
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxadendralenes in asymmetric organocatalysis for the construction of tetrahydroisochromenes†
Niels
Hammer
,
Lars A.
Leth
,
Julian
Stiller
,
Magnus E.
Jensen
and
Karl Anker
Jørgensen
*
Department of Chemistry, Aarhus University, DK-8000 Aarhus C, Denmark. E-mail: kaj@chem.au.dk
First published on 17th February 2016
Abstract
Oxadendralenes are integrated in a novel manner into a one-pot cascade utilizing synergistic catalysis for the construction of valuable and complex bicyclic heterocyclic scaffolds. The construction is based on the organocatalytic activation of the oxadendralenes generating a vinylogous iminium-ion intermediate which is set-up for a 1,6-addition with an enamine formed from an aldehyde and the same organocatalyst. This reaction generates a cyclic oxadendralenic intermediate, which acts as an electron-deficient heterodiene reacting in a Lewis-acid catalyzed hetero-Diels–Alder reaction with vinyl ethers to form tetrahydroisochromenes with five continuous stereocenters in high yields, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 99% ee. This synergistic organo- and Lewis-acid catalysed system also displays high tolerance for variation in oxadendralenes and aldehydes, which provides tetrahydroisochromenes with high diversity in the substituent pattern and the same excellent stereoselectivities. Mechanistic studies have been performed to account for the activation modes and stereochemical outcome of the reaction. The reaction concept has been extended to also include a sequential organocatalytic reaction of oxadendralenes with aldehydes, in which the enamine formed from the aldehyde and the organocatalyst act both in the first catalytic cycle forming the cyclic oxadendralenic intermediate and in a second catalytic cycle leading to tetrahydroisochromenes in good yields and excellent stereoselectivities. Mechanistic studies reveal that the stereochemistry of the organocatalyst has an influence on the diastereoselectivity of the reaction sequence. Some transformations of the tetrahydroisochromenes are also presented. The chiral tetrahydroisochromenes formed might be applied in the diversified synthesis of important drugs.
:1 dr and 99% ee. This synergistic organo- and Lewis-acid catalysed system also displays high tolerance for variation in oxadendralenes and aldehydes, which provides tetrahydroisochromenes with high diversity in the substituent pattern and the same excellent stereoselectivities. Mechanistic studies have been performed to account for the activation modes and stereochemical outcome of the reaction. The reaction concept has been extended to also include a sequential organocatalytic reaction of oxadendralenes with aldehydes, in which the enamine formed from the aldehyde and the organocatalyst act both in the first catalytic cycle forming the cyclic oxadendralenic intermediate and in a second catalytic cycle leading to tetrahydroisochromenes in good yields and excellent stereoselectivities. Mechanistic studies reveal that the stereochemistry of the organocatalyst has an influence on the diastereoselectivity of the reaction sequence. Some transformations of the tetrahydroisochromenes are also presented. The chiral tetrahydroisochromenes formed might be applied in the diversified synthesis of important drugs.
Introduction
The diversified synthesis of intricate molecular structures is becoming an increasingly applied strategy in organic synthesis.1 However, gaining easy access to important molecular scaffolds while being able to vary the structural components and the stereochemical information still constitutes a challenging task.Catalysis provides one of the most efficient ways to perform asymmetric operations and within recent years organocatalysis has evolved from being merely a proof of concept into a powerful synthetic tool for the construction of chiral compounds.2 This rapidly growing field now features a large number of examples displaying complex strategies aimed at the synthesis of natural compound-resembling targets, attaining high yields and stereoselectivities.3 The unique ability of organocatalysis to use simple starting materials to build up complex nature-inspired molecules in a “mix & hit” fashion enhances its applicability in academia and industry.4 In addition, organocatalytic methodologies are highly tolerable against other synthetic modifications, allowing one-pot strategies to be easily implemented, which enables the rapid synthesis of compounds with increased molecular complexity.5 One of the challenges remaining is the combination of metal catalysis and organocatalytic processes. Recently, the problematic compatibility of these two types of catalysis has received significant attention, as it holds the potential to facilitate unprecedented transformations.6
One of the most efficient synthetic routes towards attaining intricate cyclic frameworks is the application of cycloadditions. Dendralenes (Fig. 1, top) are well-known for their synthetic value in Diels–Alder reactions, as they have been shown to undergo multiple cycloadditions as part of reaction cascades with dienophiles.7 However, there are only a few examples featuring their heteroatom analogs (Fig. 1, top), despite the ability of heterodendralenes to facilitate hetero-Diels–Alder reactions yielding unprecedented polycyclic frameworks with exceptional atom economy.8
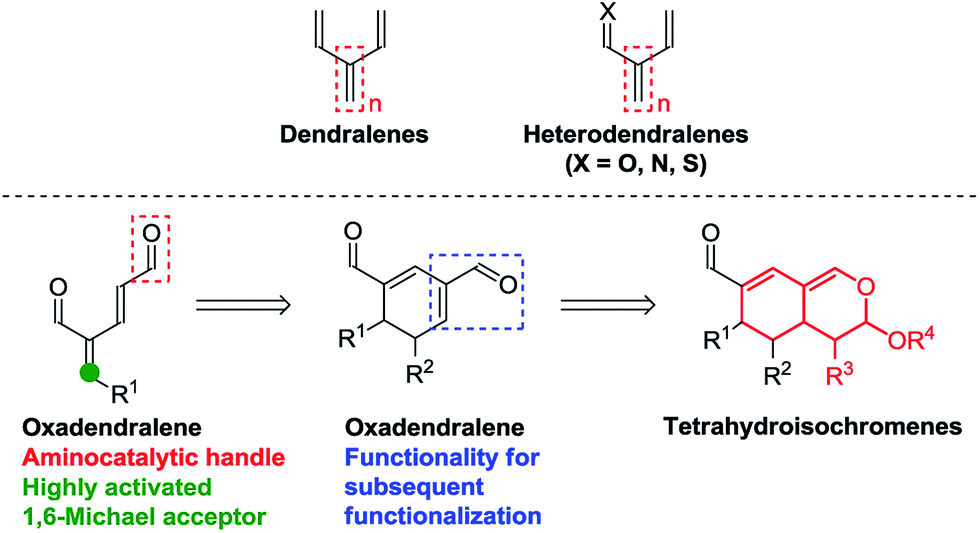 | ||
| Fig. 1 Top: Dendralenes and heterodendralenes. Bottom: Overview of the envisioned reaction sequence. | ||
The hetero-Diels–Alder reaction in particular has been subject to a vast amount of research as it offers a simple approach for the synthesis of six-membered oxygen- and nitrogen-containing heterocycles which is of great importance in medicinal chemistry.9
We envisioned the possibility of designing a reaction sequence integrating oxadendralenes in a novel manner into a one-pot cascade utilizing synergistic catalysis for the construction of valuable and complex core structures. Such a construction is envisioned to proceed via an organocatalytic annulation sequence forming a cyclic oxadendralene (Fig. 1, bottom). Inspired by the idea of constructing heterocyclic molecular complexity through utilizing hetero-Diels–Alder reactions, we sought to incorporate the oxadendralenic intermediate into a hetero-Diels–Alder reaction to generate the bicyclic tetrahydroisochromene scaffold (Fig. 1, bottom).
