
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of dynamic ligand exchange in the oxidation chemistry of cerium(III)†
Jerome R.
Robinson
,
Yusen
Qiao
,
Jun
Gu
,
Patrick J.
Carroll
,
Patrick J.
Walsh
* and
Eric J.
Schelter
*
P. Roy and Diana T. Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, PA 19104, USA. E-mail: pwalsh@sas.upenn.edu; schelter@sas.upenn.edu
First published on 23rd March 2016
Abstract
The CeIII/IV couple is useful for many applications in organic, inorganic, and materials chemistry. However, attaining a general method to access both oxidations states through reversible solution redox chemistry remains challenging. Herein we report the synthesis, characterization, and oxidation chemistry of the novel Ce/Li REMB heterochiral diastereomer, 1-Ce(het). The solution exchange processes of 1-RE(het) (RE = Ce and Yb) were investigated to estimate rates of ligand and cation exchange relevant in homochiral and heterochiral frameworks. A detailed mechanistic investigation following the solution dynamics of 1-Ce(het) revealed reactivity controlled both by ligand reorganization and redistribution processes. Ligand reorganization was responsible for the kinetics associated with the chemical oxidation reaction, whereas ligand redistribution and exchange dictated the isolated products.
Introduction
The metal–ligand bonds formed in trivalent rare earth metal (RE) complexes are ionic in nature, and show faster rates of ligand exchange compared to the more covalent metal–ligand bonds found in transition metal systems.1 The increased ligand lability can be an advantageous property for applications such as MRI contrast agents, sensors, and catalysts,2 however, only if suitable ancillary ligand design strategies are implemented to enforce specific coordination environments and control rates of ligand exchange.2c,3 Well known methods to control rates of ligand exchange with transition metal and lanthanide complexes include the use of bulky or multidentate ligands.4 More challenging, however, is development of general and predictable ligand-based strategies to control redox chemistry with molecular lanthanide complexes.5 Among the lanthanides cerium's accessible 4f1/4f0 redox couple has attracted much attention due to its diverse applications in organic, inorganic, and materials chemistry.4a,5e,6 Nonetheless, its applications in solution chemistry could be significantly broadened if a better understanding of the factors that control its redox properties, including ligand reorganization and exchange processes, could be gained.Recently, we reported an alternative strategy for controlled cerium redox chemistry making use of a heterobimetallic platform, [M3(THF)n][(BINOLate)3RE] (Fig. 1; REMB; RE = Ce, M = Li, Na, K, and Cs; B = (S)-BINOLate; RE/M/B = 1/3/3). We found that the properties and oxidation chemistry of CeIII were modulated by ligand reorganization.7 The ability of the CeIII complex to reorganize the ligand framework and coordinate neutral ligands, such as THF or TPPO (TPPO = triphenylphosphine oxide) or oxidants was controlled from the secondary coordination sphere through simple choice of the main group metal, M. We found that when M = Li, [Li3(THF)4][(BINOLate)3Ce(THF)] (1-Ce(homo)), wherein all the BINOLate ligands have the same configuration, the complex could readily reorganize to stabilize similar coordination numbers and geometries upon oxidation (Fig. 1). This characteristic facilitated general, predictable, and controlled oxidation chemistry. The net result of this ligand reorganization was striking, a seemingly small perturbation such as shifting the equilibrium constant for coordination of a Lewis basic ligand (THF, TPPO) at the RE cation led to over a 130-fold difference in chemical oxidation rates and resulted in completely different product outcomes.
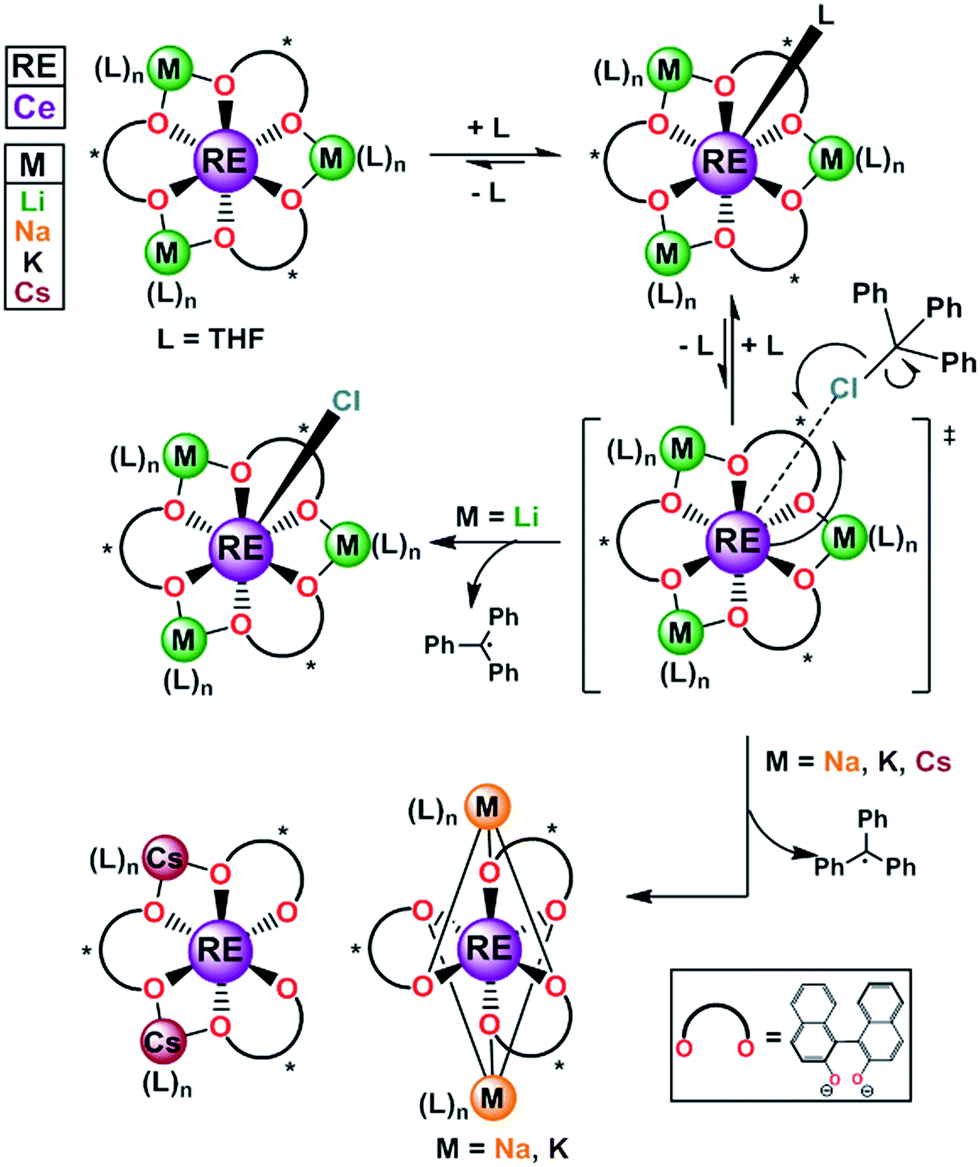 | ||
| Fig. 1 The homochiral REMB framework, 1-RE(homo) and reactivity with inner sphere oxidant trityl chloride. RE = rare earths; M = Li, Na, K, Cs; B = (S)-BINOLate; RE/M/B = 1/3/3. | ||
While the impact of ligand reorganization on cerium redox chemistry has been demonstrated experimentally,7,8 the impact of other associated dynamic processes, such as ligand exchange/redistribution, have yet to be determined. These studies are necessary to understand the prevalence of redistribution products observed for oxidation reactions of CeIII.5a,c,8b,9 As such, we set out to explore the impact of ligand exchange/redistribution, and delineate their contributions from that of ligand reorganization on the observed reactivity and properties involving oxidation of CeIII. Herein we report the synthesis and characterization of the novel Ce/Li REMB heterochiral diastereomer, 1-Ce(het), which displays reactivity that is controlled by both ligand reorganization and redistribution processes. Ligand reorganization largely contributed to the kinetics of the chemical oxidation, whereas ligand redistribution and exchange dictated the ultimate isolated products. Characterization of the ligand exchange processes of 1-RE(het) (RE = Ce and Yb) facilitated the quantification of Li+ and BINOLate exchange of these frameworks. Our study sheds light on relevant dynamic processes and provides insight into the available pathways possible in CeIII oxidation chemistry.
Results and discussion
As we set out to study and parse the effects of ligand reorganization, redistribution, and exchange on the oxidation chemistry of CeIII, we needed a framework that would enable the systematic control of these processes. We postulated that mixtures of homochiral and heterochiral diastereomers of the Ce/Li REMB framework, 1-Ce(homo) and 1-Ce(het), respectively, could meet our needs. We recently demonstrated the effects of ligand reorganization and neutral ligand exchange in the CeIII/Li+ system, 1-Ce(homo).7 Aspinall,10 Salvadori,11 and Shibasaki,12 have independently demonstrated that: (1) (SSR)- or (RRS)-[Li3(THF)6][(BINOLate)3RE], 1-RE(het) (REIII = Y or Yb), were six-coordinate RE complexes in the solid state, (2) YbIII cations in these frameworks were not accessible to additional coordination by Lewis bases, and (3) mixtures of enantiopure [Li3(THF)6][(BINOLate)3Y], (SSS)-1-Y(homo) and (RRR)-1-Y(homo), underwent ligand redistribution to form the thermodynamically preferred heterochiral diastereomer, 1-Y(het), respectively. While seminal contributions, these studies have not investigated the rates of inter- or intramolecular BINOLate and cation exchange, nor the synthesis of REIII larger than YIII. In this context, we sought to synthesize the novel heterochiral diastereomer, 1-Ce(het), characterize its dynamic exchange processes and determine the effects of those processes on the electrochemical and chemical oxidation properties of CeIII.Synthesis and characterization of 1-Ce(het)
Initially, we examined salt-metathesis synthetic routes with Ce(OTf)3, (rac)-BINOL, and LiN(SiMe3)2 or Li(OtBu), which resulted in formation of 1-Ce(het). Unfortunately, isolation of analytically pure heterobimetallic product was complicated due to the similar solubilities of 1-Ce(het) and the salt by-products (LiOTf). Similar to 1-Ce(homo), the synthesis of [Li3(THF)6][(BINOLate)3Ce], 1-Ce(het), was accomplished through protonolysis by adding 3 equiv. each of racemic BINOL and Li(N(SiMe3)2 to a stirring THF solution of Ce[N(SiMe3)2]3. Layering concentrated THF solutions of 1-Ce(het) with hexanes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v) at −35 °C produced bright yellow-orange crystals in 74% yield (Scheme 1).
:4 v/v) at −35 °C produced bright yellow-orange crystals in 74% yield (Scheme 1).
