheme 1 Synthesis of spiroadamantyl-1,3-dioxane-4,6-dione (SPIAd). | ||
The stabilities of iodonium ylides were evaluated under conditions analogous to radiofluorination, in the absence of radioactivity. Solutions of iodonium ylides (5 mM) in DMF-d7 with TEAB (25 mM) were monitored by 1H NMR prior to and between intermittent heating to the reaction temperatures for radiofluorination (120 °C). In this way, the stabilities of different precursors under radiofluorination conditions could be evaluated and plotted. We examined the influence of the ylide auxiliary on precursor stability, testing ortho-benzyloxyphenyl SPIAd (4d), dimedone (4j), and MA (4k) iodonium ylides (Fig. 6).
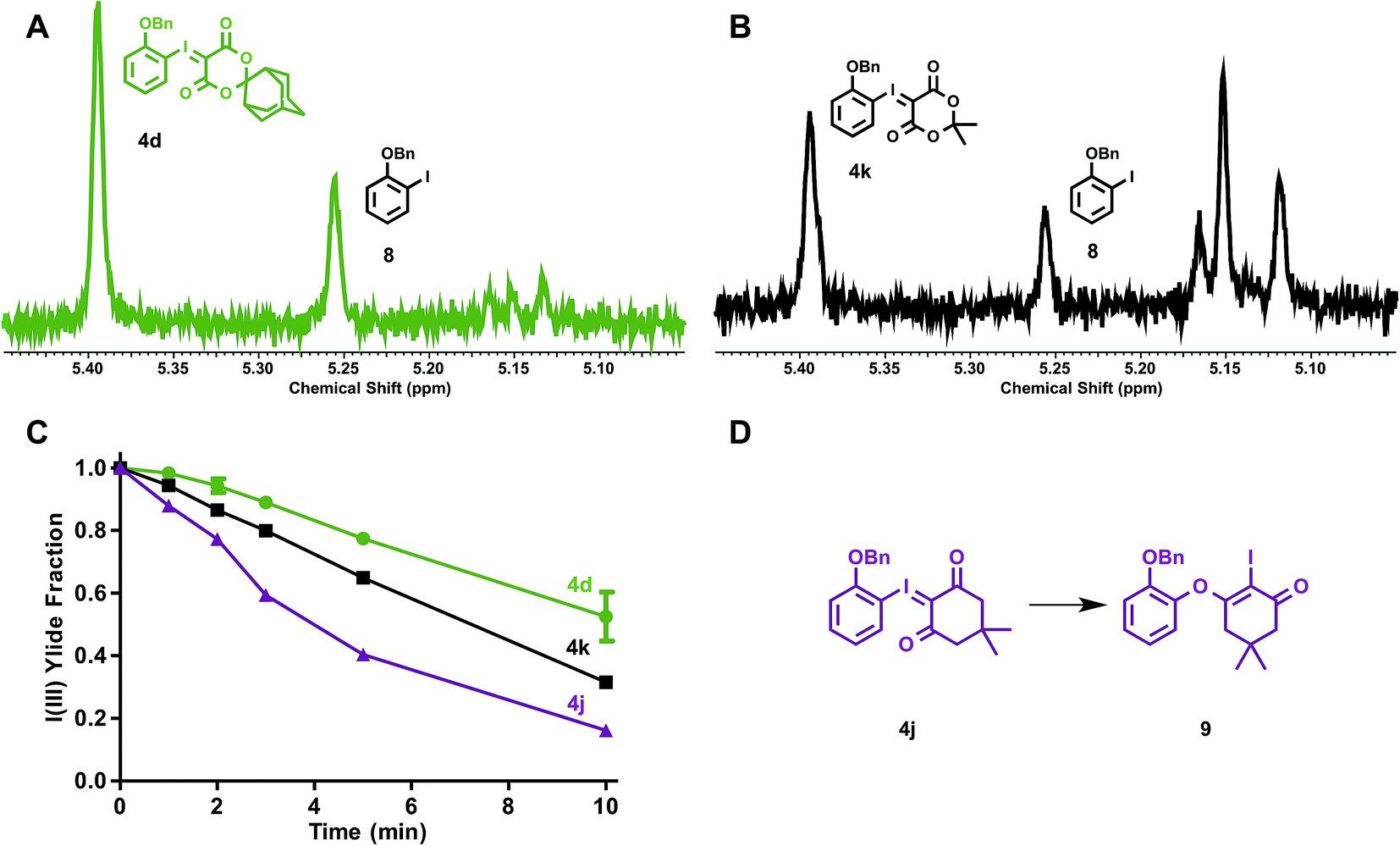 | ||
| Fig. 6 Stability of electron rich iodonium ylides (SPIAd-4d, Meldrum's acid-4k, and dimedone-4j) under reaction conditions in absence of [18F]fluoride. A–B, Representative 1H NMR spectra of 4d and 4k reaction mixtures at 10 min. C, Time course stability plot of iodonium ylides. D, Rearrangement of 4j → 9L. Conditions: 3.5 µmol iodonium ylide in 0.7 mL DMF-d7 (5 mM) + TEAB (4.8 mg mL−1, 25 mM), heated to 120 °C (n = 2). | ||
After collecting baseline 1H NMR spectra of each reaction mixture, solutions were heated to 120 °C, and spectra were acquired at 1, 2, 3, 5, and 10 min. For each substrate, the benzyl methylene of the parent iodonium(III) ylide gave rise to signal at 5.39–5.40 ppm (relative DMF-d7 solvent residual peak referenced to 8.01 ppm). At least four distinct sets of product signals in the 1H NMR could be identified at consistent chemical shifts across the substrates evaluated, including 5.25, 5.16, 5.15, and 5.12 ppm, with other minor species giving rise to additional signals between 5.11 and 5.16 ppm (Fig. 6A–B). This suggests that multiple decomposition pathways are likely to be operative. An integral region from 5.36–5.42 ppm was defined as the parent fraction, and two regions (5.24–5.27 and 5.09–5.17 ppm) were applied as product integral regions. The parent fraction of the total integral, corrected for parent fraction at t0, is plotted for ylides 4d, 4j, and 4k in Fig. 6C. The rate of decomposition appeared to remain constant over the course of the study, and does not depend on the concentration of precursor in solution. No measurable decomposition could be observed in the absence of base heating dioxodione-substituted iodonium ylides to 120 °C for over 60 min. For challenging, electron-rich iodonium ylides, the presence of the