Bicyclic tetrahydroisochromenes are interesting and valuable motifs as they are present in some very important natural products. For instance, they can be found in the core structure of the anti-malarial artemisinin (Fig. 2), which was recently awarded the Nobel Prize in Physiology or Medicine.10 Furthermore, they resemble promising intermediates towards the total synthesis of artemisinin and its analogs.11 As of today, artemisinin and its analogs are among the most widely used anti-malarial drugs, displaying a very high potency against the malaria-causing Plasmodium parasite; however, examples of resistant strains are beginning to emerge, stressing the importance of analog-development.12 Additionally, the anti-cancer agent oridonin also includes the tetrahydroisochromene core structure. Oridonin has been found to exhibit a broad spectrum of remarkable anti-cancer and anti-bacterial properties, such as displaying selective induction of apoptosis in leukemia cells and general anti-proliferative activity in tumors.13
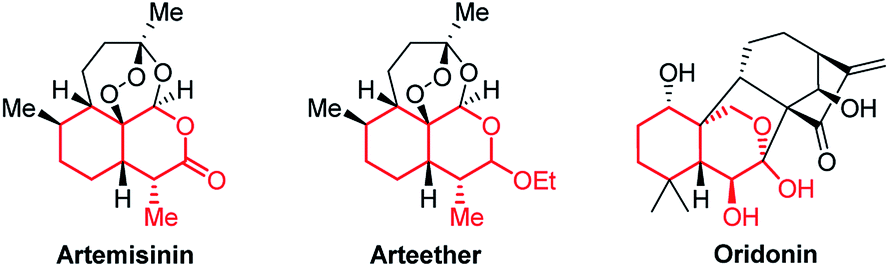 | ||
| Fig. 2 Structure of artemisinin, arteether and oridonin, containing the tetrahydroisochromene scaffold (marked in red). | ||
Herein, we present two novel organocatalysis-initiated strategies, enabling the enantioselective formation of a comprehensive tetrahydroisochromene library with full stereocontrol of all sp3-carbon centers (five stereocenters). The two-step one-pot cascades employ novel oxadendralenes in a sequential catalysis system, giving rise to the products in excellent enantio- and diastereoselectivities.
Strategic design
The synthetic strategy integrating oxadendralenes for the enantioselective construction of bicyclic tetrahydroisochromenes is presented in Scheme 1. We envisioned that the activation of oxadendralenic dienal 1 by an organocatalyst A would generate a vinylogous iminium-ion set-up for a 1,6-addition reaction14 with an enamine formed from an aldehyde 2 and the same organocatalyst. Thus, we assume the possible involvement of the unprecedented organocatalytic double activation of both the oxadendralenic dienal and aldehyde, in order to explain the stereochemical reaction outcome as outlined at the top of cycle 1, Scheme 1.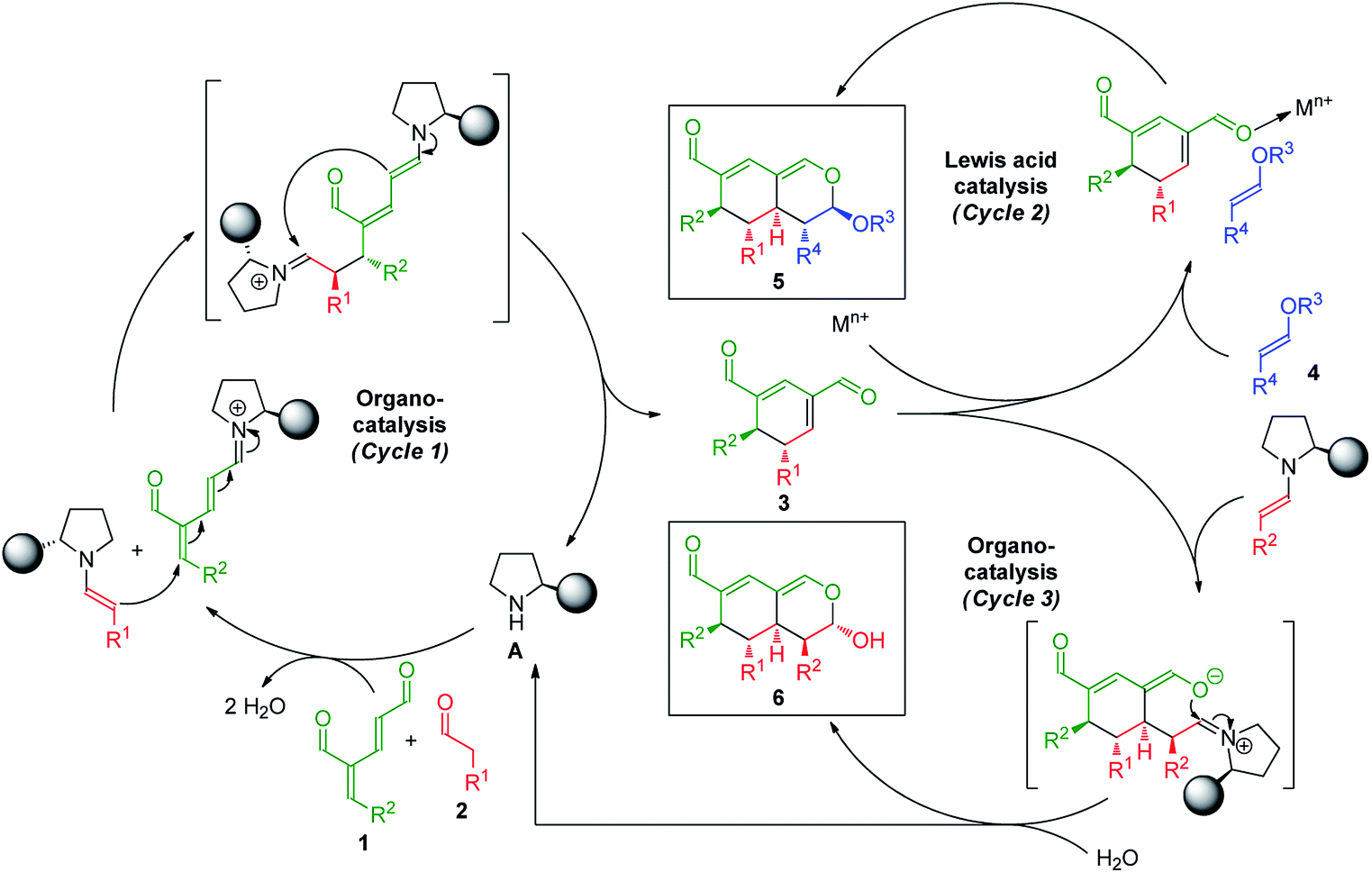 | ||
| Scheme 1 Synthetic strategy integrating oxadendralene 1 for the construction of the bicyclic tetrahydroisochromenes 5 and 6via a combination of organocatalysis and Lewis-acid catalysis (cycles 1 and 2) or sequential organocatalysis (cycles 1 and 3). | ||
The outcome of the first catalytic cycle in Scheme 1 is a new cyclic oxadendralenic intermediate 3 which is susceptible to a hetero-Diels–Alder reaction as an electron-deficient heterodiene (an inverse-electron demand hetero-Diels–Alder reaction). By adding a Lewis-acid compatible with the organocatalytic reaction conditions, the LUMO energy of the electron-deficient oxadendralenic intermediate 3 will be lowered,15 favoring a regioselective hetero-Diels–Alder cycloaddition with vinyl ether 4 to form tetrahydroisochromene 5 with five continuous stereocenters (Scheme 1, cycle 2).
Furthermore, we envisioned an alternative reaction pathway in which the cyclic oxadendralenic intermediate 3 might function as an acceptor in an organocatalytic enamine facilitated cycloaddition. This concept is based on the condensation of aldehyde 2 with organocatalyst A raising the HOMO energy, which enables an enantioselective cycloaddition16 generating tetrahydroisochromene product 6 with very high enantioselectivity (Scheme 1, cycle 3).