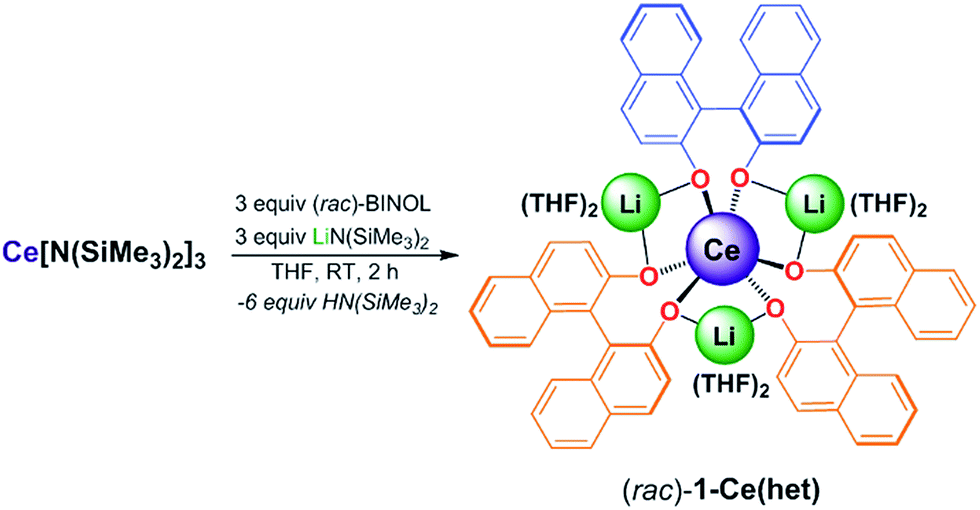 | ||
| Scheme 1 Synthesis of (rac)-1-Ce(het). The BINOLate ligands have been colored blue and orange to clearly indicate the heterochiral configuration of the BINOLate ligand fragments as found in the X-ray crystal structure. | ||
Solid-state characterization of 1-Ce(het)
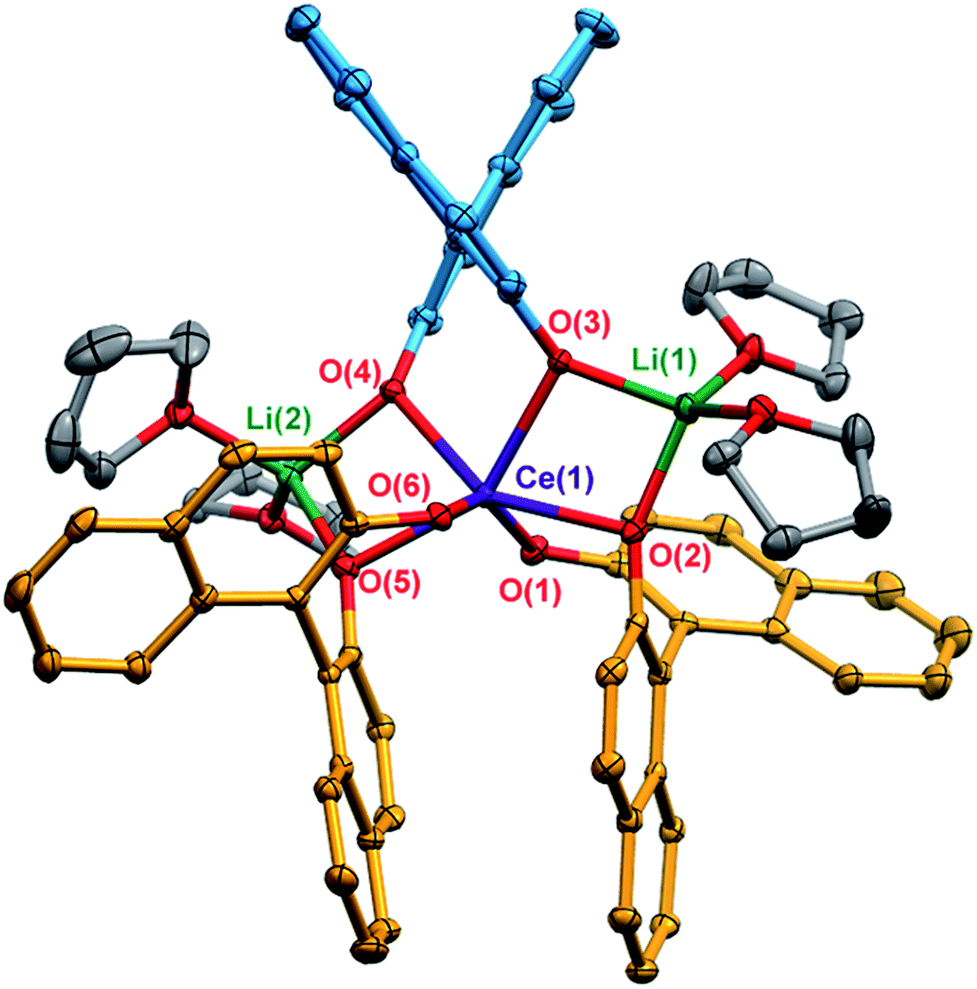
X-ray diffraction studies confirmed the formation of the expected racemic heterochiral diastereomer, 1-Ce(het) (RRS/SSR, Fig. 2), where the coordination environment of the CeIII cation was isostructural to the known complexes 1-Y(het) and 1-Yb(het).10 Orientation of the RRS/SSR BINOLate fragments lead to a different coordination environment for 1-Ce(het) compared to 1-Ce(homo). Whereas the CeIII cation in 1-Ce(homo) readily accommodated a seventh ligand (THF or TPPO), the heterochiral arrangement of the BINOLate fragments sterically block the CeIII cation in 1-Ce(het) toward interaction with donor solvents such as THF. Average CeIII–OBINOLate bond distances for 1-Ce(het) were shorter by 0.0380(1) Å compared to 1-Ce(homo), and reflect the difference in ionic radii between six- and seven-coordinate CeIII cations.13 The decreased coordination number at the CeIII cation in 1-Ce(het)versus1-Ce(homo) was accompanied by increased solvation at the Li+ cations, providing further stabilization for 1-Ce(het). Similar to solid state structures of 1-Y(het) and 1-Yb(het),10 a weak C–H to π hydrogen bond was observed between a BINOLate C–H and the neighboring BINOLate naphthyl π system (Fig. S21†).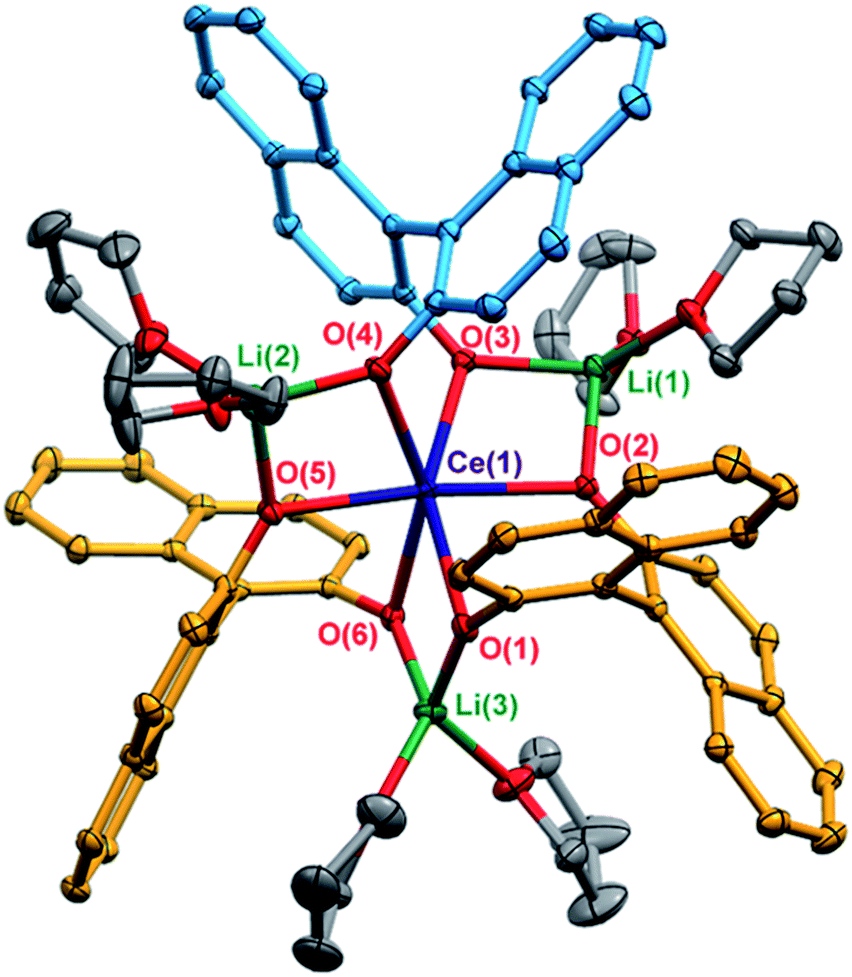 | ||
| Fig. 2 Thermal ellipsoid plot of 1-Ce(het) shown at 30% probability. The BINOLate ligands have been colored blue and orange to indicate the SS/RR and R/S fragments respectively. Selected bond distances (Å): Ce(1)–O(1) 2.3908(16), Ce(1)–O(2) 2.3924(16), Ce(1)–O(3) 2.3900(17), Ce(1)–O(4) 2.3947(16), Ce(1)–O(5) 2.3743(16), Ce(1)–O(6) 2.3909(17), Li(1)–O(2) 1.888(5), Li(1)–O(3) 1.946(4), Li(2)–O(4) 1.927(5), Li(2)–O(6) 1.899(5), Li(3)–O(1) 1.946(4), Li(3)–O(5) 1.955(4). | ||
Solution NMR characterization of 1-Ce(het)
While 1-Ce(het) was identified as the only diastereomer in the solid state, 1H-, 7Li-, and 13C{1H}-NMR spectroscopy revealed a varied energy landscape in solution (Fig. 3). Dissolving crystals of 1-Ce(het) in THF-d8 resulted in two major solution species, 1-Ce(het) and 1-Ce(homo). These diastereomers were readily identified using 1D 1H-, 7Li-, and 13C{1H}-NMR (Fig. 3) and 2D-NMR correlation spectroscopy (COSY and HSQC; Fig. S5 and S6†) techniques. As determined by integration of 1H and 7Li-NMR spectra, 1-Ce(het) and 1-Ce(homo) were present in a ∼6:1 ratio (K = 0.17), which corresponded to an energy difference ΔG (300 K) of 0.63 kcal mol−1 between the two diastereomeric forms. In comparison, ΔG (300 K) must be greater than 1.79 kcal mol−1 (K ≈ 0.05) for the smaller REs, YIII and YbIII, as the homochiral diastereomers were reportedly not observed using solution NMR spectroscopy.10,12
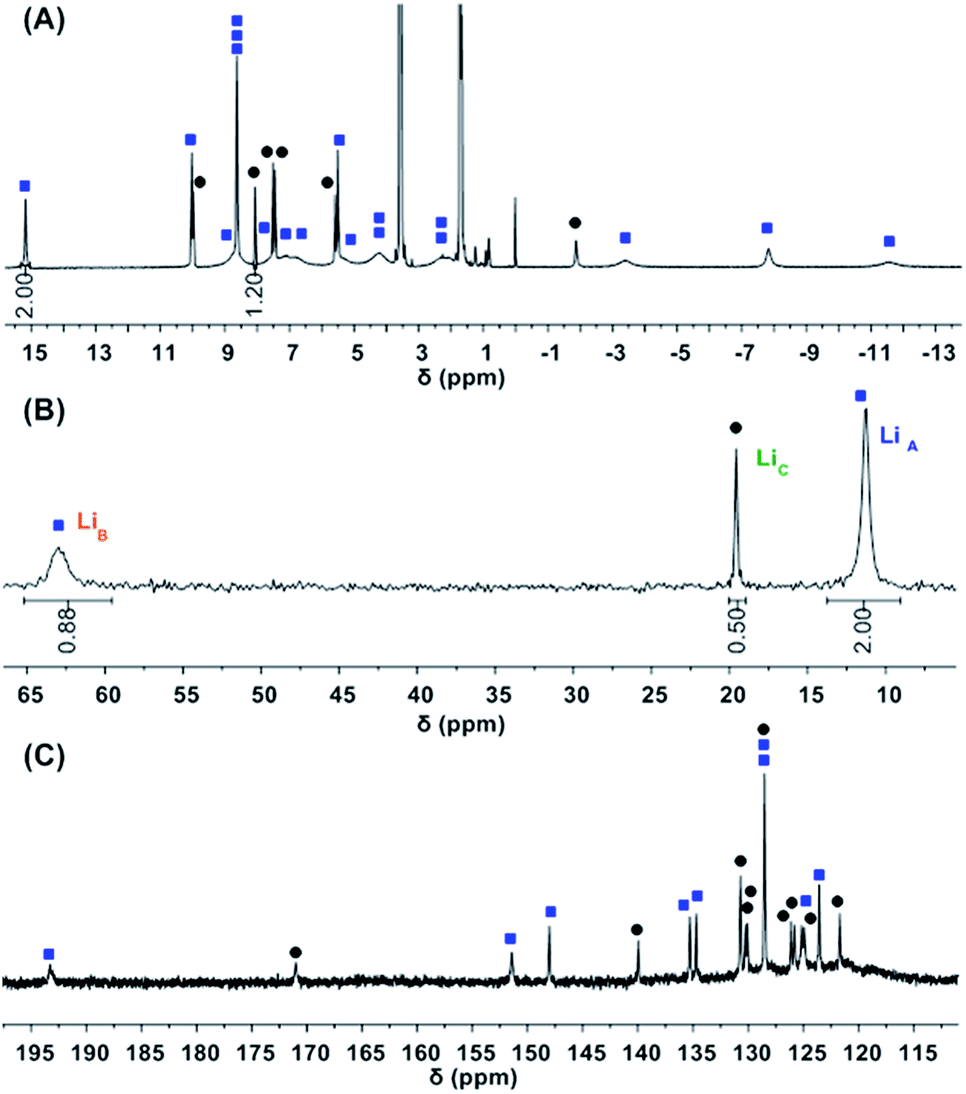 | ||
Fig. 3 (A) 1H- (top), (B) 7Li-NMR (middle), and (C) 13C{1H}-NMR (bottom) of 1-Ce(het) (1-Ce = 0% ee).  = 1-Ce(het), = 1-Ce(het),  = 1-Ce(homo). = 1-Ce(homo). | ||
The energy differences for the two diastereomers, 1-RE(homo) and 1-RE(het), are a consequence of the ionic radii of the REIII cation. CeIII, a large REIII cation, can more readily accommodate a seven-coordinate geometry than a small RE like YbIII due to increased steric accessibility.7,14 In turn, this enthalpic gain from coordination of a seventh ligand stabilizes formation of the homochiral diastereomer, and is more favorable for the early REIII. This observation was further supported by 1H and 7Li-NMR spectroscopy performed in a more strongly coordinating solvent, pyridine-d5 (pyr-d5). In pyr-d5 only the homochiral diastereomer [Li3(sol)n][(BINOLate)3Ce(sol)] (sol = pyr; n = 5), 1-Ce(homo), was observed by NMR (Fig. S3†). These results indicated that the thermodynamic preference of the diastereomeric forms in solution cannot only be controlled by the size of REIII cation, but also through choice of solvent.