bulkier auxiliary, SPIAd, conferred considerably improved stability under the reaction conditions. The product signal at 5.25 ppm can be assigned to a single species, the ortho-benzyloxyiodobenzene (8), whose formation is largely insensitive to auxiliary substitution. For all dioxodione ylide auxiliaries, the ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 at 10 minutes was approximately 1.0:0.4. Dimedone substituted 4j gave rise to a second species in this region, 9, product of the known rearrangement (Fig. 6D).24c We found no evidence for the formation of analogous species derived from dioxodione-substituted auxiliaries, nor O-monobenzylcatechol. Differences between the stabilities of dioxodione-substituted ylides are associated with products in the 5.09–5.17 ppm region, which include the proto-deiodinated arene among other species. Given the considerable extent of decomposition over 10 min (30% vs. 50% precursor remaining for 4kvs.4d, respectively), and the predicted insensitivity of reductive elimination to the sterics of the spirocyclic auxiliary, improved precursor persistence in the radiolabeling reaction mixture is a compelling explanation for the advantages of SPIAd iodonium ylides.
:8 at 10 minutes was approximately 1.0:0.4. Dimedone substituted 4j gave rise to a second species in this region, 9, product of the known rearrangement (Fig. 6D).24c We found no evidence for the formation of analogous species derived from dioxodione-substituted auxiliaries, nor O-monobenzylcatechol. Differences between the stabilities of dioxodione-substituted ylides are associated with products in the 5.09–5.17 ppm region, which include the proto-deiodinated arene among other species. Given the considerable extent of decomposition over 10 min (30% vs. 50% precursor remaining for 4kvs.4d, respectively), and the predicted insensitivity of reductive elimination to the sterics of the spirocyclic auxiliary, improved precursor persistence in the radiolabeling reaction mixture is a compelling explanation for the advantages of SPIAd iodonium ylides.
Synthesis and radiofluorination of heteroaryl iodonium ylides
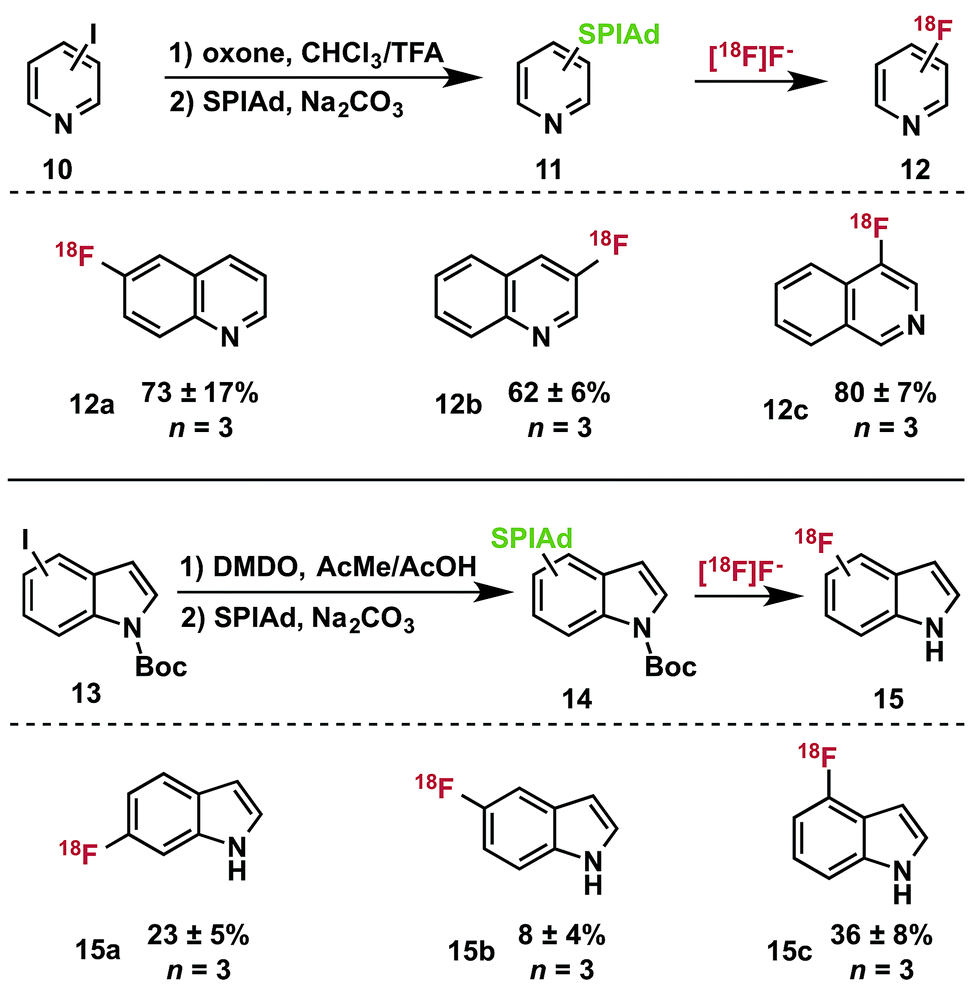