Results and discussion
To develop the reaction concept, we started by focusing on the organocatalytic annulation reaction between oxadendralenic dienal 1a and isovaleric aldehyde 2a to investigate the formation of the envisaged cyclic oxadendralenic intermediate 3a (Scheme 2).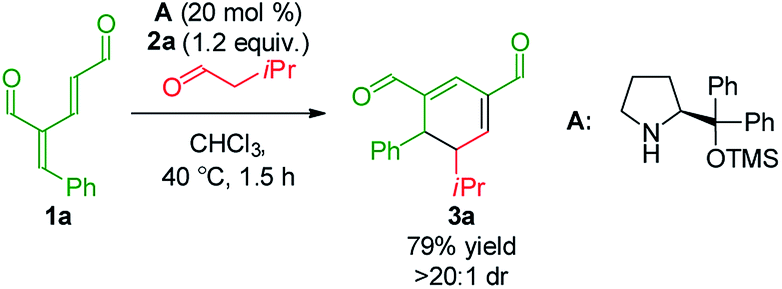 | ||
| Scheme 2 Testing the viability of the organocatalytic annulation for the formation of the oxadendralenic intermediate 3. | ||
Upon the addition of isovaleric aldehyde 2a to oxadendralenic dienal 1a and 20 mol% diphenylprolinol-silyl ether catalyst A,17 we were delighted to discover that the enamine underwent exclusive 1,6-conjugate addition, facilitating full conversion of the envisioned annulation of the cyclic oxadendralenic intermediate 3a within only 1.5 h. Compound 3, formed by the organocatalytic reaction, is a new cyclic oxadendralene, designed to be the intermediate required for the hetero-Diels–Alder reaction leading to the formation of the desired tetrahydroisochromenes. The absolute stereochemistry of the optically active cyclic oxadendralene 3b was obtained through X-ray analysis of the reaction product derived from the reaction between para-brominated oxadendralenic dienal 1b and 2a with the addition of 20 mol% A (Fig. 3, top).
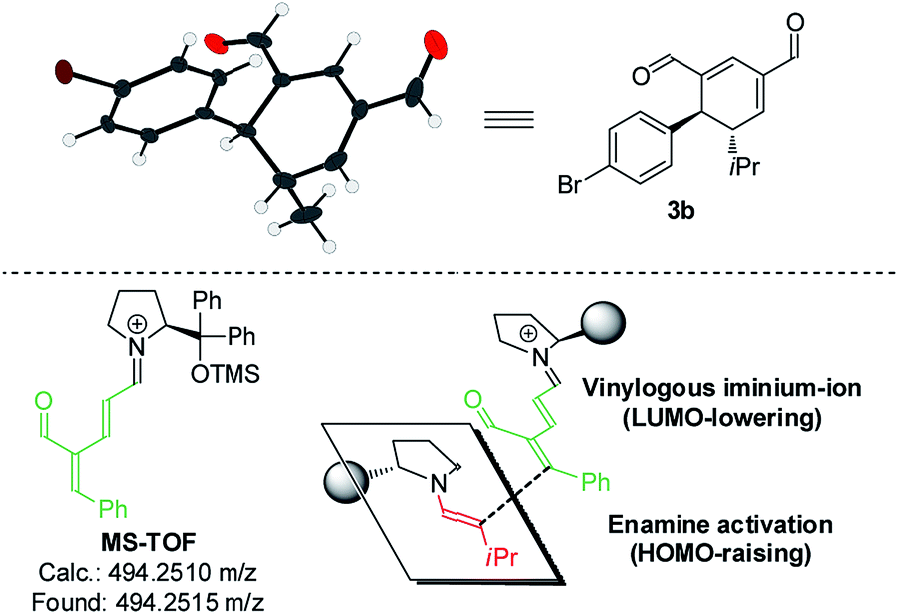 | ||
| Fig. 3 Top: X-ray structure of intermediate 3b. Bottom: MS-TOF analysis of 1a condensed with catalyst A and the enamine approach to account for the observed stereochemistry of the double activation strategy. | ||
Notably, the absolute configuration of compound 3b corresponds to the stereochemistry obtained if the enamine approaches the vinylogous iminium-ion activated oxadendralenic dienal from the bottom face, corresponding to the synergistic double condensation of both compounds with organocatalyst A (Fig. 3, bottom). There are examples of Michael acceptors undergoing organocatalytic enamine addition to conjugated esters and sulfones. However, the reaction times of these are in the range of 40–120 h, employing an equal or larger catalyst loading than in the present case.18 The short reaction time under the present reaction conditions might support the proposed double activation; however, we set out to provide evidence for the hypothesis. The investigations into the compatibility of oxadendralenic dienals with a secondary aminocatalyst demonstrated that upon mixing oxadendralenic dienal 1a with organocatalyst A the corresponding vinylogous iminium-ion was identified as the most abundant ion in MS-TOF (Fig. 3, bottom). Essentially, we propose that the reaction is initiated by a classic enamine performing a 1,6-conjugate addition to the vinylogous iminium-ion-activated species of the oxadendralenic dienal 1, followed by an intramolecular aldol condensation forming a reactive oxadendralenic intermediate in situ (Scheme 1, cycle 1).
Combined organo- and Lewis-acid catalysis
The investigation towards the formation of substituted tetrahydroisochromenes was initiated by the reaction of oxadendralenic dienal 1a with isovaleric aldehyde 2a. Using the previously mentioned conditions, full conversion into intermediate 3a was reached within 1.5 h at 40 °C in the presence of 20 mol% A in CHCl3. Gratifyingly, we discovered that upon the one-pot addition of ethyl vinyl ether 4a the inverse-electron demand hetero-Diels–Alder reaction proceeded smoothly forming 5a in a 7:1 dr and 99% ee, with a modest 61% yield (Table 1, entry 1). Encouraged by these findings we turned our attention towards the effect of solvents. Performing the reaction in Et2O and CH3CN resulted in a significant decrease in the yield; however, with a slight improvement in the diastereomeric ratio (Table 1, entries 2 and 3). Notably, EtOH increased the diastereomeric ratio to 11:1, nevertheless the yields of 3a and 5a were lower (Table 1, entry 4). Upon thorough analysis of the solvent screening we learned that the oxidation of intermediate 3a into an aromatic 2,4-dicarbaldehyde species was competing with the inverse-electron demand hetero-Diels–Alder reaction, consequently decreasing the yield substantially. To solve this problem we decided to explore the application of the catalytic Lewis-acid activation of the hetero-Diels–Alder cycloaddition. The addition of MgCl2 and Yb(fod)3 showed no improvements in the reaction (Table 1, entries 5 and 6), whereas the addition of 10 mol% Eu(fod)319 gave 5a in 75% yield, a 15:1 dr and 99% ee (Table 1, entry 7). A further improvement was observed when changing the number of equivalents of 1a, furnishing the desired tetrahydroisochromene 5a in a diasteromerically pure 88% isolated yield, with a 14:1 dr and 99% ee (Table 1, entry 8).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R | Solvent | Co-catalyst | t (h) | Equiv. of 1a | Yield of (3a/5a and b) (%)b | dr of 5a and bd | ee (%) of 5a and be |