Solution concentrations of the heterochiral and homochiral diastereomers of 1-RE (RE = Ce and Yb) were determined by 1H- and 7Li-NMR spectroscopy over a range of optical purities (0–100% ee). Non-linear relationships between the concentration of 1-RE(homo) and optical purity of BINOL were observed for both CeIII (Fig. 4) and YbIII cations (Fig. S16†). Ligand redistribution/exchange reactions between 1-RE(het) and 1-RE(homo) (RE = Ce, Yb) were completed before the time needed to prepare and record the NMR spectra (<2 min). Variable-temperature NMR (VT-NMR) studies indicated that the equilibrium constant for 1-Ce(het)/1-Ce(homo) was effectively temperature independent over the range of 215 to 315 K (Fig. S7†).
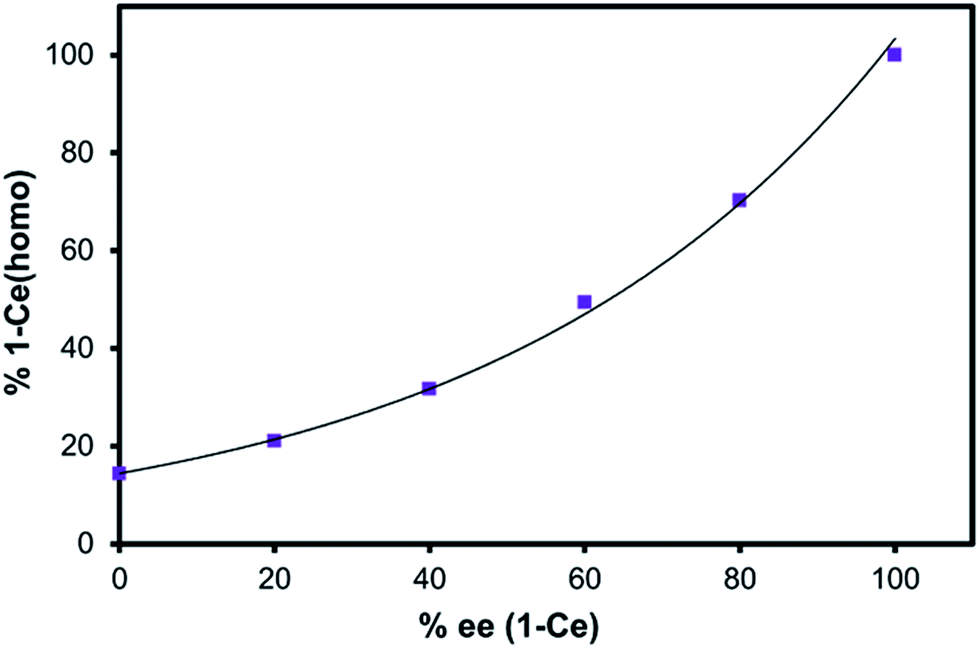 | ||
| Fig. 4 Enantiomeric excess of BINOLate versus percent 1-Ce(homo) in solution (7Li-NMR integration). Line provided as a guide for the eye. | ||
Solution NMR characterization – ligand exchange and redistribution of 1-RE(het)/1-RE(homo) (RE = Yb, Ce)
After establishing that an equilibrium between 1-Ce(het) and 1-Ce(homo) existed in solution, we set out to establish rates of ligand and cation exchange for the system. While the groups of Aspinall,10 Shibasaki,12 and Salvadori11 have independently investigated solution behavior of heterochiral [Li3(THF)6][(BINOLate)3RE] complexes, 1-RE(het), limited investigations have been made towards characterizing the exchange processes of these complexes. Only Shibasaki and coworkers have demonstrated that the thermodynamically preferred heterochiral diastereomer, 1-Y(het), can be formed through ligand exchange between mixtures of enantiopure (SSS)-1-Y(homo) and (RRR)-1-Y(homo). Quantification of ligand exchange rates, however, have yet to be established.Two-dimensional exchange spectroscopy (2D-EXSY) has emerged as a useful technique to determine rates of chemical exchange that are slow on the NMR timescale.15 The technique has been successfully applied to a number of diverse chemical systems and nuclei, including 1H and 7Li nuclei and paramagnetic metal cations.15b,16 Recently, we have employed this technique to determine BINOLate and Li+ exchange for 1-RE(homo) under catalytically relevant conditions.17
2D 1H- and 7Li-NMR EXSY experiments were performed on a 19.3 mM solution of 1-Yb(het) and were collected at 300 K with a mixing time (tmix) of 5 ms. Proposed Li+ (Scheme 2A) and BINOLate (Scheme 2B) exchange processes are depicted in Scheme 2. The two inequivalent Li+ cations, LiA and LiB, readily underwent exchange at 300 K, where the pseudo-first order rate constants for exchange of LiA → LiB and LiB → LiA were 34.0 and 80.1 s−1 respectively (Fig. 5 and Table 1). Comparable rates of exchange were observed for SS/RR and R/S BINOLate fragments (kobs = 78.3 and 79.5 s−1, Table 1 and Fig. S12†), which were ∼100-fold faster than we previously observed for rates of intermolecular BINOLate exchange between 1-Pr(homo)/1-Eu(homo).17
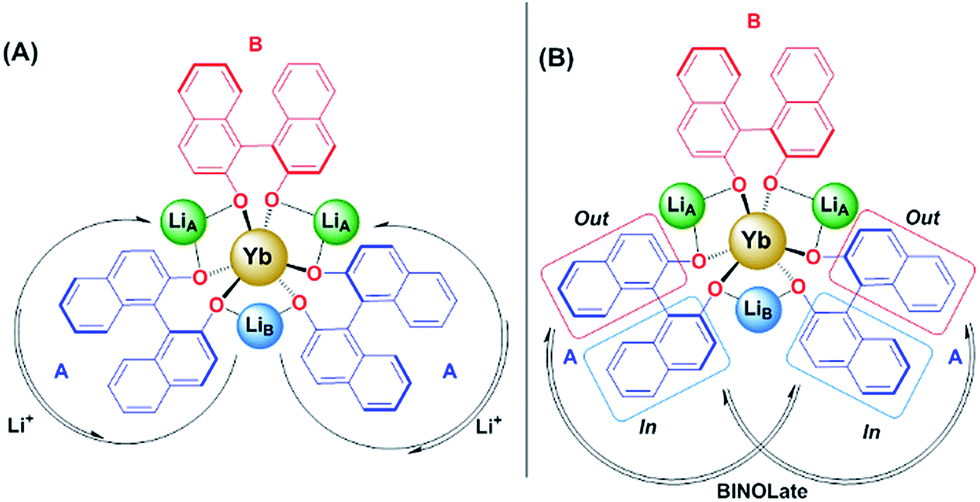 | ||
| Scheme 2 Proposed (A) Li+ and (B) BINOLate exchange processes for 1-Yb(het) (19.3 mM; 1-Yb = 0% ee) as determined from 2D 1H and 7Li{1H}-NMR EXSY experiments. Bound solvents not shown. | ||
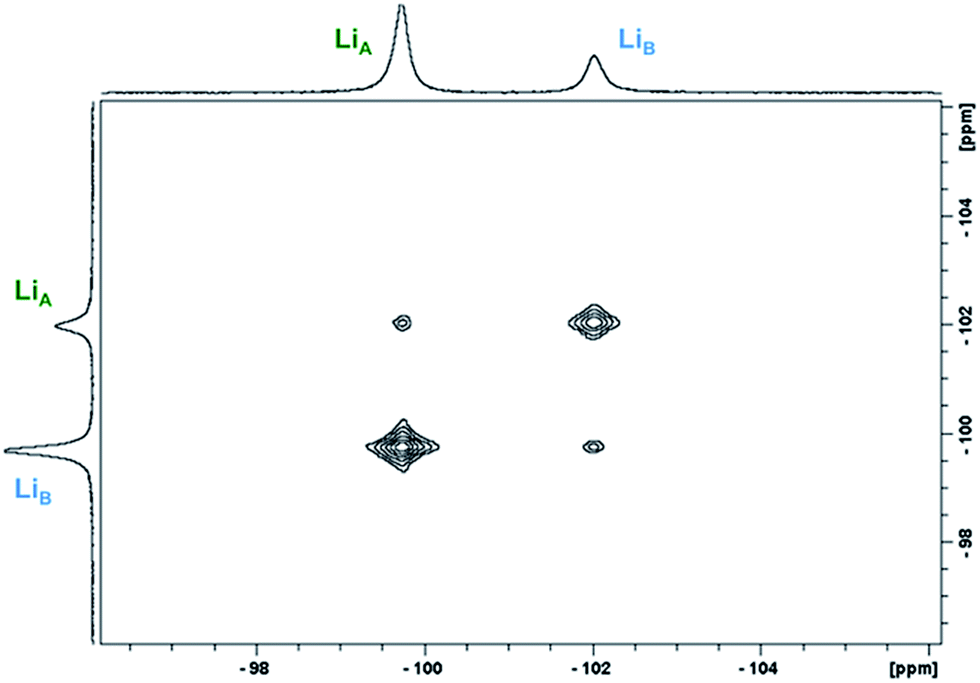 | ||
| Fig. 5 Representative 2D 7Li-NMR EXSY experiment of 1-Yb(het) (19.3 mM; tmix = 5 ms, 300 K; 1-Yb = 0% ee) taken in THF-d8. | ||
| Exchange process | k obs , (s−1) | ΔH‡c (kcal mol−1) | ΔS‡c (eu) | ΔG‡c (kcal mol−1) |