As iodonium ylides are synthesized by oxidation of aryl iodides, heteroaryl iodonium ylides have presented a challenge for selective oxidation protocols. We have developed and applied a set of strategies for preparing these precursors and successfully utilized the products in radiofluorination. Nucleophilic and aprotic iodine-substituted heterocycles, such as quinolines (10, Table 2) can be selectively oxidized in acidic media that protonate the ring-nitrogen as a protecting group strategy. Oxidation of the iodoarene using oxone in trifluoroacetic acid:chloroform (3:1) at room temperature33 furnished the [bis(trifluoroacetoxy)iodo]arene, which can be converted directly to iodonium ylides 11 by treatment with the acid auxiliary under basic conditions.14 Indoles are susceptible to oxidation in the presence of hypervalent iodonium reagents.34 Nevertheless, we were able to selectively oxidize several isomers of N-Boc-iodoindole 13 by treatment with dimethyldioxirane (DMDO) to prepare diacetoxyiodoarenes, which could be directly converted into iodonium ylides 14 for radiofluorination.
| a Radiochemical conversions by rTLC; identities confirmed by coinjection on radioHPLC; conditions: precursor (3.5 µmol), anhydrous DMF (0.4 mL), TEAB (0.6 mg), [18F]fluoride (ca. 50 µCi), 120 °C, 10 min. |
|---|
|
We prepared the SPIAd ylides of a series of quinolines (11), isoquinolines, and indoles (13) and subjected them to radiofluorination conditions (Table 2). In all cases, the expected ipso-substitution 18F-heteroarene was the sole product. RCCs ranged from moderate to high, with the 18F-fluoroquinolines (12a–c) displaying generally good yields for historically difficult to access products (not optimized).35N-Boc-indoles 14 underwent quantitative deprotection during the labeling reactions, yielding 18F–NH-indoles (15a–c) as the sole radioactive products. Unoptimized RCCs for 15 were useful though generally lower than those of 12, and highly dependent on the site of substitution. These products represent highly desirable 18F-heterocycles that have proven extremely challenging to access in high specific activity, and could serve as valuable scaffolds for radiotracer and drug development.36
Radiofluorination of drug scaffolds
To demonstrate the utility of iodonium ylides for radiofluorination of biologically active molecules that could serve as the basis for PET radiopharmaceuticals or radiolabeled drugs to be used for in vivo dynamic pharmacokinetic studies, we selected fragments of a number of drug compounds that contain a fluorine atom for isotopic substitution and which would present a significant challenge for labeling using traditional methods for incorporation of [18F]fluoride. Substrates were prepared using selective oxidation strategies and protecting groups as needed to facilitate radiofluorination. The radiofluorinated products are depicted in Table 3, and preparation of SPIAd precursors is described in detail in the ESI.