| a Experiments performed on a 0.1 mmol scale. See the ESI for details. b Yields were determined using 1,3,5-tris(trifluoromethyl)benzene as an internal standard unless otherwise noted. c Isolated yield determined after FC. d Determined using 1H NMR analysis of the crude mixture, see dr of the isolated compound in brackets. e Determined using chiral stationary phase UPC2. f Co-catalyst and dienophile were added after 1.5 h of reaction time. g All reactants were added simultaneously. h The reaction yielded a complex mixture of unidentified products. | ||||||||
| 1 | H | CHCl3 | — | 24 | 1 | 10/61 | 7:1 |
99 |
| 2 | H | Et2O | — | 24 | 1 | 37/34 | 8:1 |
99 |
| 3 | H | CH3CN | — | 24 | 1 | 14/33 | 10:1 |
99 |
| 4 | H | EtOH | — | 24 | 1 | 0/53 | 11:1 |
99 |
| 5f | H | CHCl3 | MgCl2 | 24 | 1 | 12/60 | 8:1 |
nd |
| 6f | H | CHCl3 | Yb(fod)3 | 24 | 1 | ndh | nd | nd |
| 7f | H | CHCl3 | Eu(fod)3 | 24 | 1 | 0/75 | 15:1 |
99 |
| 8f | H | CHCl3 | Eu(fod)3 | 30 | 1.5 | 0/88c | 14:1 (>20:1) |
99 |
| 9 | Me | CHCl3 | — | 24 | 1.5 | 65/29 | >20:1 |
99 |
| 10f | Me | CHCl3 | Eu(fod)3 | 26 | 1.5 | 0/90c | >20:1 (>20:1) |
99 |
| 11g | Me | CHCl3 | Eu(fod)3 | 24 | 1.5 | 18/40 | >20:1 |
nd |
| 12g | Me | CHCl3 | — | 24 | 1.5 | 60/28 | >20:1 |
99 |
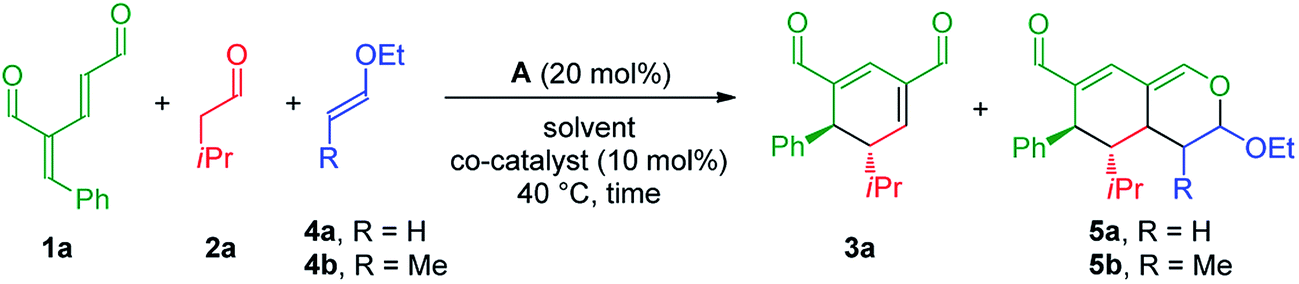
Having established the optimal reaction conditions with unsubstituted vinyl ether 4a as the substrate for the inverse-electron demand hetero-Diels–Alder reaction, we began exploring the possible employment of substituted vinyl ether 4b, which would potentially generate a fifth stereocenter in the tetrahydroisochromene scaffold. Applying similar conditions as for the reaction utilizing the unsubstituted vinyl ether 4a, the initial results revealed promising reactivity, as 5b was formed in 29% yield after 24 h with complete diastereoselectivity and 99% ee (Table 1, entry 9). These observations indicated a considerably longer reaction time for the cycloaddition as high amounts of intermediate 3a were still present in the crude reaction mixture, attributable to the more bulky dienophile, which could lead to the increased oxidation of intermediate 3a. Consequently, to accelerate the second reaction step a Lewis acid was added to catalyze the cycloaddition. In accordance with the prior screening results, the addition of Eu(fod)3 facilitated the formation of 5b with an isolated yield of 90%, >20:1 dr and 99% ee (Table 1, entry 10). Finally, we explored the possibility of adding all of the reactants simultaneously, potentially simplifying the reaction procedure while strengthening the synthetic applicability. Encouragingly, the unoptimized results demonstrated the potential yield of 5b to be high, without the addition of Eu(fod)3 (Table 1, entries 11 and 12).
We also tested the reaction for the formation of 5a with different loadings of organocatalyst A. The formation of 5a with 10 mol% of A gave 69% yield and the same stereoinduction, while with 5 mol% of A, only 36% yield of 5a was achieved. Performing the same reactions in the presence of Eu(fod)3 leads to a significant lowering of the yields compared to the use of 20 mol%. Using 5 mol% of A and 10 mol% Eu(fod)3 only gave 29% yield of 5a.
With the optimized reaction conditions in hand, the scope of the vinyl ether dienophiles 4 in the organocatalytic cascade reaction, using oxadendralenic dienal 1a and isovaleric aldehyde 2a as the reaction partners, was investigated (Scheme 3).
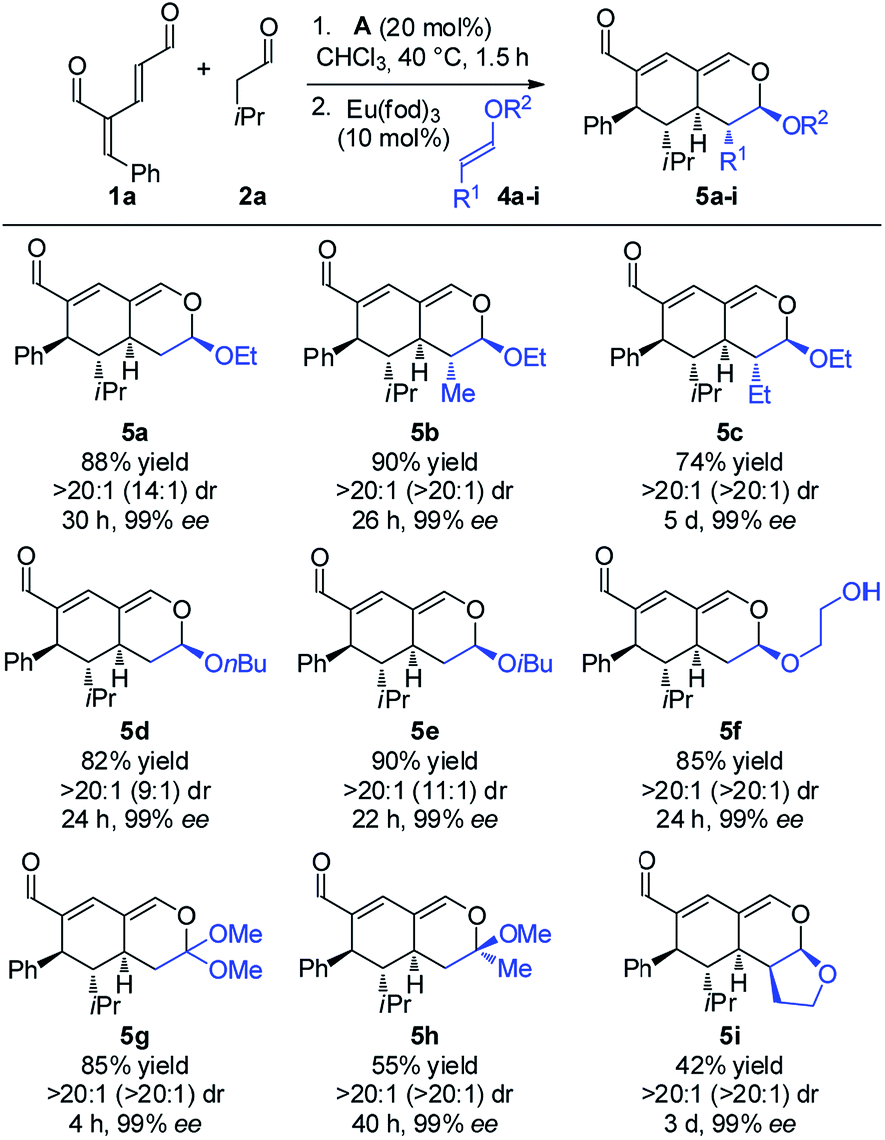 | ||
| Scheme 3 Scope of vinyl ethers 4a–i. | ||
Using unsubstituted vinyl ether 4a, tetrahydroisochromene 5a was isolated after 30 h of reaction time in a very good yield with perfect diastereoselectivity and an excellent enantioselectivity of 99% ee. Employing substituted vinyl ethers generated products (5b and c) with five consecutive stereocenters in 99% ee, >20:1 dr and high yields. Interestingly, extending the substituent of the vinyl ether by one carbon reduced the reaction rate of the inverse-electron demand hetero-Diels–Alder reaction significantly; however, variations of the ether substituent into branched or linear carbon chains were easily tolerated (5d and e). Furthermore, it was possible to incorporate a free hydroxy group, showing the robustness of the reaction (5f). By utilizing a double activated vinyl ether the reaction rate of the cycloaddition was accelerated considerably, reaching full conversion within 4 h and forming a fully substituted carbon center (5g). By means of a similar strategy, a tetrasubstituted stereocenter was installed (5h). Delightfully, it proved possible to use a cyclic dienophile, which proceeded to form the tricyclic product 5i in 42% yield. To demonstrate the utility of the procedure, a scaled-up experiment was performed on a gram scale (3.0 mmol) for the formation of 5b following the general conditions (see the ESI†).