|---|---|---|---|---|
| a At 310 K. b Determined using EXSYCalc.18 c At 298 K. d 2-fold dilution of 1-Yb(het). e Exchange between Ain and Aout (Scheme 2B) correspond to peak assignments A(3) and A(10) (see ESI, Fig. S1 and S10–S12). | ||||
| Li + | ||||
| A → B | 34.0 (31.4)d | 15.1 | −12.2 | 16 |
| B → A | 80.1 (79.8)d | 16.9 | 19.0 | 15.5 |
|
||||
| BINOLate | ||||
| Ain → Aoute | 78.3 (82.5)d | 14.2 | −17.7 | 15.4 |
| Aout → Aine | 79.5 (79.6)d | 14.9 | −8.3 | 15.5 |
Both BINOLate and Li+ exchange processes were assigned as intramolecular due to: (1) observed exchange rates being independent of the concentration of 1-Yb(het), and (2) activation parameters that were significantly different compared to the intermolecular processes observed between 1-Pr(homo) and 1-Eu(homo). Values of kobs were insensitive to the concentration of 1-Yb(het), where a 2-fold dilution resulted in minimal changes of rate (Table 1). Further support for the assignment of an intramolecular BINOLate exchange observed in 1-Yb(het) is the ∼100-fold increase in rate compared to the previously reported intermolecular BINOLate exchange between 1-Pr(homo)/1-Eu(homo). While the magnitude of ΔS‡ was non-zero for BINOLate and Li+ exchange processes, ΔS‡ was significantly smaller than that determined for intermolecular exchange (∼50 and 105 eu respectively) and support assignment as an intramolecular exchange process.
:1.4 ratio of 1-Yb(het):1-Yb(homo)) over the temperature range of 300–330 K (Table 2 and Fig. 6). In addition to the previously established intramolecular exchange processes, 7Li-NMR EXSY experiments performed at 310 K (tmix = 10 ms) revealed intermolecular Li+ exchange processes (Table 2 and Fig. 6). Exchange occurred between the three possible sites, LiA, LiB, and LiC, where the rate of intermolecular exchange was an order of magnitude slower than intramolecular exchange.
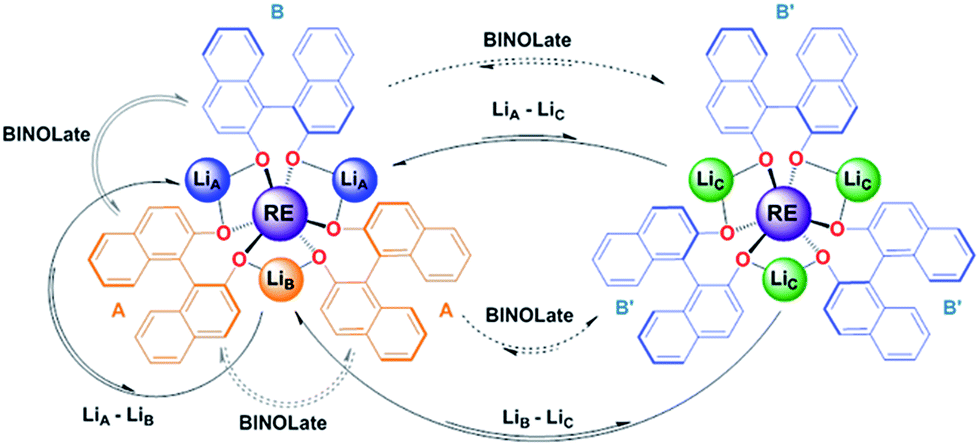 | ||
| Scheme 3 Representative intra- and inter-molecular exchange processes for (rac)-1-RE(het) and 1-RE(homo) (RE = Ce and Yb) from 2D 1H and 7Li-NMR EXSY experiments. Dashed lines indicate equilibria that were not directly observable by EXSY experiments, but have been observed through simple 1D mixing experiments (vide supra) or 2D EXSY experiments with 1-RE(homo).17 | ||
| Exchange process | k , (s−1) | ΔH‡c (kcal mol−1) | ΔS‡c (eu) | ΔG‡c (kcal mol−1) |
|---|---|---|---|---|
| a At 310 K. b Determined using EXSYCalc.18 c At 298 K. d Values for 1-Yb(het) (19.3 mM). e Exchange between Ain and Aout (Scheme 2B) correspond to peak assignments A(3) and A(10) (see ESI, Fig. S1 and S10–S12). | ||||
| Li + | ||||
| A → B | 44.3 (34.0)d | 12.0 | −52.3 | 15.7 |
| B → A | 92.8 (80.1)d | 13.4 | −25.3 | 15.2 |
| A → C | 3.37 | 12.5 | −66.8 | 17.3 |
| C → A | 2.78 | 11.8 | −76.8 | 17.3 |
| B → C | 7.12 | 8.06 | −120 | 16.6 |
| C → B | 3.16 | 4.55 | −175 | 17.0 |
|
||||
| BINOLate | ||||
| Ain → Aoute | 77.4 (78.3)d | 14.2 | −17.7 | 15.4 |
| Aout → Aine | 80.3 (79.5)d | 14.9 (78.3)d | −8.3 | 15.5 |
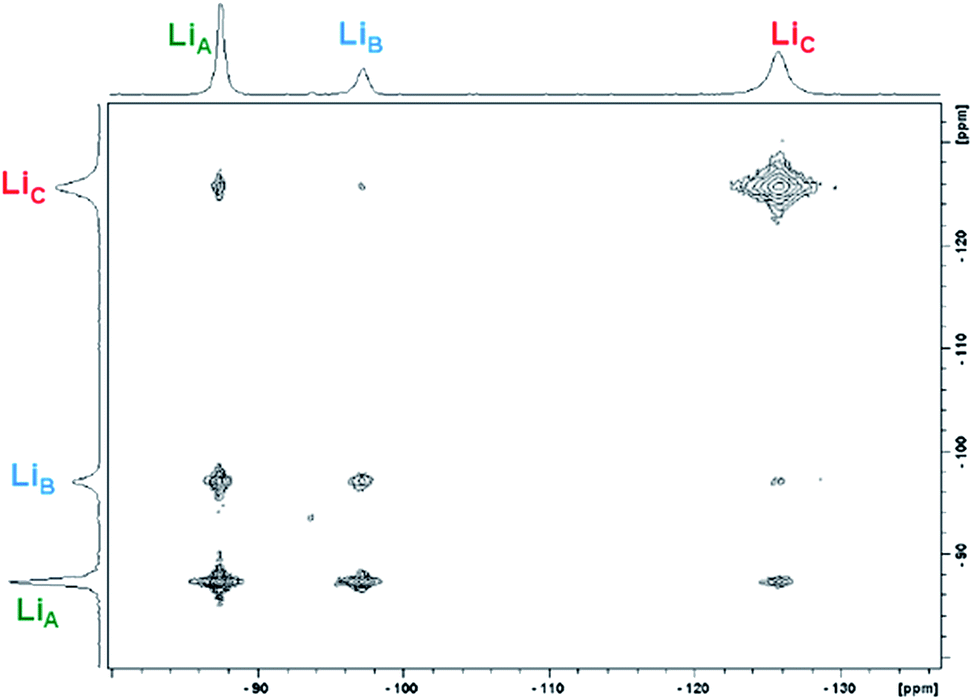 | ||
| Fig. 6 Representative 2D 7Li-NMR EXSY experiment of 1-Yb(het)/1-Yb(homo) (9.64 mM/32.7 mM; tmix = 15 ms, 300 K; 1-Yb = 77% ee) taken in THF-d8. | ||
Intermolecular BINOLate exchange processes were not observed by 1H 2D EXSY NMR experiments, despite previous observation of intermolecular exchange between different 1-RE(homo) (RE = Pr and Eu).17 Qualitatively, we have demonstrated that ligand redistribution is a facile process at RT (optical purity experiments, vide supra). To explain the absence of observable exchange using 1H 2D EXSY NMR experiments, we propose that BINOLate exchange between the diastereomers is either slow on the EXSY NMR time scale or at least slower than the relaxation rates rendered by the paramagnetic YbIII cation.