† In all cases, the precursors were efficiently prepared for radiofluorination using oxidation methods germane to all synthetic laboratories and without the use of specialized apparatus, such as a glovebox. Several protecting group strategies including acid- and hydrogenation-sensitive groups have been applied using a variety of selective oxidation strategies, which may require substrate-specific refinement for targets of greater complexity. Typically, oxidation of aryl iodides can be followed by ylide formation in a one-pot process by addition of the SPIAd auxiliary acid and adjustment of the pH of the reaction mixture. Importantly, all precursors could be purified by normal phase flash chromatography and isolated in high purity. SPIAd precursors are typically solid, colorless, and stable to storage at reduced temperatures.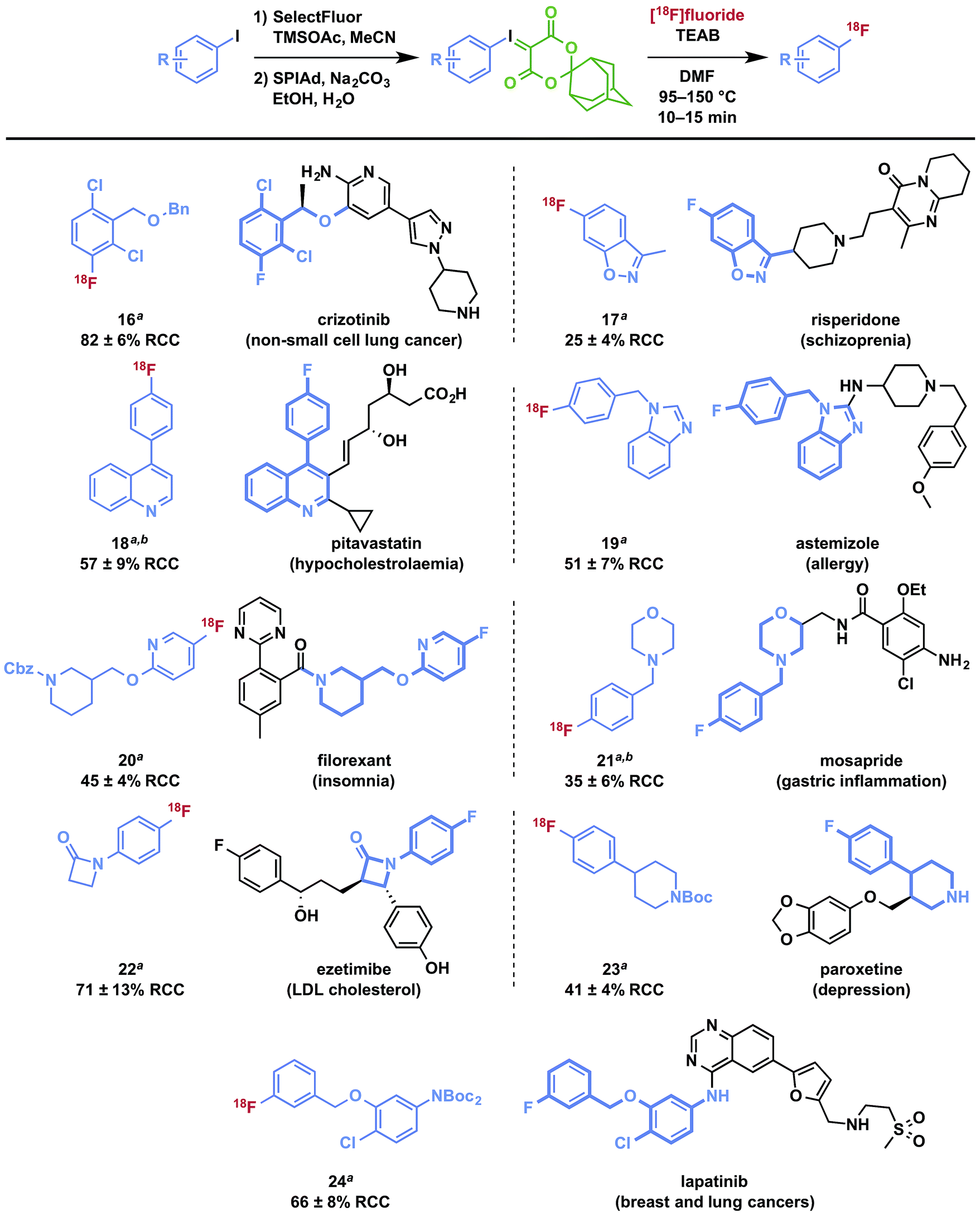
Radiofluorination to prepare drug fragments 16–24 was conducted with minor modifications to our general procedure, using TEAB in DMF as a reaction medium at elevated temperatures for 10–15 minutes. Under these conditions, labeling with [18F]fluoride was accomplished for the highly hindered 16, which represents a fragment of the dual c-MET and ALK inhibitor, crizotinib. Such encumbered positions are highly challenging for SNAr radiofluorination, but especially activated in the context of iodonium(III) ylides (vide supra). Radiofluorination of aromatic heterocycles, including isoxazoles, pyridines, and imidazoles 17–20 are demonstrated for fragments of risperidone, pitavastatin, astemizole, and filorexant. Similarly, key fluorine-containing drug fragments with saturated heterocycles and basic amines can be labeled, such as piperidines, morpholines, β-lactams, and anilines have been radiolabeled (20–24), and include fragments of mosapride, ezetimibe, paroxetine, and lapatinib. These functional groups do not interfere with preparation of SPIAd ylides or their radiofluorination. Carbamate-based protecting groups are well tolerated for primary and secondary amines, and offer versatility for multistep synthesis by orthogonal protection strategies.37 [18F]Safinamide (26), a reversible monoamine oxidase B (MAO-B) inhibitor was also prepared from a protected precursor by a two-step synthesis involving radiofluorination of the SPIAd precursor, followed by global acid deprotection (Scheme 2A).