Next, we focused the investigation towards the oxadendralenic dienals 1 and saturated aldehydes 2, using the substituted vinyl ether 4b as the dienophile to ensure better diastereoselectivity and the formation of a fifth stereocenter (Scheme 4).
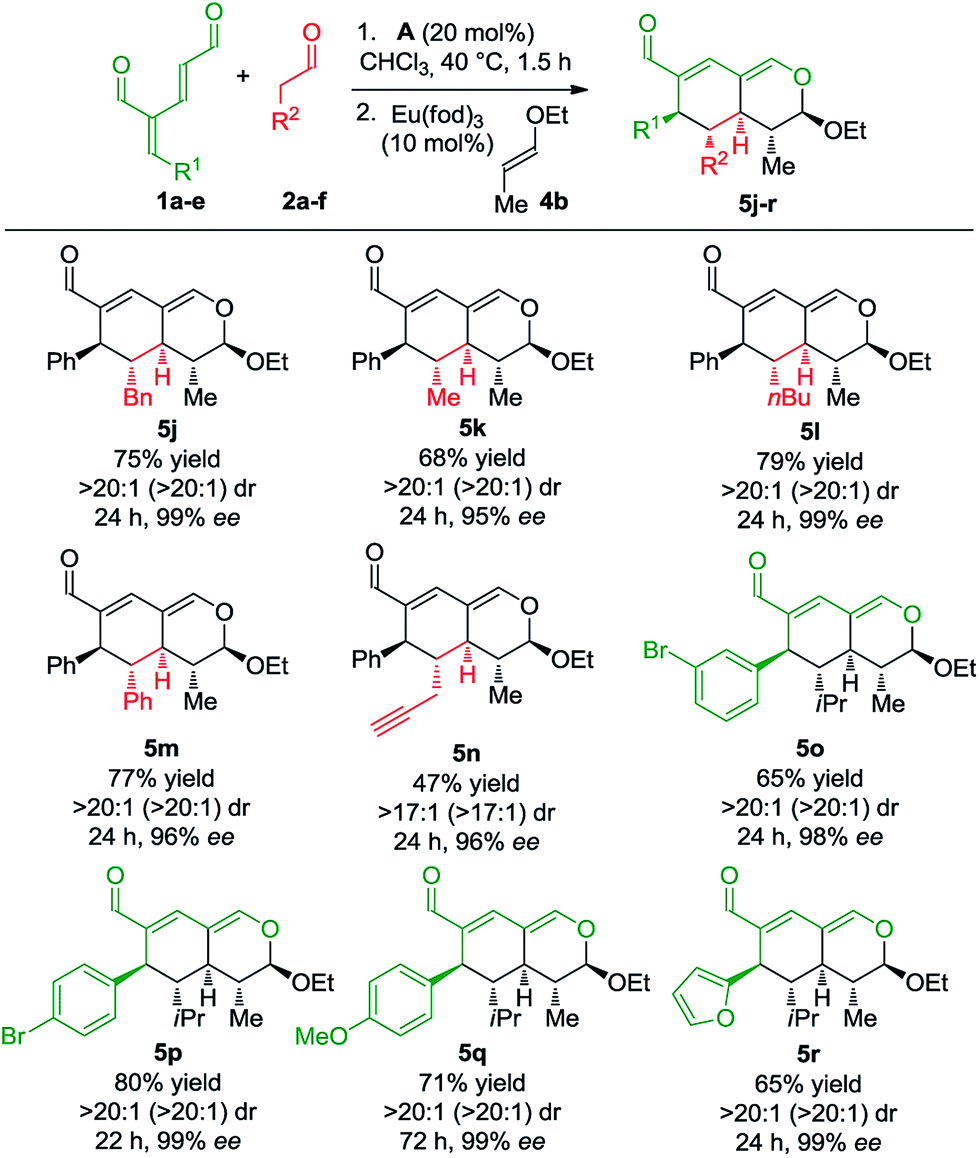 | ||
| Scheme 4 Scope of oxadendralenic dienals 1a–e and saturated aldehydes 2a–f. | ||
Various saturated aldehydes were then employed in the reaction outlined in Scheme 4. Both aliphatic and aromatic substituents on the saturated aldehydes afforded the desired products with perfect diastereoselectivity, high enantioselectivity and good yields (5j–m). Furthermore, an alkyne substituent was introduced forming 5n in 47% yield with >17:1 dr and 96% ee. The oxadendralenic dienals carrying meta- or para-substituted bromine underwent the reaction smoothly (5o and p), and the products displayed results comparable to the other scope entries. An electron-donating group was also well tolerated as the para-methoxy substituted oxadendralenic dienal 1d easily underwent the reaction (5q). Furthermore, it was possible to introduce a furan, forming 5r in 65% yield, >20:1 dr and 99% ee.
With a comprehensive scope in hand, we turned our attention towards establishing the absolute configuration of the obtained tetrahydroisochromenes 5. The relative configuration of tetrahydroisochromenes 5b and 5i (Fig. 4) was attained by means of X-ray analysis, from which the absolute configuration was assigned relative to 3b (Fig. 3). The configuration of the remaining tetrahydroisochromenes 5 was determined analogously.
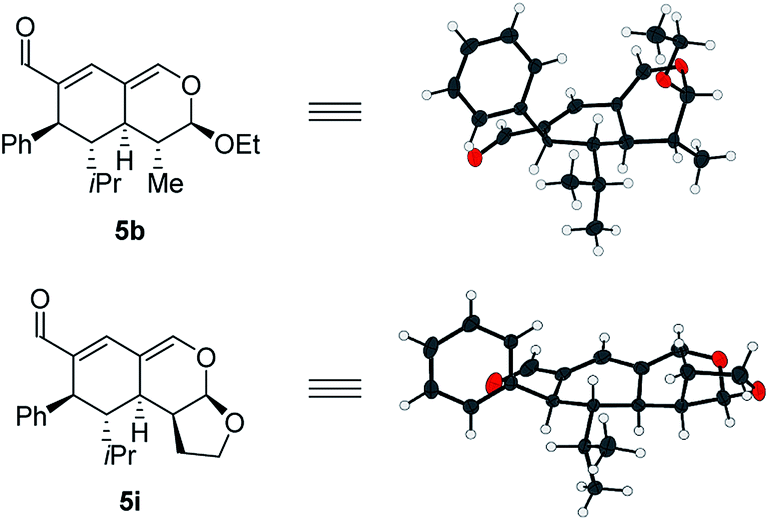 | ||
| Fig. 4 Absolute configuration of tetrahydroisochromenes 5b and i assigned with regards to 3b. | ||
The stereochemical outcome of the tetrahydroisochromenes 5b and 5i can be explained by assuming that the dienophile approaches from the top face due to the steric bulk of the R1-substituent (Scheme 5), and proceeds through an endo-transition state.