Quantification of Li+ and BINOLate exchange rates between 1-Ce(het) and 1-Ce(homo) proved more challenging. The peak shape of the BINOLate C–H of 1-Ce(het) are significantly broadened in the presence of 1-Ce(homo) (full width at half maximum, νFWHM ∼ 25–180 Hz). The broadened peaks, coupled with rapid relaxation rates and insufficient spectral resolution precluded clear observation of intramolecular and intermolecular BINOLate exchange through EXSY techniques. Similar to 1-Yb(het)/1-Yb(homo), intra- and intermolecular Li+ exchange occurred between all three possible sites – LiA, LiB, and LiC (Scheme 3 and Fig. S15†). Intramolecular Li+ exchange occurred at much faster rates, and prevented a full quantitative treatment under experimental conditions investigated. Notably, intramolecular Li+ exchange for 1-RE(het) in the presence of 1-RE(homo) occurred ∼5 times faster for CeIII compared to the smaller YbIII cation (115 and 23 s−1 at 300 K respectively), and indicates increased Li+ lability with a larger REIII cation.
To summarize, by using 2D 1H- and 7Li-NMR EXSY we have established that intramolecular and intermolecular exchange processes occurred readily for 1-RE(het) and mixtures of 1-RE(het)/1-RE(homo) (RE = Ce and Yb). For YbIII, activation parameters provided experimental support for the assignment of intramolecular and intermolecular exchange processes. Quantitation of intermolecular BINOLate exchange was not possible using EXSY techniques, however, rapid rates of ligand redistribution at RT were established qualitatively through NMR spectroscopy. Rapid equilibration was confirmed by mixing enantiomers of 1-RE(homo) at varying optical purities, where the enantiomers rapidly equilibrated to 1-RE(het) and 1-RE(homo).
Electrochemical and chemical oxidation behavior of 1-Ce(het)
After synthesizing 1-Ce(het) and establishing that ligand exchange/redistribution occurred readily between 1-Ce(het) and 1-Ce(homo), the electrochemical and chemical oxidation behavior of 1-Ce(het) and 1-Ce(homo) was investigated to determine the role of these processes on the oxidation chemistry of CeIII.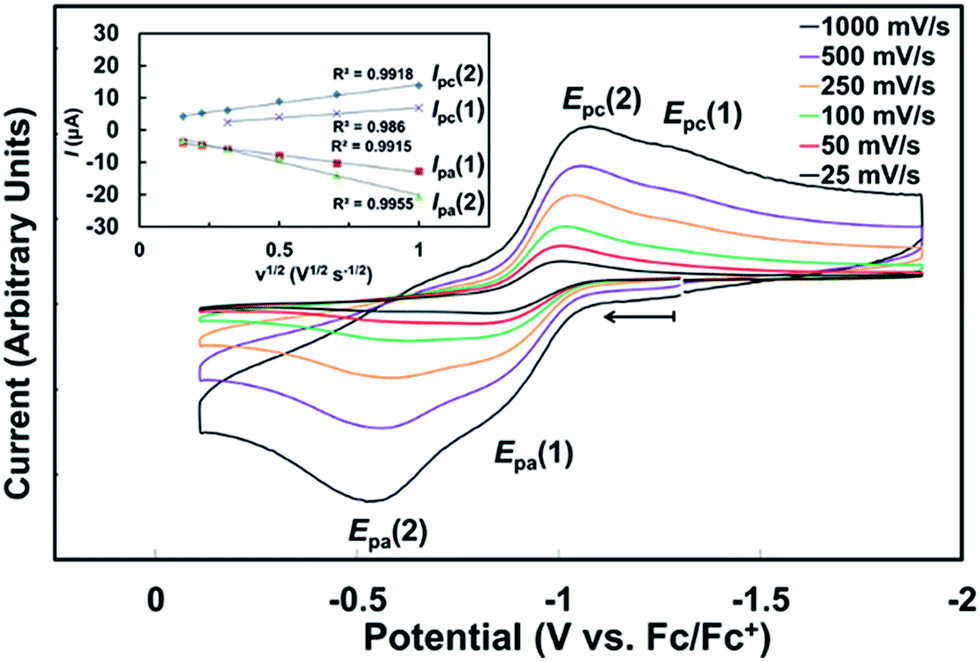 | ||
| Fig. 7 Cyclic voltammogram of 1-Ce(het) ([Ce] = ∼1 mM; THF, [NPr4][B(ArF)4] = 100 mM, ν = 500 mV s−1). | ||
For a racemic solution of 1-Ce(het)/1-Ce(homo), oxidation of 1-Ce(het) (Epa(1) = −0.860 V vs. Fc) occurred at 275 mV more reducing than that of 1-Ce(homo) (Ep(2) = −0.585 V). This is consistent with a more electron rich CeIII cation in 1-Ce(het), which could be expected from the shorter Ce–OBINOLate distances observed in the solid state.5e,7,19 In addition to a higher driving force, the heterogeneous electron transfer occurs at a slightly faster rate for 1-Ce(het), which can be qualitatively determined by the separation of the oxidation and reduction events, ΔEp, for 1-Ce(het) and 1-Ce(homo) (450 mV versus 490 mV). At low scan rates preferential oxidation of 1-Ce(het) could be achieved; however, the reduction current (Ipc) for the electrochemically generated CeIV complexes display similar ratios across the scan rates investigated (Fig. 7, inset of Ipc(1) vs. (2)). This suggests that BINOLate exchange occurs quickly on the CV collection time scale, and is consistent with our observations regarding facile BINOLate exchange in solution in our NMR studies. The two distinct redox events characterized for 1-Ce(het) and 1-Ce(homo) coupled with the different coordination numbers supported at the respective Ce cations provided an opportunity to directly compare the complexes' electrochemical properties and their chemical oxidation reactivities.
:4 v/v) led to the isolation of an analytically pure product in 97% yield. X-ray diffraction studies confirmed the identity of the oxidation product as [Li2(THF)4][(BINOLate)3Ce]·1/2(C5H12) (2-Ce(het)), the product of salt-elimination (Fig. 8 and Scheme 4).
 | ||
| Fig. 8 Thermal ellipsoid plot of 2-Ce(het) shown at 30% probability. The BINOLate ligands have been colored blue and orange to indicate the SS/RR and R/S fragments respectively. One interstitial molecule of THF has been removed for clarity. Selected bond distances (Å): Ce(1)–O(1) 2.2685(17), Ce(1)–O(2) 2.1958(18), Ce(1)–O(3) 2.2709(18), Ce(1)–O(4) 2.2773(17), Ce(1)–O(5) 2.1755(18), Ce(1)–O(6) 2.2917(17), Li(1)–O(1) 1.921(5), Li(1)–O(3) 1.954(5), Li(2)–O(4) 1.960(5), Li(2)–O(6) 1.913(5). | ||
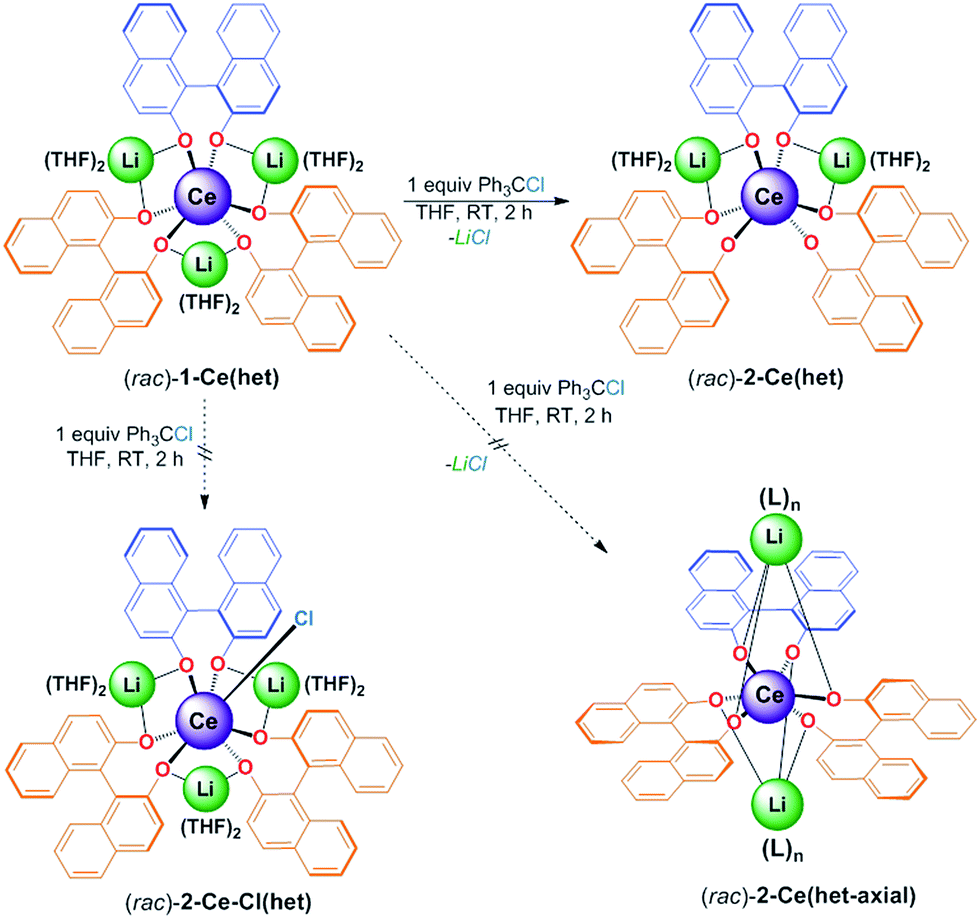 | ||
| Scheme 4 Oxidation of 1-Ce(het) with Ph3CCl to produce salt-elimination product, 2-Ce(het). Other previously observed oxidation products in the CeMB framework such as oxidative functionalization, (rac)-2-Ce-Cl(het), and M+ axially capped salt-elimination products, (rac)-2-Ce(het-axial), were not observed. | ||
Unlike the salt-elimination products of the heavier alkali metal homochiral frameworks (M = Na, K, Cs), 2-Ce(het) did not undergo a large structural rearrangement (Scheme 4, (rac)-2-Ce(het)versus (rac)-2-Ce(het-axial)). The primary coordination sphere of 2-Ce(het) was composed of anionic BINOLate oxygens arranged in a distorted trigonal prismatic geometry. The Li+ cations remained bridging in a pseudo-equatorial fashion between BINOLate ligands rather than migrating to axially cap the tris(BINOLate) framework, as was the case for homochiral CeIV/M (M = Na, K) frameworks.7 The preference for the heterochiral diastereomer was maintained for 2-Ce(het) due to the smaller ionic radius of the CeIV cation, while the conformation of the heterochiral BINOLate ligands disfavored axial Li–OBINOLate coordination. Ce–OBINOLate and Li–OBINOLate bond distances for 2-Ce(het) were similar to the previously reported CeIV terminal and bridging aryloxides in the Ce/Li framework, and supported the CeIV oxidation-state assignment.7a,19 Additional support for the CeIV oxidation state was provided by cyclic voltammetry, which displayed clear metal-based reduction features (Fig. S18†).