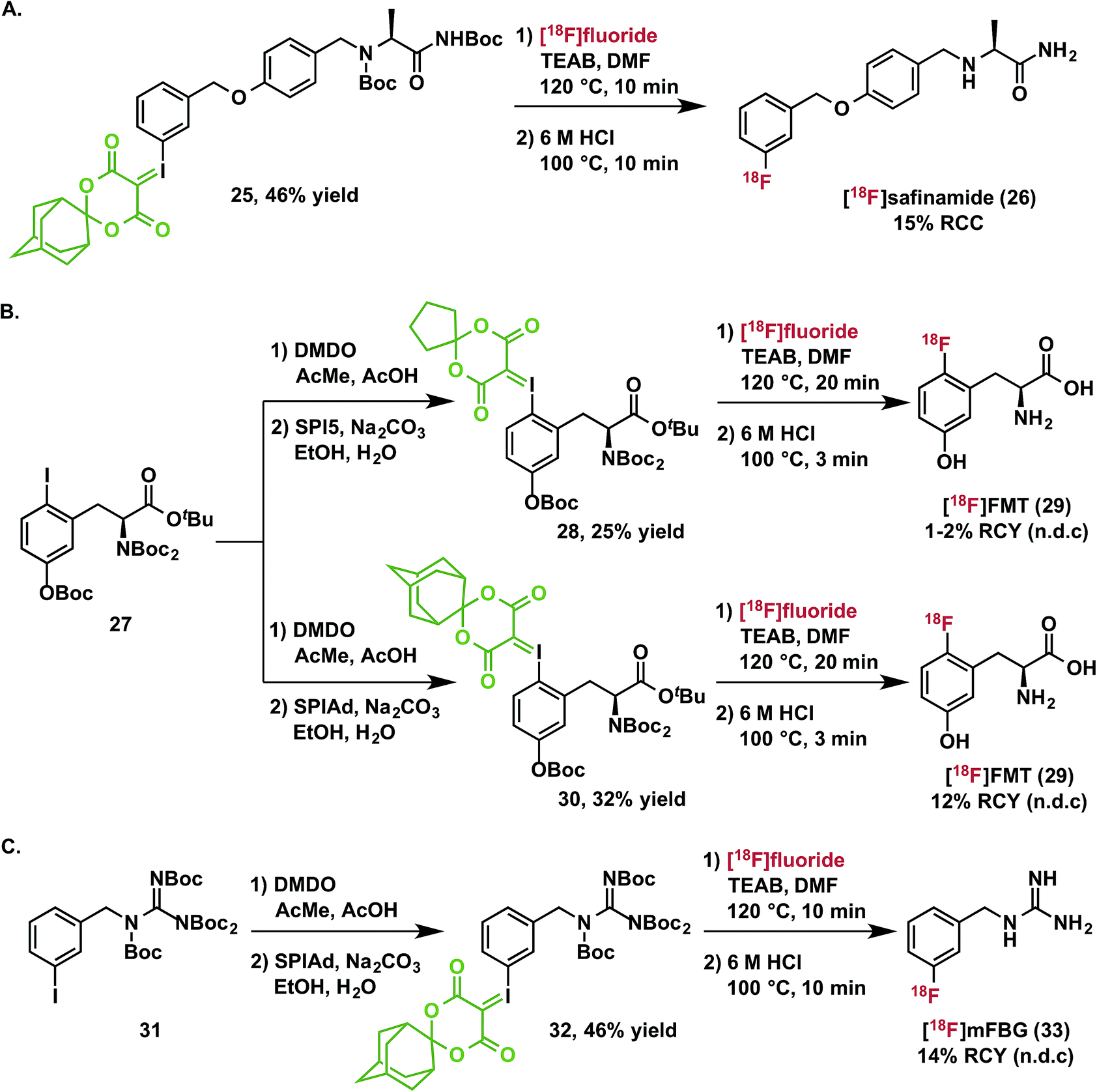 | ||
| Scheme 2 Synthesis and isolation of [18F]safinamide, [18F]FMT, and [18F]mFBG. | ||
Synthesis of clinically relevant radiopharmaceuticals
The practical utility of spirocyclic iodonium ylides has been established since our initial communication14 by the preparation of 3-[18F]fluoro-5-[(pyridin-3-yl)ethynyl]benzonitrile ([18F]FPEB), which has been validated as a radiopharmaceutical suitable for human administration.20 We now report the isolation of two additional clinically relevant radiopharmaceuticals: 6-[18F]fluoro-meta-tyrosine and meta-[18F]fluorobenzylguanidine ([18F]mFBG).Because it is not a substrate for catechol-O-methyltransferase, 6-[18F]fluoro-L-meta-tyrosine ([18F]FMT, 29) is a more metabolically stable radiopharmaceutical than 6-[18F]fluoro-L-DOPA and is also used for clinical research imaging of cerebral dopamine transport and neuroendocrine tumors.38 [18F]FMT has previously been prepared in low specific activity.39–42 Due to the high electron-density on the fluoroarene, SNAr radiofluorination with less activated leaving groups has proven challenging. We initially prepared a spirocyclic iodonium(III) ylide precursor 28 for 6-fluoro-meta-tyrosine using the auxiliary 6,10-dioxaspiro[4.5]decane-7,9-dione (SPI5) and a fully protected amino acid 27. Radiofluorination of this species was indeed possible and radiochemical conversions reached a plateau around 30% after 10 minutes at 120 °C (Fig. S1†). After deprotection and semi-preparative HPLC purification, we were able to isolate the product 29 in 1–2% non-decay corrected isolated radiochemical yield from starting [18F]fluoride. As a comparison to the corresponding SPIAd precursor (30), the extent of radiofluorination for this precursor increased over a 20 minute reaction time to reach ca. 60% radiochemical conversion. Rapid quantitative deprotection and semi-preparative HPLC purification yielded 12% of 6-[18F]fluoro-meta-tyrosine (29) in approximately 1 hour (n = 3) (Scheme 2B).