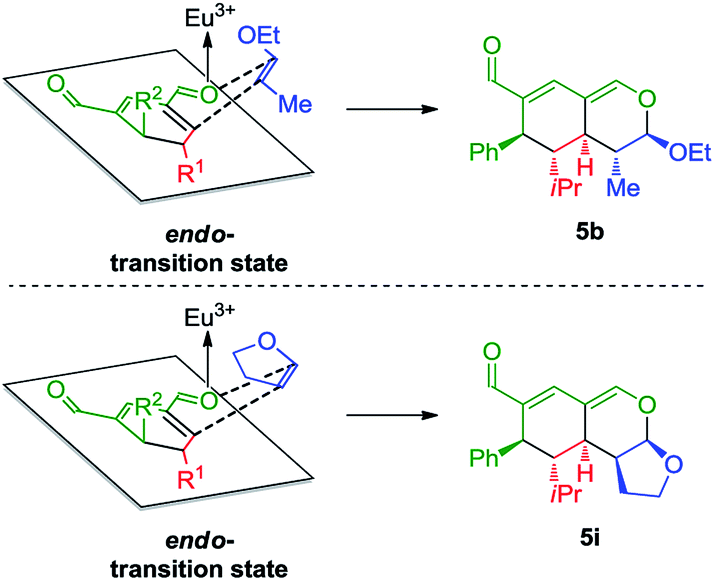 | ||
| Scheme 5 Transition state of the Lewis-acid catalyzed inverse-electron demand hetero-Diels–Alder reaction displaying preferred endo-selectivity. | ||
Sequential organocatalysis
Having established the scope and mechanistic aspects of the organocatalytic cascade featuring a sequential metal catalyzed inverse-electron demand hetero-Diels–Alder reaction, we set out to investigate the viability of utilizing aminocatalytic enamines as alternatives to vinyl ethers (see Scheme 1). We designed a preliminary experiment as a simple procedure, in which an excess of aldehyde 2 would be added to oxadendralenic dienal 1a with diphenylprolinol-silyl ether A in CHCl3. Encouragingly, when applying propionaldehyde 2c with oxadendralenic dienal 1a in the presence of 20 mol% A, the desired tetrahydroisochromene 6a was procured in 73% yield, a 7:1 dr and 99% ee (Table 2, entry 1). Furthermore, the reaction was successfully scaled up, providing 75% yield in a 2.0 mmol reaction. The observed diastereoselectivity is assumed to be controlled by the substituent in the R1-position of aldehyde 2, as the product most likely exists in an equilibrium between the two anomers of 6a. If isovaleric aldehyde 2a and hydrocinnamaldehyde 2b were employed in the envisioned procedure, only the formation of the corresponding cyclic oxadendralenic intermediate 3 would be observed. It is assumed that the lack of reactivity results from steric interactions. Hence, we reasoned that an aldehyde with a smaller substituent was needed for the reaction to proceed. Thus, we explored the hypothesis of incorporating acetaldehyde 2g as part of the two-step organocatalytic cascade. Following the formation of the cyclic oxadendralenic intermediate 3, the organocatalytic cycloaddition proceeded well, yielding 6b in 60% yield, 99% ee and a 2:1 dr (Table 2, entry 2).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | A (step 1) | A (step 2) | Temp. (step 2) | t (h) | Product | Yieldb (%) | drc | eed (%) |
| a Experiments performed on a 0.2 mmol scale following general procedure C. See ESI Section 5.1 for details. b Isolated yield determined after FC. c Determined using 1H NMR analysis of the isolated compound. d Determined using chiral stationary phase UPC2. | ||||||||||
| 1 | Me | Me | (S)-A | — | 40 °C | 24 | 6a | 73 | 7:1 |
99 |
| 2 | Me | H | (S)-A | — | 40 °C | 24 | 6b | 60 | 2:1 |
99 |
| 3 | Bn | Bn | (S)-A | (R)-A | 40 °C | 36 | 6c | 49 | >20:1 |
99 |
| 4 | nBu | nBu | (S)-A | (R)-A | rt | 72 | 6d | 45 | >20:1 |
99 |
| 5 | Et | Et | (S)-A | (R)-A | rt | 48 | 6e | 47 | >20:1 |
99 |
In addition to these results, the relative configuration of 6a (Fig. 5) was obtained using X-ray analysis and the absolute configuration was assigned relative to 3b (Fig. 3). Interestingly, the second enamine addition provides 6a with the opposite diastereoselectivity in the dihydropyran ring compared to the formation of tetrahydroisochromene 5b. As the bottom-face of the cyclic oxadendralenic intermediate 3 is assumed to be sterically blocked, the enamine has to approach from above and proceed through an exo-transition state in the inverse-electron demand hetero-Diels–Alder reaction to obtain the observed stereochemistry. This is peculiar as the exo-transition state with an enamine intermediate appearing in an (E-s-trans)-conformation would be unfavorable as a result of the steric clash between 3 and the bulk of the aminocatalyst (S)-A (Fig. 6, left). However, calculations have shown that a methyl-substituted enamine may just as well exist in the (E-s-cis)-form, and that this conformation is as favorable as the corresponding (E-s-trans)-enamine.20 Conversely, the (E-s-cis)-geometry of an enamine substituted with a larger substituent is less favorable. Hence, it is possible that the enamine intermediate of propionaldehyde 2c reacts through the (E-s-cis)-conformation, which also explains why isovaleric aldehyde 2a and hydrocinnamaldehyde 2b did not work in the reaction as their substituents are too bulky to exist in this form (Fig. 6, left). Based on these considerations, we envisioned adding the other enantiomer of aminocatalyst A after the formation of cyclic oxadendralenic intermediate 3, which should allow for more bulky aldehydes to react in an exo-transition state through the (E-s-trans)-conformation (Fig. 6, right).
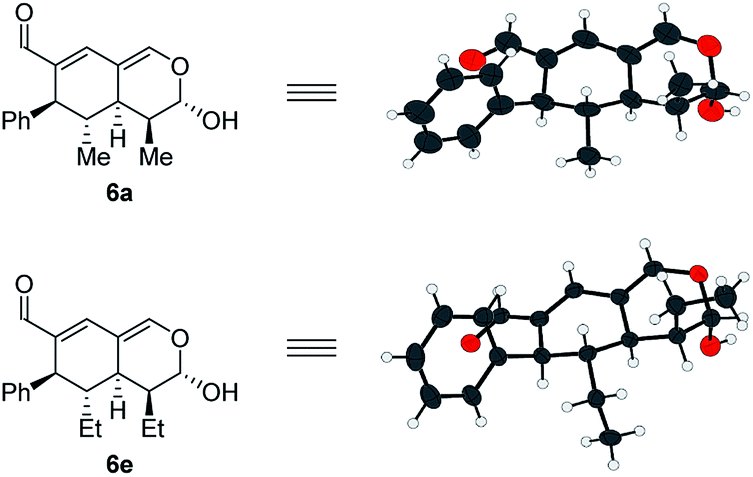 | ||
| Fig. 5 Absolute configuration of tetrahydroisochromenes 6a and 6e assigned with regards to 3b. | ||
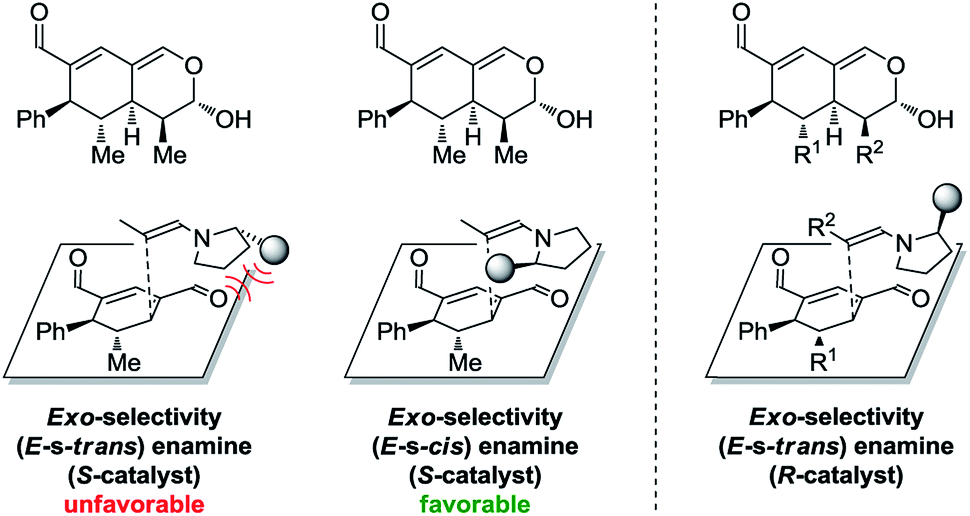 | ||
| Fig. 6 Left: The mechanistic reasoning behind the stereochemical configuration of 6a. Right: Proposed solution to extend the scope. | ||
We tested this hypothesis by adding 20 mol% (R)-A to a reaction mixture containing the cyclic oxadendralenic intermediate 3 formed in the reaction between an excess of hydrocinnamaldehyde 2b, 20 mol% (S)-A and oxadendralenic dienal 1a. Full conversion was achieved within 36 h and 6c was isolated in 49% yield, >20:1 dr and 99% ee (Table 2, entry 3). This led us to extend the scope of the reaction, and it was demonstrated that saturated aldehydes bearing linear carbon substituents were tolerated, giving tetrahydroisochromenes 6d and e in >20:1 dr, 99% ee and 45–47% yield. The absolute configuration of 6e (Fig. 5) was determined by analogy with 3b (Fig. 3), and the observed stereochemistry is consistent with the proposed theory (Fig. 6). To the best of our knowledge this is one of the very rare cases where the enamine intermediate reacts through the (E-s-cis)-conformer. It should be noted that the implementation of achiral secondary amines such as pyrrolidine in the second step (as a cheap alternative to (R)-A) was unsuccessful, as their increased basicity resulted in an increased rate of the undesired oxidation of intermediate 3a into the aromatic 2,4-dicarbaldehyde species.