Formation of 2-Ce(het) was surprising, as previous chemical oxidation studies of 1-Ce(homo) revealed an exclusive preference for oxidative functionalization over salt-elimination (Scheme 4, (rac)-2-Ce-Cl(het)).7,19 In those studies we demonstrated that the accessibility of the CeIII cation, which was governed by the main group metal (Li, Na, or K), controlled the rate of inner-sphere oxidation with trityl chloride. The ability of the CeIV complex to reorganize and maintain a seven-coordinate geometry, however, appeared to dictate the selectivity of oxidative functionalization versus salt elimination. We observed that 1-Ce(homo) showed the highest level of reactivity with trityl chloride and formed only oxidative functionalized products (Scheme 5, and 2-Ce-Cl(homo)). In contrast, other Ce/M complexes (M = Na, K, Cs) exhibited slower chemical oxidation with trityl chloride and formed salt-elimination products.
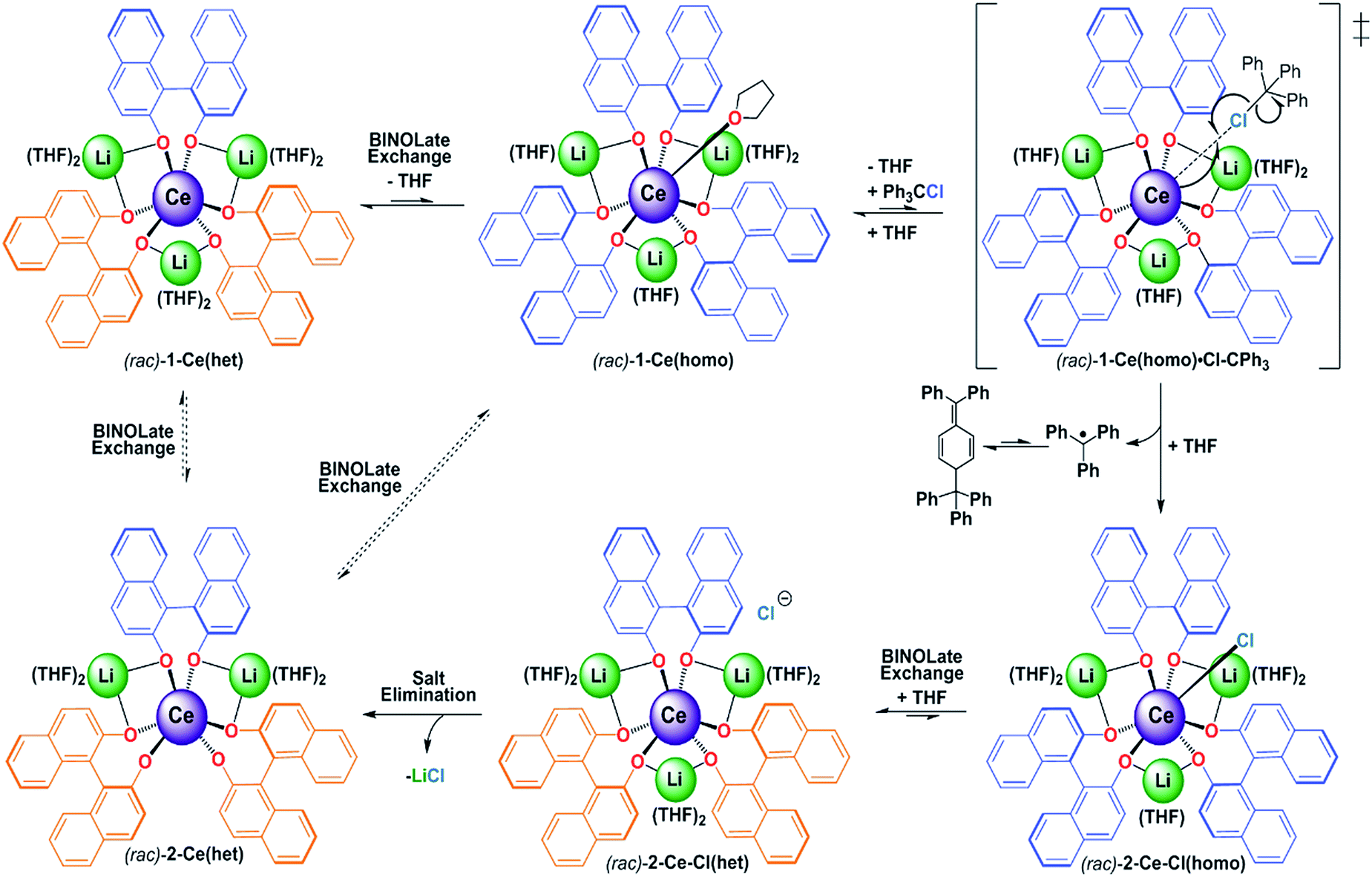 | ||
| Scheme 5 Proposed mechanism and ligand exchange processes for the oxidation reaction of 1-Ce(het). Dashed equilibria are exchange pathways that have not been directly measured, but likely can occur under reaction conditions. | ||
Rate studies were performed for 1-Ce with varying optical purities under pseudo-first order conditions using a 10-fold excess of trityl chloride (Fig. 9). The initial concentration of 1-Ce(het) and 1-Ce(homo) were independently confirmed for each sample using 7Li-NMR (Table S2†). Oxidation of 1-Ce with trityl chloride was monitored by UV-Vis absorption spectroscopy following the growth of the characteristic ligand to metal charge transfer (LMCT) band at 487 nm in the Ce(IV) products 2-Ce(homo) and 2-Ce(het).7,19,20
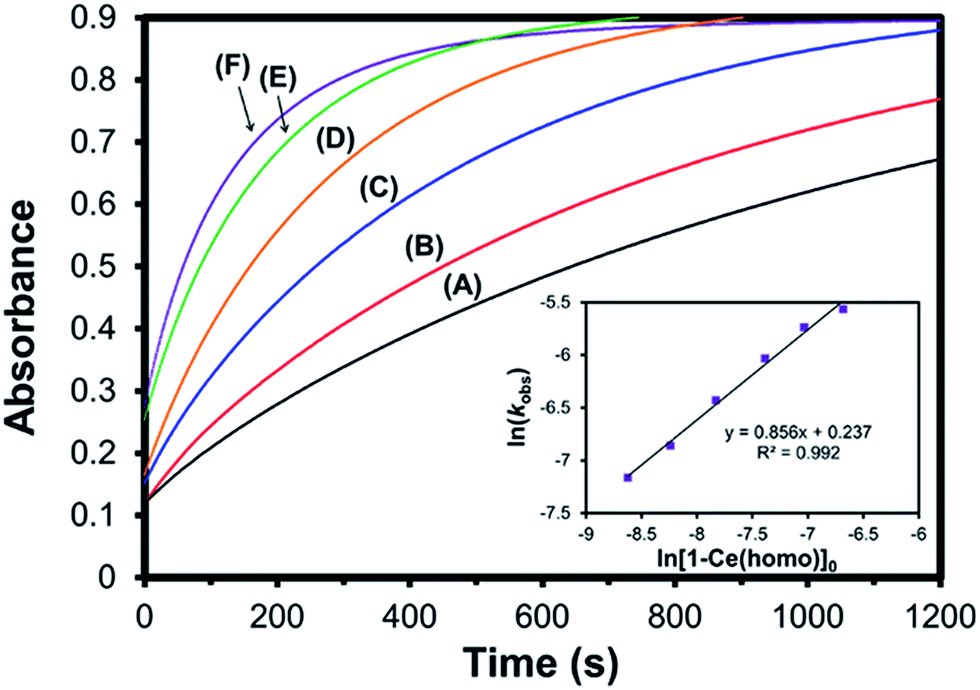 | ||
| Fig. 9 Kinetic profile of 1-Ce(het)/1-Ce(homo) + Ph3CCl in THF under pseudo-first order conditions (10 equiv. Ph3Cl; [1-Ce] = 1.26 mM) at different optical purities of 1-Ce. (A) 0% ee (black trace), (B) 20% ee (red trace), (C) 40% ee (blue trace), (D) 60% ee (orange trace), (E) 80% ee (green trace), and (F) 100% ee (purple trace). Inset shows ln(kobs) versus the ln[1-Ce(homo)]0 in solution, confirming a first order relationship of 1-Ce(homo) on observed oxidation rate. | ||
Increased optical purity of 1-Ce resulted in increased rates of chemical oxidation (kobs), where a ∼5 fold rate enhancement was observed by employing optically pure 1-Ce(homo) rather than racemic 1-Ce (3.37 versus 0.713 s−1; Fig. 9, traces F and A). Values of kobs did not agree with an outer sphere mechanism, where it would be expected that 1-Ce(het) would undergo oxidation at a faster rate due to the more facile oxidation of 1-Ce(het) than 1-Ce(homo). Instead, a first order dependence was observed for the initial concentration of 1-Ce(homo) in solution (Fig. 9, inset), and is consistent with an inner sphere oxidation mechanism involving 1-Ce(homo) in the rate determining step.7
Similar to our studies investigating the impact of M+ on the kinetics of chemical oxidation with trityl chloride,7kobs for 1-Ce followed the ability of the CeIII cation to adopt a seven-coordinate geometry rather than the thermodynamics or kinetics associated with an outer sphere electron transfer process. The configuration of the RRS/SSR BINOLate fragments disfavor ligand reorganization in 1-Ce(het) to bind an additional ligand and achieve a seven-coordinate geometry. As a result, the chemical oxidation of 1-Ce(het) proceeds much slower than that of 1-Ce(homo). Given the rapid chemical oxidation rates and the observation that ligand redistribution/exchange is facile at RT in solution between 1-Ce(het) and 1-Ce(homo), we propose that the chemical oxidation reaction proceeds through the higher energy, but more reactive diastereomer, 1-Ce(homo) (Scheme 5).