[18F]mFBG is a PET radiotracer for peripheral imaging of the norepinephrine transporter with applications in oncology and cardiac imaging, and is a derivative of the clinically important SPECT agent [123I]mIBG. Currently, the manual multistep radiosynthesis of [18F]mFBG involves SNAr of a meta-trimethylammoniumbenzonitrile salt, followed by reduction with LiAlH4, and finally guanylation.43 Very recently, an automated radiosynthesis of [18F]mFBG has been reported using a diaryliodonium triflate precursor.44 We prepared the SPIAd precursor 32 featuring a protected benzylguanidine from the iodoarene 31, and subjected it to radiofluorination under our standard conditions in the presence of 2.5 mg mL−1 TEAB (Scheme 2C). Radiochemical conversion to the partially protected 18F-intermediate is highly reproducible and exceeds 70%, more than double that accessible using the analogous SPI5 precursor. Subsequent acid deprotection and semi-preparative HPLC purification yielded [18F]mFBG (33) in 14% non-decay-corrected radiochemical yield based on aqueous [18F]fluoride and 98% radiochemical purity in an overall synthesis time of <75 minutes (n = 3).
Conclusions
Spirocyclic iodonium(III) ylides are highly selective and reactive precursors for the preparation of 18F-fluoroarenes, including electron-rich and sterically-hindered substrates. These radiofluorination precursors are easily prepared without the need for transition metals or counterions, which can complicate automation, purification procedures, and clinical translation for injectables. Furthermore, we have developed a computational approach using PBE0-D3 that is benchmarked to the well-established G2[ECP] method for reductive elimination pathways of aryl iodonium(III) fluoride complexes and confirmed that iodonium(III) ylides are comparatively more reactive and regioselective than diaryliodonium(III) salts. Regioselectivity of iodonium ylide radiofluorination can be explained in light of an unfavourable anionic intermediate that would result during reductive elimination of the auxiliary. These theoretical methods further explain the electronic and steric influences of arene substituents on radiofluorination, and should prove generally valuable for theoretical studies of I(III) compounds. Sterically hindered spirocyclic auxiliaries, and particularly SPIAd, impart superior precursor stability under radiofluorination conditions and are therefore highly advantageous for radiofluorination of deactivated and electron-rich substrates, which require heating to facilitate 18F-incorporation. The promise of these newly developed reactants for radiofluorination is demonstrated for the wide range of nitrogen and oxygen-containing heterocycles and drug fragments that have been labeled with [18F]fluoride, including basic amine functional groups which have been a longstanding challenge in 18F-radiochemistry. Furthermore, two PET radiopharmaceuticals, [18F]FMT and [18F]mFBG, which are both challenging to prepare for routine clinical research, have been isolated in high radiochemical yields by this methodology.Acknowledgements
We would like to thank the staff at the radiochemistry program, Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital, MA, USA for their support with cyclotron operation, radioisotope production and radiosynthesis. We thank Dr Thomas J. Brady and Dr Thomas Lee Collier for helpful discussions. S.H.L is a recipient of an NIH career development award (NIH/NIDA K01DA038000).Notes and references
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Chem. Sci., 2014, 5, 4545–4553 RSC.
- M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed.
- Z. Gao, Y. H. Lim, M. Tredwell, L. Li, S. Verhoog, M. Hop-kinson, W. Kaluza, T. L. Collier, J. Passchier, M. Huiban and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 6733–6737 CrossRef CAS PubMed.
- E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Bour-salian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639–642 CrossRef CAS PubMed.
- E. Lee, J. M. Hooker and T. Ritter, J. Am. Chem. Soc., 2012, 134, 17456–17458 CrossRef CAS PubMed.
- M. Tredwell, S. M. Preshlock, N. J. Taylor, S. Gruber, M. Huiban, J. Passchier, J. Mercier, C. Génicot and V. Gouverneur, Angew. Chem., Int. Ed., 2014, 53, 7751–7755 CrossRef CAS PubMed.