Transformations
With the intention of strengthening the utility of the developed one-pot cascades even further, transformations of the tetrahydroisochromenes were performed. In particular, functionalization of the lactol moiety in 6a was explored. First, it was anticipated that the lactol could be oxidized to the corresponding lactone. Using a modified procedure wherein NaHCO3 was added to buffer the acidity of the Dess–Martin periodinane, the oxidized product 7 was obtained in 38% yield after only 5 h (Scheme 6).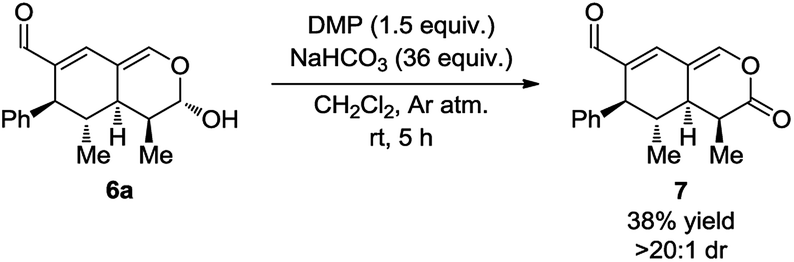 | ||
| Scheme 6 Modified Dess–Martin oxidation of the lactol moiety. | ||
Next, it was envisioned that the lactol moiety in 6a could be converted into an acetal, attaining a product complimentary to tetrahydroisochromene 5k; however, with the opposite stereochemistry of the substituent positioned next to the acetal. A DMAP-catalyzed acetylation of the hydroxyl group afforded the acetylated intermediate, which subsequently underwent a TMSOTf promoted coupling, yielding the desired product 8 in a 7:1 dr and 57% yield (Scheme 7).
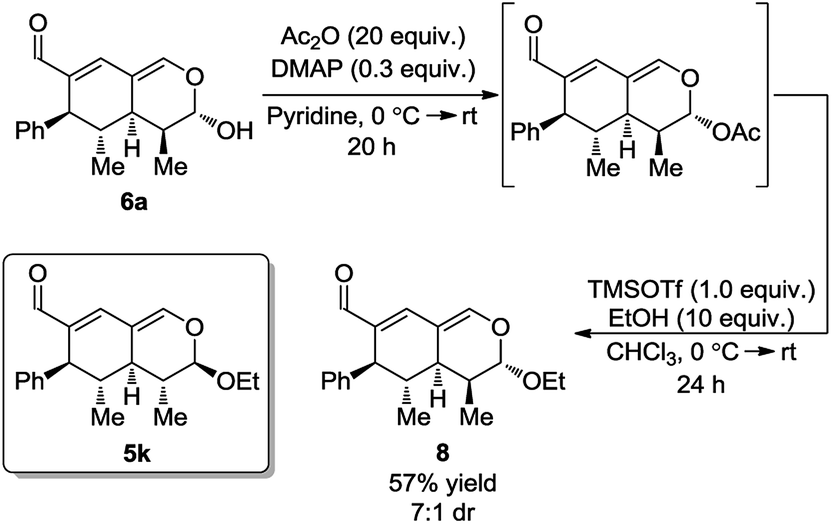 | ||
| Scheme 7 DMAP-catalyzed acetylation and coupling. | ||
Conclusion
In conclusion, oxadendralenes are introduced in asymmetric catalysis for the construction of chiral bicyclic heterocyclic scaffolds by applying synergistic catalysis. The development is based on the dual activation of the oxadendralene and an aldehyde which forms a vinylogous iminium-ion intermediate and an enamine, respectively. These two intermediates are set-up for the 1,6-addition of the enamine to the vinylogous iminium-ion. This reaction generates a novel cyclic oxadendralenic intermediate, which in a Eu(fod)3-catalyzed hetero-Diels–Alder reaction with vinyl ethers forms tetrahydroisochromenes with five continuous stereocenters.The scope of the reaction is demonstrated for a great variety of oxadendralenes, aldehydes and vinyl ethers giving high substituent diversity of the tetrahydroisochromenes formed in high yields and >20:1 dr and up to 99% ee. The dual activation concept is supported by characterization of the vinylogous iminium-ion intermediate formed by reaction of the oxadendralene and the organocatalyst, and the stereochemical outcome of the reaction. The reaction concept has been extended to a double organocatalytic reaction, in which the enamine formed as an intermediate in the first catalytic cycle can be utilized in a second catalytic cycle providing tetrahydroisochromenes with a different stereochemical outcome in moderate to good yields, >20:1 dr and up to 99% ee. Finally, the formation of an attractive lactone moiety and diastereoselective acetalization has been demonstrated. The presented novel developments might have the potential to be applied for the formation of novel anti-malaria drug candidates, as the formed tetrahydroisochromene core structure is the central skeleton in artemisinin and arteether. Furthermore, the reaction concept might also be integrated in the synthesis of anti-cancer candidates related to oridonin.
Acknowledgements
This work was financially supported by the Aarhus University, FNU and the Carlsberg Foundation.Notes and references
- (a) S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS PubMed; (b) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef PubMed; (c) M. Garcia-Castro, L. Kremer, C. D. Reinkemeier, C. Unkelbach, C. Strohmann, S. Ziegler, C. Ostermann and K. Kumar, Nat. Commun., 2015, 6, 6516 CrossRef CAS PubMed; (d) D. S. Tan, Nat. Chem. Biol., 2005, 1, 74 CrossRef CAS PubMed.