To test the feasibility of the ligand redistribution step, both the SSS and RRR enantiomers of 2-Ce-Cl(homo) were independently synthesized and then mixed in equimolar quantities in THF-d8 (Fig. 10). Immediate ligand redistribution was observed, where the 1H- and 7Li-NMR spectra matched those of 2-Ce(het) in the presence of LiCl (Fig. 10D). Given these observations, we propose that intermolecular BINOLate ligand redistribution of 2-Ce-Cl(homo) would generate (rac)-2-Ce-Cl(het). As the larger CeIII cation in 1-Ce(het) only adopted a six-coordinate geometry, it is likely that the Cl− ligand would be dissociated as an outer sphere counterion for the smaller CeIV cation in 2-Ce-Cl(het). Salt-elimination of LiCl would then readily occur, producing 2-Ce(het). The thermodynamic preference of the CeIV cation for the heterochiral diastereomer is likely associated with the size of the ion, as the smaller REIII cations, YbIII and YIII, have similar ionic radii to CeIV (0.900 and 0.870 versus 0.868 Å respectively)13 and also prefer the heterochiral diastereomer.10,12
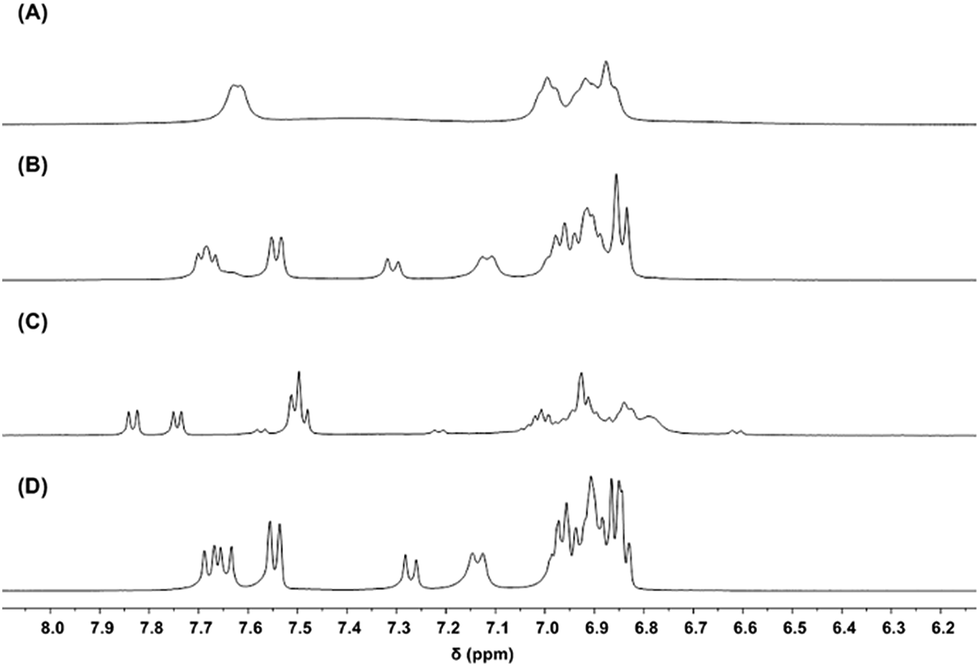 | ||
| Fig. 10 1H-NMR (400 MHz, THF-d8) spectra of (A) (RRR)-2-Ce-Cl, (B) (RRR)-2-Ce-Cl (19.3 mM) + (SSS)-2-Ce-Cl (21.2 mM), (C) 2-Ce(het), and (D) 2-Ce(het) + 1 equiv. LiCl. | ||
Our proposed mechanism accounts for both the unique kinetics and product selectivity observed in the chemical oxidation of (rac)-1-Ce(het)/1-Ce(homo) (Scheme 5). Ligand reorganization and cation accessibility to Lewis base coordination dictate the rate of inner-sphere chemical oxidation, which occurs through the faster reacting diastereomer, (rac)-1-Ce(homo). Oxidation will lead to the formation of (rac)-2-Ce-Cl(homo); however, we have demonstrated that enantiomers of (rac)-2-Ce-Cl(homo) readily participate in ligand redistribution and undergo salt-elimination to form (rac)-2-Ce(het).21 We have shown that while ligand reorganization controls the kinetics of the oxidation process, ligand redistribution is responsible for the observed product selectivity. Rapid ligand redistribution allows equilibration between kinetic and thermodynamic products, and facilitates the ultimate formation of the observed thermodynamic product.
The key findings of our mechanistic studies help address observed oxidative reactivity preferences of CeIII in various ligand frameworks towards ligand redistribution. For example, the simple homoleptic amides, CeIII(NR2)3 (R = Cy,9b SiHMe2,5a,22 SiMe35c,5d,23) display strikingly different oxidation chemistry with only subtle changes in ligand sterics. Similar to 1-Ce(homo), the CeIII cations in CeIII(NR2)3 are accessible to the coordination of an additional ligand; however the identity of the ultimate isolated product depends on the thermodynamics associated with a redistributed product.
In the case of the bulkier SiMe3 substituents, binding a fourth equivalent of NR2 is disfavored, leading to oxidative functionalized products CeIV[N(SiMe3)2]3X over the redistributed homoleptic amide CeIV[N(SiMe3)2]4. Alternatively, with slightly smaller R groups, R = Cy and SiHMe2, a fourth amide will readily bind to a trivalent or tetravalent cerium cation.5a,9b,22 Similar to 1-Ce(homo)/1-Ce(het), Ce[(NR2)]3X may form during the course of the reaction when R = Cy and SiHMe2; however, the redistributed product, CeIV(NR2)4, is ultimately favored. This is due to increased stabilization by the more electron-rich ligand group, which was also seen for 1-Ce(homo)/1-Ce(het). In addition to heterobimetallic complexes such as 1-Ce(homo), tripodal frameworks8a,24 show great promise towards minimizing ligand reorganization (i.e. increased accessibility of the CeIII cation), while minimizing the favorability of redistributed products.
Conclusion
In summary, we have delineated the roles of ligand reorganization7,8 and redistribution processes5a,c,8b,9 in the observed oxidation chemistry of CeIII complexes. Synthesis and characterization of the heterochiral Ce/Li REMB complex, 1-Ce(het), was achieved. In the solid state, formation of the heterochiral diastereomer, 1-Ce(het), was exclusively preferred, whereas in solution a mixture of 1-Ce(het) and 1-Ce(homo) existed. Rapid exchange of the BINOLate ligands and Li cations between the two diastereomers was observed, and the thermodynamic preference for diastereomeric forms could be manipulated by choice of coordinating solvent. Electrochemical studies indicated that the CeIII/IV redox couple for 1-Ce(het) was more accessible than 1-Ce(homo) by ∼250 mV. Despite the increased driving force, chemical oxidations using trityl chloride revealed that 1-Ce(homo) is oxidized more rapidly than 1-Ce(het). Furthermore, kobs displayed a linear dependence on the concentration of 1-Ce(homo). Similar to our previous studies on ligand reorganization, the oxidation occurred through the most accessible CeIII ion and not the one with the most accessible CeIII/IV couple.Ligand exchange and redistribution processes were found to be directly responsible for product selectivity. Even though ligand reorganization facilitated rapid oxidation from the higher energy diastereomer, 1-Ce(homo), to yield the oxidative functionalization product, 2-Ce-Cl(homo), subsequent ligand redistribution processes resulted in the exclusive formation of the salt-eliminated product, 2-Ce(het) (Scheme 5). The reactivity of 1-Ce can mirror that of other CeIII frameworks that are prone to ligand redistribution, where subtle changes in ligand sterics can lead to dramatically different oxidation outcomes.
Even in systems where ligand reorganization can be controlled, active approaches to minimize ligand redistribution are needed to facilitate more predictable reaction outcomes. One approach would be to alter the thermodynamics of ligand redistribution pathways by minimizing specific conformational possibilities with a particular supporting ligand framework, such as chiral ligands or constrained tripodal frameworks. We believe that heterobimetallic complexes should continue to be useful CeIII starting materials, as salt-elimination can serve as a suitable driving force, while “-ate” complex formation readily occurs with a number of ligand frameworks.
Acknowledgements
E. J. S. and P. J. W. acknowledge the University of Pennsylvania and the U.S. NSF (CHE-1026553 and CHE-0840438 for an X-ray diffractometer) for financial support of this work.Notes and references
- D. T. Richens, The Chemistry of Aqua Ions: Synthesis, Structure, and Reactivity, John Wiley & Sons, New York, 1997 Search PubMed.
- (a) M. Shibasaki, H. Sasai and T. Arai, Angew. Chem., Int. Ed. Engl., 1997, 36, 1236–1256 CrossRef; (b) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187–2209 CrossRef CAS PubMed; (c) J.-C. G. Bünzli and C. Piguet, Chem. Rev., 2002, 102, 1897–1928 CrossRef; (d) H. C. Aspinall, Chem. Rev., 2002, 102, 1807–1850 CrossRef CAS PubMed; (e) D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977–2010 CrossRef CAS PubMed; (f) Y. Horiuchi, V. Gnanadesikan, T. Ohshima, H. Masu, K. Katagiri, Y. Sei, K. Yamaguchi and M. Shibasaki, Chem.–Eur. J., 2005, 11, 5195–5204 CrossRef CAS PubMed; (g) S.-y. Tosaki, R. Tsuji, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 2147–2155 CrossRef CAS PubMed; (h) J. Andrews and A.-S. Chauvin, Lanthanides: Luminescence Applications, in The Rare Earth Elements Fundamentals and Applications, ed. D. A. Atwood, John Wiley & Sons, Chichester, West Sussex, UK, 2012, pp. 135–152 Search PubMed; (i) Y. Mori and S. Kobayashi, Organic Synthesis, in The Rare Earth Elements Fundamentals and Applications, ed. D. A. Atwood, John Wiley & Sons, Chichester, West Sussex, UK, 2012, pp. 437–458 Search PubMed; (j) Y. Yao and K. Nie, Homogeneous Catalysis, in The Rare Earth Elements Fundamentals and Applications, ed. D. A. Atwood, John Wiley & Sons, Chichester, West Sussex, UK, 2012, p. 475 Search PubMed; (k) C. F. G. C. Geraldes, Lanthanide Shift Reagents, in The Rare Earth Elements Fundamentals and Applications, ed. D. A. Atwood, John Wiley & Sons, Chichester, West Sussex, UK, 2012, pp. 501–520 Search PubMed; (l) S. Laurent, L. V. Elst, S. Boutry and R. N. Nuller, Lanthanides: Magnetic Resonance Imaging, in The Rare Earth Elements Fundamentals and Applications, ed. D. A. Atwood, John Wiley & Sons, Chichester, West Sussex, UK, 2012, pp. 521–534 Search PubMed; (m) A.-S. Chauvin, Luminescent Bioprobes, in The Rare Earth Elements Fundamentals and Applications, ed. D. A. Atwood, John Wiley & Sons, Chichester, West Sussex, UK, 2012, pp. 535–560 Search PubMed.
- (a) E. Pershagen and K. E. Borbas, Coord. Chem. Rev., 2014, 273–274, 30–46 CrossRef CAS; (b) J.-C. G. Bünzli, J. Coord. Chem., 2014, 67, 3706–3733 CrossRef; (c) P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed; (d) A. D. Sherry, P. Caravan and R. E. Lenkinski, J. Magn. Reson. Imag., 2009, 30, 1240–1248 CrossRef PubMed; (e) J.-M. Idée, M. Port, C. Robic, C. Medina, M. Sabatou and C. Corot, J. Magn. Reson. Imag., 2009, 30, 1249–1258 CrossRef PubMed; (f) S. Viswanathan, Z. Kovacs, K. N. Green, S. J. Ratnakar and A. D. Sherry, Chem. Rev., 2010, 110, 2960–3018 CrossRef CAS PubMed; (g) K. N. Green, S. Viswanathan, F. A. Rojas-Quijano, Z. Kovacs and A. D. Sherry, Inorg. Chem., 2011, 50, 1648–1655 CrossRef CAS PubMed; (h) W. T. K. Chan and W.-T. Wong, Polyhedron, 2014, 83, 150–158 CrossRef CAS.