- N. Ichiishi, A. F. Brooks, J. J. Topczewski, M. E. Rodnick, M. S. Sanford and P. J. H. Scott, Org. Lett., 2014, 16, 3224–3227 CrossRef CAS PubMed.
- V. W. Pike and F. I. Aigbirhio, J. Chem. Soc., Chem. Commun., 1995, 2215–2216 RSC.
- T. L. Ross, J. Ermert, C. Hocke and H. H. Coenen, J. Am. Chem. Soc., 2007, 129, 8018–8025 CrossRef CAS PubMed.
- N. Satyamurthy and J. R. Barrio, World. Pat., WLO2010117435 (A2), 2010.
- J. Cardinale, J. Ermert, S. Humpert and H. H. Coenen, RSC Adv., 2014, 4, 17293–17299 RSC.
- B. H. Rotstein, N. A. Stephenson, N. Vasdev and S. H. Liang, Nat. Commun., 2014, 5, 4365–4371 CrossRef CAS PubMed.
- L. Mu, C. R. Fischer, J. P. Holland, J. Becaud, P. A. Schubiger, R. Schibli, S. M. Ametamey, K. Graham, T. Stellfeld, L. M. Dinkelborg and L. Lehmann, Eur. J. Org. Chem., 2012, 889–892 CrossRef CAS.
- K. Sander, T. Gendron, E. Yiannaki, K. Cybulska, T. L. Kalber, M. F. Lythgoe and E. Årstad, Sci. Rep., 2015, 5, 9941–9945 CrossRef CAS PubMed.
- B. S. Moon, H. S. Kil, J. H. Park, J. S. Kim, J. Park, D. Y. Chi, B. C. Lee and S. E. Kim, Org. Biomol. Chem., 2011, 9, 8346–8355 CAS.
- W.-J. Kuik, I. P. Kema, A. H. Brouwers, R. Zijlma, K. D. Neu-mann, R. A. J. O. Dierckx, S. G. DiMagno and P. H. Elsinga, J. Nucl. Med., 2015, 56, 106–112 CrossRef CAS PubMed.
- F. Kügler, J. Ermert, P. Kaufholz and H. Coenen, Molecules, 2014, 20, 470–486 CrossRef PubMed.
- N. A. Stephenson, J. P. Holland, A. Kassenbrock, D. L. Yokell, E. Livni, S. H. Liang and N. Vasdev, J. Nucl. Med., 2015, 56, 489–492 CrossRef CAS PubMed.
- L. Wang, O. Jacobson, D. Avdic, B. H. Rotstein, I. D. Weiss, L. Collier, X. Chen, N. Vasdev and S. H. Liang, Angew. Chem., Int. Ed., 2015, 54, 12777–12781 CrossRef CAS PubMed.
- O. Jacobson, I. D. Weiss, L. Wang, Z. Wang, X. Yang, A. Dewhurst, Y. Ma, G. Zhu, G. Niu, D. O. Kiesewetter, N. Vasdev, S. Liang and X. Chen, J. Nucl. Med., 2015, 56, 1780–1785 CrossRef PubMed.
- S. Calderwood, T. L. Collier, V. Gouverneur, S. H. Liang and N. Vasdev, J. Fluorine Chem., 2015, 178, 249–253 CrossRef CAS.
- (a) E. G. Bakalbassis, S. Spyroudis and E. Tsiotra, J. Org. Chem., 2006, 71, 7060 CrossRef CAS PubMed; (b) F. Mocci, G. Uccheddu, A. Frongia and G. Cerioni, J. Org. Chem., 2007, 72, 4163 CrossRef CAS PubMed; (c) R. M. Moriarty, S. Tyagi, D. Ivanov and M. Constantinescu, J. Am. Chem. Soc., 2008, 130, 7564 CrossRef CAS PubMed; (d) L. Pouységu, S. Chassaing, D. Dejugnac, A.-M. Lamidey, K. Miqueu, J.-M. Sotiropoulos and S. Quideau, Angew. Chem., Int. Ed., 2008, 47, 3552 CrossRef PubMed; (e) Y.-S. Lee, M. Hodošček, J.-H. Chun and V. W. Pike, Chem.–Eur. J., 2010, 16, 10418 CrossRef CAS PubMed; (f) H. P. de Magalhães, H. P. Lüthi and A. Togni, Org. Lett., 2012, 14, 3830 CrossRef PubMed; (g) P. K. Sajith and C. H. Suresh, Inorg. Chem., 2013, 52, 6046 CrossRef CAS PubMed; (h) M. M. Konnick, B. G. Hashiguchi, D. Devarajan, N. C. Boaz, T. B. Gunnoe, J. T. Groves, N. Gunsalus, D. H. Ess and R. A. Periana, Angew. Chem., Int. Ed., 2014, 53, 10490 CrossRef CAS PubMed; (i) R. Frei, M. D. Wodrich, D. P. Hari, P.-A. Borin, C. Chauvier and J. Waser, J. Am. Chem. Soc., 2014, 136, 16563 CrossRef CAS PubMed; (j) A. Sreenithya and R. B. Sunoj, Org. Lett., 2014, 16, 6224 CrossRef CAS PubMed; (k) C. Zhu, Y. Liang, X. Hong, H. Sun, W.-Y. Sun, K. N. Houk and Z. Shi, J. Am. Chem. Soc., 2015, 137, 7564 CrossRef CAS PubMed.