- For general reviews regarding asymmetric organocatalysis, see: (a) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed; (b) Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, ed. P. I. Dalko, Wiley-VCH, Weinheim, Germany, 2013 Search PubMed; (c) M. E. Abbasov and D. Romo, Nat. Prod. Rep., 2014, 31, 1318 RSC; (d) F. Giacalone, M. Gruttadauria, P. Agrigento and R. Noto, Chem. Soc. Rev., 2012, 41, 2406 RSC. For reviews regarding general organocatalytic systems, see: (e) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312 CrossRef CAS PubMed; (f) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS; (g) C. Palomo, M. Oiarbide and R. López, Chem. Soc. Rev., 2009, 38, 632 RSC; (h) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040 RSC; (i) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS PubMed; (j) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588 RSC.
- For reviews, see: (a) E. Marqués-López, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138 RSC; (b) J. Alemán and S. Cabrera, Chem. Soc. Rev., 2013, 42, 774 RSC; (c) R. M. de Figueiredo and M. Christmann, Eur. J. Org. Chem., 2007, 2575 CrossRef CAS; (d) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed; (e) B.-F. Sun, Tetrahedron Lett., 2015, 56, 2133 CrossRef CAS.
- (a) I. Atodiresei, C. Vila and M. Rueping, ACS Catal., 2015, 5, 1972 CrossRef CAS; (b) I. R. Shaikh, J. Catal., 2014 DOI:10.1155/2014/402860. For selected examples of organocatalysis used in industrial processes, see: (c) G. Sedelmeier, New Methods, WIPO Patent, WO 2008119804 A1, Novartis AG, 2008; (d) F. Xu, M. Zacuto, N. Yoshikawa, R. Desmond, S. Hoerrner, T. Itoh, M. Journet, G. R. Humphrey, C. Cowden, N. Strotman and P. Devine, J. Org. Chem., 2010, 75, 7829 CrossRef CAS PubMed.
- (a) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef PubMed; (b) R. C. Wende and P. R. Schreiner, Green Chem., 2012, 14, 1821 RSC; (c) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS PubMed; (d) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211 RSC. For an example of an organocatalytic one-pot strategy yielding an important drug, see: (e) H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304 CrossRef CAS PubMed.
- For reviews, see: (a) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337 RSC; (b) C. C. J. Loh and D. Enders, Chem.–Eur. J., 2012, 18, 10212 CrossRef CAS PubMed; (c) A. Gualandi, L. Mengozzi, C. M. Wilson and P. G. Cozzi, Chem.–Asian J., 2014, 9, 984 CrossRef CAS PubMed.
- (a) H. Hopf and M. S. Sherburn, Angew. Chem., Int. Ed., 2012, 51, 2298 CrossRef CAS PubMed; (b) M. S. Sherburn, Acc. Chem. Res., 2015, 48, 1961 CrossRef CAS PubMed.
- (a) C. Spino and G. Liu, J. Org. Chem., 1993, 58, 817 CrossRef CAS; (b) C. Spino, G. Liu, N. Tu and S. Girard, J. Org. Chem., 1994, 59, 5596 CrossRef CAS; (c) C. Spino and N. Tu, Tetrahedron Lett., 1994, 35, 3683 CrossRef CAS; (d) F. Tripoteau, T. Verdelet, A. Hercouet, F. Carreaux and B. Carboni, Chem.–Eur. J., 2011, 17, 13670 CrossRef CAS PubMed; (e) S. Kobayashi, K. Kudo, A. Ito, T. Honjo, M. Yata, T. Otani, N. Kutsumura, T. Saito, F. Berrée, E. Romain, F. Tripoteau and B. Carboni, Eur. J. Org. Chem., 2015, 4367 CrossRef CAS.
- (a) V. Eschenbrenner-Lux, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2014, 53, 11146 CrossRef CAS PubMed; (b) H. Pellissier, Tetrahedron, 2009, 65, 2839 CrossRef CAS; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- The 2015 Nobel Prize in Physiology or Medicine – Press Release, http://www.nobelprize.org/nobel_prizes/medicine/laureates/2015/press.html, accessed Dec 1, 2015.
- (a) C. Zhu and S. P. Cook, J. Am. Chem. Soc., 2012, 134, 13577 CrossRef CAS PubMed; (b) S. P. Cook, Synlett, 2014, 25, 751 CrossRef CAS.
- (a) N. J. White, Science, 2008, 320, 330 CrossRef CAS PubMed; (b) B. Carol and H. Sibley, Science, 2015, 347, 373 CrossRef PubMed; (c) A. M. Dondorp, S. Yeung, L. White, C. Nguon, N. P. J. Day, D. Socheat and L. von Seidlein, Nat. Rev. Microbiol., 2010, 8, 272 CAS.
- (a) T. Zhen, C.-F. Wu, P. Liu, H.-Y. Wu, G.-B. Zhou, Y. Lu, J.-X. Liu, Y. Liang, K. K. Li, Y.-Y. Wang, Y.-Y. Xie, M.-M. He, H.-M. Cao, W.-N. Zhang, L.-M. Chen, K. Petrie, S.-J. Chen and Z. Chen, Sci. Transl. Med., 2012, 4, 127ra38 Search PubMed; (b) C. Y. Li, E. Q. Wang, Y. Cheng and J. K. Bao, Int. J. Biochem. Cell Biol., 2011, 43, 701 CrossRef CAS PubMed.
- (a) R. C. Fuson, Chem. Rev., 1935, 16, 1 CrossRef CAS; (b) L. Dell'Amico, Ł. Albrecht, T. Naicker, P. H. Poulsen and K. A. Jørgensen, J. Am. Chem. Soc., 2013, 135, 8063 CrossRef PubMed; (c) M. Silvi, I. Chatterjee, Y. Liu and P. Melchiorre, Angew. Chem., Int. Ed., 2013, 52, 10780 CrossRef CAS PubMed.
- (a) Lewis Acids and Selectivity in Organic Synthesis, ed. M. Santelli and J.-M. Pons, CRC Press, Boca Raton, Florida, 1996 Search PubMed; (b) K. A. Jørgensen, in Cycloaddition Reactions in Organic Synthesis, ed. S. Kobayashi and K. A. Jørgensen, Wiley-VCH, Weinheim, Germany, 2002, ch. 4, pp. 151–185 Search PubMed.
- First example of an organocatalytic enamine-facilitated inverse-electron demand hetero-Diels–Alder reaction: (a) K. Juhl and K. A. Jørgensen, Angew. Chem., Int. Ed., 2003, 42, 1498 CrossRef CAS PubMed. For selected examples, see: (b) S. Samanta, J. Krause, T. Mandal and C.-G. Zhao, Org. Lett., 2007, 9, 2745 CrossRef CAS PubMed; (c) D. Xu, Y. Zhang and D. Ma, Tetrahedron Lett., 2010, 51, 3827 CrossRef CAS.
- (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794 CrossRef CAS PubMed; (b) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS PubMed; (c) B. S. Donslund, T. K. Johansen, P. H. Poulsen, K. S. Halskov and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 13860 CrossRef CAS PubMed . Testing other diarylprolinol-silyl ethers as catalysts gave less satisfactory results.
- (a) J. J. Murphy, A. Quintard, P. McArdle, A. Alexakis and J. C. Stephens, Angew. Chem., Int. Ed., 2011, 50, 5095 CrossRef CAS PubMed; (b) B. Pezzati, M. F. Chellat, J. J. Murphy, C. Besnard, G. Reginato, J. C. Stephens and A. Alexakis, Org. Lett., 2013, 15, 2950 CrossRef CAS PubMed.
- This is according to the best of our knowledge the first application of europium-catalyzed inverse-electron demand hetero-Diels–Alder reaction in combination with organocatalysis and heterodendralenes.
- P. Dinér, A. Kjærsgaard, M. A. Lie and K. A. Jørgensen, Chem.–Eur. J., 2008, 14, 122 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and full compound characterization, including NMR data, UPC2 traces and crystallographic data. CCDC 1419567, 1408759, 1405309, 1405275, 1405277 and 1419566. For the ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00185h |
| This journal is © The Royal Society of Chemistry 2016 |