- (a) H. C. Aspinall, Chemistry of the f-block Elements, Overseas Publishing Company, UK, 2001, vol. 1 Search PubMed; (b) S. A. Cotton and J. M. Harrowfield, Lanthanides: Coordination Chemistry, in The Rare Earth Elements: Fundamentals and Applications, ed. D. A. Atwood, John Wiley & Sons, Chichester, West Sussex, UK, 2012 Search PubMed; (c) S. Cotton, Lanthanide and Actinide Chemistry, John Wiley & Sons Ltd, West Sussex, England, 2006 Search PubMed.
- (a) A. R. Crozier, A. M. Bienfait, C. Maichle-Mossmer, K. W. Tornroos and R. Anwander, Chem. Commun., 2013, 49, 87–89 RSC; (b) P. B. Hitchcock, Q. G. Huang, M. F. Lappert and X. H. Wei, J. Mater. Chem., 2004, 14, 3266–3273 RSC; (c) P. B. Hitchcock, A. G. Hulkes and M. F. Lappert, Inorg. Chem., 2004, 43, 1031–1038 CrossRef CAS PubMed; (d) O. Eisenstein, P. B. Hitchcock, A. G. Hulkes, M. F. Lappert and L. Maron, Chem. Commun., 2001, 1560–1561 RSC; (e) N. A. Piro, J. R. Robinson, P. J. Walsh and E. J. Schelter, Coord. Chem. Rev., 2014, 260, 21–36 CrossRef CAS.
- (a) G. A. Molander, Chem. Rev., 1992, 92, 29–68 CrossRef CAS; (b) V. Nair, L. Balagopal, R. Rajan and J. Mathew, Acc. Chem. Res., 2004, 37, 21–30 CrossRef CAS PubMed; (c) T. L. Ho, Synthesis, 1973, 347–354 CrossRef CAS; (d) T. Imamoto, Y. Koide and S. Hiyama, Chem. Lett., 1990, 1445–1446 CrossRef CAS; (e) A. K. Das, Coord. Chem. Rev., 2001, 213, 307–325 CrossRef CAS; (f) N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed; (g) W. Rüttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1–24 CrossRef; (h) J. L. Fillol, Z. Codola, I. Garcia-Bosch, L. Gomez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed; (i) W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef CAS PubMed; (j) R. Tagore, H. Y. Chen, H. Zhang, R. H. Crabtree and G. W. Brudvig, Inorg. Chim. Acta, 2007, 360, 2983–2989 CrossRef CAS PubMed; (k) S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS; (l) T. Kreuzer, E. S. Lox, D. Lindner and J. Leyrer, Catal. Today, 1996, 29, 17–27 CrossRef CAS; (m) H. Inaba and H. Tagawa, Solid State Ionics, 1996, 83, 1–16 CrossRef CAS; (n) A. Trovarelli, Catal. Rev., 1996, 38, 439–520 CrossRef CAS; (o) A. Trovarelli, C. de Leitenburg, M. Boaro and G. Dolcetti, Catal. Today, 1999, 50, 353–367 CrossRef CAS; (p) A. Trovarelli, Catalysis by Ceria and Related Materials, Imperial College Press, London, 2002 Search PubMed.
- (a) J. R. Robinson, Z. Gordon, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 19016–19024 CrossRef CAS PubMed; (b) J. R. Robinson, P. J. Carroll, P. J. Walsh and E. J. Schelter, Angew. Chem., Int. Ed., 2012, 51, 10159–10163 CrossRef CAS PubMed.
- (a) C. Morton, N. W. Alcock, M. R. Lees, I. J. Munslow, C. J. Sanders and P. Scott, J. Am. Chem. Soc., 1999, 121, 11255–11256 CrossRef CAS; (b) I. J. Casely, S. T. Liddle, A. J. Blake, C. Wilson and P. L. Arnold, Chem. Commun., 2007, 5037–5039 RSC.
- (a) M. DelaRosa, K. Bousman, J. Welch and P. Toscano, J. Coord. Chem., 2002, 55, 781–793 CrossRef CAS; (b) P. B. Hitchcock, M. F. Lappert and A. V. Protchenko, Chem. Commun., 2006, 3546–3548 RSC; (c) M. Ciampolini, F. Mani and N. Nardi, Dalton Trans., 1977, 1325–1328 RSC; (d) F. M. A. Sroor and F. T. Edelmann, Tetravalent Chemistry: Organometallic, in The Rare Earth Elements: Fundamentals and Applications, D. A. Atwood, ed. John Wiley & Sons, Chichester, West Sussex, UK, 2012, pp. 321–333 Search PubMed; (e) J. A. Bogart, A. J. Lewis, S. A. Medling, N. A. Piro, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2013, 52, 11600–11607 CrossRef CAS PubMed; (f) T. Behrsing, A. M. Bond, G. B. Deacon, C. M. Forsyth, M. Forsyth, K. J. Kamble, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2003, 352, 229–237 CrossRef CAS; (g) D. Werner, G. B. Deacon, P. C. Junk and R. Anwander, Chem.–Eur. J., 2014, 20, 4426–4438 CrossRef CAS PubMed; (h) G.-C. Wang, Y.-M. So, K.-L. Wong, K.-C. Au-Yeung, H. H. Y. Sung, I. D. Williams and W.-H. Leung, Chem.–Eur. J., 2015, 21, 16126–16135 CrossRef CAS PubMed.
- H. C. Aspinall, J. F. Bickley, J. L. M. Dwyer, N. Greeves, R. V. Kelly and A. Steiner, Organometallics, 2000, 19, 5416–5423 CrossRef CAS.
- (a) L. Di Bari, M. Lelli and P. Salvadori, Chem.–Eur. J., 2004, 10, 4594–4598 CrossRef CAS PubMed; (b) L. Di Bari, M. Lelli, G. Pintacuda, G. Pescitelli, F. Marchetti and P. Salvadori, J. Am. Chem. Soc., 2003, 125, 5549–5558 CrossRef CAS PubMed.
- (a) N. Yamagiwa, H. B. Qin, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 13419–13427 CrossRef CAS PubMed; (b) N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2004, 43, 4493–4497 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef.
- (a) A. J. Wooten, P. J. Carroll and P. J. Walsh, Angew. Chem., Int. Ed., 2006, 45, 2549–2552 CrossRef CAS PubMed; (b) A. J. Wooten, P. J. Carroll and P. J. Walsh, Org. Lett., 2007, 9, 3359–3362 CrossRef CAS PubMed; (c) A. J. Wooten, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2008, 130, 7407–7419 CrossRef CAS PubMed.
- (a) K. G. Orrell, Two-Dimensional Methods of Monitoring Exchange, in eMagRes, John Wiley & Sons, Ltd, 2007 Search PubMed; (b) C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935–967 CrossRef CAS; (c) J. Jeener, B. H. Meier, P. Bachmann and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546–4553 CrossRef CAS; (d) B. S. Brunschwig and N. Sutin, Coord. Chem. Rev., 1999, 187, 233–254 CrossRef CAS.
- (a) S. P. Babailov, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 52, 1–21 CrossRef CAS; (b) L. S. Cahill, R. P. Chapman, J. F. Britten and G. R. Goward, J. Phys. Chem. B, 2006, 110, 7171–7177 CrossRef CAS PubMed; (c) A. Pastor and E. Martinez-Viviente, Coord. Chem. Rev., 2008, 252, 2314–2345 CrossRef CAS; (d) A. P. Kalverda, J. Salgado, C. Dennison and G. W. Canters, Biochemistry, 1996, 35, 3085–3092 CrossRef CAS PubMed; (e) S. Fischer, P. D. J. Grootenhuis, L. C. Groenen, W. P. Vanhoorn, F. Vanveggel, D. N. Reinhoudt and M. Karplus, J. Am. Chem. Soc., 1995, 117, 1611–1620 CrossRef CAS; (f) M. C. Chen, J. A. S. Roberts and T. J. Marks, J. Am. Chem. Soc., 2004, 126, 4605–4625 CrossRef CAS PubMed; (g) T. Brotin, A. Lesage, L. Emsley and A. Collet, J. Am. Chem. Soc., 2000, 122, 1171–1174 CrossRef CAS; (h) L. Banci, I. Bertini, F. Briganti, C. Luchinat, A. Scozzafava and M. V. Oliver, Inorg. Chem., 1991, 30, 4517–4524 CrossRef CAS; (i) F. A. Dunand, S. Aime and A. E. Merbach, J. Am. Chem. Soc., 2000, 122, 1506–1512 CrossRef CAS; (j) S. Aime, M. Botta and G. Ermondi, Inorg. Chem., 1992, 31, 4291–4299 CrossRef CAS; (k) R. Willem, Prog. Nucl. Magn. Reson. Spectrosc., 1988, 20, 1–94 CrossRef; (l) J. D. Heise, D. Raftery, B. K. Breedlove, J. Washington and C. P. Kubiak, Organometallics, 1998, 17, 4461–4468 CrossRef CAS; (m) L. Di Bari, S. Di Pietro, G. Pescitelli, F. Tur, J. Mansilla and J. M. Saá, Chem.–Eur. J., 2010, 16, 14190–14201 CrossRef CAS PubMed.
- J. R. Robinson, J. Gu, P. J. Carroll, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2015, 137, 7135–7144 CrossRef CAS PubMed.
- J. C. Cobas and M. Martin-Pastor, EXSYCalc, 1.0, Mestrelab Research, Santiago De Compostela Search PubMed.
- J. R. Robinson, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem.–Eur. J., 2013, 19, 5996–6004 CrossRef CAS PubMed.
- Our proposed mechanism implies the formation of 2-Ce-Cl(homo) as an intermediate to the formation of 2-Ce(het). Although ε values of the two compounds are within a 5% of one another at 487 nm (ε(2-Ce(het)) = 8691 M−1 cm−1; ε(2-Ce-Cl(homo)) = 8270 M−1 cm−1), if 2-Ce-Cl(homo) were to build up during the rate studies an underestimation of kobs would occur. In the worst case scenario, which would assume that all of the CeIV species in solution corresponded to 2-Ce-Cl(homo), a 12% error in kobs would be expected.
- It should be noted that exchange between the CeIV product, (rac)-2-Ce(het), and the CeIII starting materials, (rac)-1-Ce(het) and (rac)-1-Ce(homo), cannot be excluded in our current proposal, given the ease of the ligand redistribution processes for (rac)-1-Ce and (rac)-2-Ce (Scheme 5).
- U. J. Williams, D. Schneider, W. L. Dorfner, C. Maichle-Mossmer, P. J. Carroll, R. Anwander and E. J. Schelter, Dalton Trans., 2014, 43, 16197–16206 RSC.
- (a) U. J. Williams, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 6338–6345 CrossRef CAS PubMed; (b) U. J. Williams, J. R. Robinson, A. J. Lewis, P. J. Carroll, P. J. Walsh and E. J. Schelter, Inorg. Chem., 2014, 53, 27–29 CrossRef CAS PubMed.
- J. A. Bogart, C. A. Lippincott, P. J. Carroll, C. H. Booth and E. J. Schelter, Chem.–Eur. J., 2015, 21, 17850–17859 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, electrochemical data, UV-vis data, and crystallographic data (CIF). CCDC 1442465 and 1442466. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04897d |
| This journal is © The Royal Society of Chemistry 2016 |