- (a) J. W. Graskemper, B. Wang, L. Qin, K. D. Neumann and S. G. DiMagno, Org. Lett., 2011, 13, 3158 CrossRef CAS PubMed; (b) D. E. Hill and J. P. Holland, Comput. Theor. Chem., 2015, 1066, 34 CrossRef CAS.
- M. A. Carroll, S. Martín-Santamaría, V. W. Pike, H. S. Rzepa and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 2, 1999, 2707–2714 RSC.
- M. N. Glukhovtsev, A. Pross, M. P. McGrath and L. Radom, J. Chem. Phys., 1995, 103, 1878–1885 CrossRef CAS.
- V. V. Grushin and I. I. Demkina, J. Chem. Soc., Perkin Trans. 2, 1992, 505–511 RSC.
- B. Wang, L. Qin, K. D. Neumann, S. Uppaluri, R. L. Cerny and S. G. DiMagno, Org. Lett., 2010, 12, 3352–3355 CrossRef CAS PubMed.
- S. Martín-Santamaría, M. A. Carroll, V. W. Pike, H. S. Rzepa and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 2, 2000, 2158–2161 RSC.
- J.-H. Chun, S. Lu, Y.-S. Lee and V. W. Pike, J. Org. Chem., 2010, 75, 3332–3338 CrossRef CAS PubMed.
- Under radiofluorination conditions at higher temperatures (120–140 °C) nitro–substituted precursor 1d forms multiple nonpolar radioactive products, which are not observed at ambient temperature.
- A. A. Zagulyaeva, M. S. Yusubov and V. V. Zhdankin, J. Org. Chem., 2010, 75, 2119–2122 CrossRef CAS PubMed.
- Q. Liu, Q. Y. Zhao, J. Liu, P. Wu, H. Yi and A. Lei, Chem. Commun., 2012, 48, 3239–3241 RSC.
- M. A. Carroll, J. Nairne and J. L. Woodcraft, J. Labelled Compd. Radiopharm., 2007, 50, 452–454 CrossRef CAS.
- P. S. Weiss, J. Ermert, J. C. Meleán, D. Schäfer and H. H. Coe-nen, Bioorg. Med. Chem., 2015, 23, 5856–5869 CrossRef CAS PubMed.
- S. H. Liang, T. L. Collier, B. H. Rotstein, R. Lewis, M. Steck and N. Vasdev, Chem. Commun., 2013, 49, 8755–8757 RSC.
- O. T. Dejesus, C. J. Endres, S. E. Shelton, R. J. Nickles and J. E. Holden, Synapse, 2001, 39, 58–63 CrossRef CAS PubMed.
- O. T. DeJesus, J. J. Sunderland, J. R. Nickles, J. Mukherjee and E. H. Appelman, Int. J. Radiat. Appl. Instrum., Part A, 1990, 41, 433–437 CrossRef CAS.
- M. Namavari, N. Satyamurthy, M. E. Phelps and J. R. Barrio, Appl. Radiat. Isot., 1993, 44, 527–536 CrossRef CAS PubMed.
- H. F. VanBrocklin, M. Blagoev, A. Hoepping, J. P. O'Neil, M. Klose, P. A. Schubiger and S. Ametamey, Appl. Radiat. Isot., 2004, 61, 1289–1294 CrossRef CAS PubMed.
- J. Castillo Meleán, J. Ermert and H. H. Coenen, J. Labelled Compd. Radiopharm., 2015, 58, 133–140 CrossRef PubMed.
- H. Zhang, R. Huang, N. Pillarsetty, D. L. J. Thorek, G. Vaidya-nathan, I. Serganova, R. G. Blasberg and J. S. Lewis, Eur. J. Nucl. Med. Mol. Imaging, 2014, 41, 322–332 CrossRef CAS PubMed.
- B. Hu, A. L. Vāvere, K. D. Neumann, B. L. Shulkin, S. G. Di-Magno and S. E. Snyder, ACS Chem. Neurosci., 2015, 6, 1870–1879 CrossRef CAS PubMed.
- A. J. Hoover, M. Lazari, H. Ren, M. K. Narayanam, J. M. Murphy, R. M. van Dam, J. M. Hooker and T. Ritter, Organometallics, 2016 DOI:10.1021/acs.organomet.6b00059.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization of compounds, NMR spectra and computational studies. See DOI: 10.1039/c6sc00197a |
| ‡ These authors contributed equally to this work. |
| § While our manuscript was under review, a paper describing the nickel-mediated synthesis of [18F]5-fluorouracil suitable for human use was published.45 |
| This journal is © The Royal Society of Chemistry 2016 |