
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comment on “A quantitative definition of hypervalency” by M. C. Durrant, Chem. Sci., 2015, 6, 6614
Richard D.
Harcourt
*a and
Thomas M.
Klapötke
b
aSchool of Chemistry, The University of Melbourne, Victoria 3010, Australia. E-mail: r.harcourt@unimelb.edu.au; Fax: +61 3 93475180
bDepartment of Chemistry, Ludwig-Maximilian University of Munich, Butenandstr. 5-13(D), D-81377 Munich, Germany
First published on 25th February 2016
Abstract
Consideration is given to (electronically) hypervalent increased-valence structures, which possess 2c–1e bonds, fractional 2c–2e bonds, and usually normal 2c–2e bonds. For singlet-spin electron-rich systems, increased-valence structures, with Heitler–London 2c–2e bond wavefunctions, are equivalent to resonance between non-hypervalent Kekulé and Dewar (or singlet diradical) type Lewis structures. Dewar structures are not considered in the Chem. Sci. 2015, 6, 6614 Edge article on hypervalency. Using one-electron delocalizations from lone-pair atomic orbitals into separate bonding molecular orbitals, increased-valence structures for PCl5, O3, SO42−, NO3−, N2O4 and SN2 reactions are derived from the Edge-article's Kekulé-type Lewis structures, and compared with the Edge article's hypervalent structures with 2c–2e bonds. It is also shown that Durrant's method to determine the γ parameter for XAY-type systems that possess a symmetrical 3c–4e bonding unit is related to the A-atom charge density.
Introduction
Durrant1 has provided a quantitative definition of hypervalency via reference to atomic charges, which were obtained either from experimental or from theoretical electron densities using Bader's quantum theory of atoms in molecules (QTAIM) methodology.3 Without calculation it was not usually possible to conclude whether a given molecule was hypervalent. For example, the hypercoordinate molecules CLi6 and SiH62− were calculated1 to be hypervalent and non-hypervalent, respectively.4For this comment on hypervalence (primarily for electron-rich molecules), we use and discuss types of hypervalent VB structures that were not considered in ref. 1, and which, since 1968,8,9 have been designated as “increased-valence” structures without expansion of valence shells. They involve 2-centre, 1-electron (2c–1e) bonds and fractional 2c–2e bonds (with bond-numbers less than unity, and represented by thin bond lines),6,8,9 and usually non-fractional 2c–2e bonds.
In Schemes 1, 2 and 4–6, increased-valence structures for PCl5, O3, SO42−, NO3− and N2O4 are generated from Lewis octet VB structures and compared with the non-octet hypervalent VB structures displayed in ref. 1, namely structures 1a, 2c, 4c, 5c and 6c here. Brief consideration is also given to increased-valence structures for SN2 reactions.
 | ||
| Scheme 1 Hypercoordinate PCl5 valence-bond (VB) structures. Here and in Schemes 2–6, (non-variationally best) atomic formal charges for increased-valence structures are assigned on the assumption that bonding electrons are shared equally by pairs of adjacent atoms,9 and mirror-image structures are not displayed. Of course the extent of delocalization differs for non-equivalent 3c, 4e bonding units. | ||
 | ||
| Scheme 2 O3 VB structures. | ||
For illustrative purposes, in the Appendix we show that Durrant's method to determine the γ parameter of ref. 1 for XAY-type systems with a symmetrical 3c–4e bonding unit is related to the A-atom charge density obtained from a 3c–4e molecular orbital (MO) configuration. Consideration is also given to the A-atom valence.
Increased-valence structures and hypervalence
For PCl5, O3, SO42−, NO3− and N2O4, Durrant1 has displayed two types of Lewis VB structures that involve non-fractional (2c–2e) bonds between pairs of adjacent atoms, namely familiar, non-hypervalent octet Kekulé type structures 1c, 2a, 3a, 4a, 5a and 6a, and the hypervalent structures 1a, 2c, 4c, 5c, and 6c that violate the octet rule. For electron-rich hypercoordinate molecules, such as PCl5 (cf. Scheme 1 of ref. 1 and Scheme 1 here) and for the electron-rich non-hypercoordinate molecules and ions O3, SO42−, NO3− and N2O4, 2c–2e bonds are the only types of bonds that are present in both types of Lewis VB structures.Dewar/singlet diradical types of Lewis VB structures, such as 1b and 3b–3d, were not considered in ref. 1. In ref. 6, it is indicated that hypervalency for electron-rich systems arises when Dewar/type structures participate in resonance with the Kekulé type structures.
When relevant atomic orbitals (AOs) overlap, Lewis-type VB structures for electron-rich species can be stabilized via 1-electron delocalizations from separate lone-pair AOs into 2-centre bonding MOs or bond orbitals (BOs), as is shown, for example, in structures 1c for PCl5 in Scheme 1, and 2a, 4a, 5a and 6a for O3, SO42−, NO3− and N2O4 in Schemes 2 and 4–6. The resulting VB structures, 1d, 2b, 4b, 5b and 6b possess (thin-bond line) fractional 2c–2e bonds and 2c–1e bonds, as well as normal 2c–2e bonds.
By inspection, one can see that more electrons participate in bonding in VB structures 1d, 2b, 4b, 5b and 6b than does occur in the Lewis Kekulé structures 1c, 2a, 4a, 5a and 6a. Therefore 1d, 2b, 4b, 5b and 6b are examples of “increased-valence” structures6,8,9 without expansion of the valence shell. Because relative to the octet Lewis structures, increased-valence structures involve additional electrons in both nearest-neighbour and non-neighbour bonding, increased-valence structures are hypervalent relative to the Lewis structures.6,8,9
Some properties of increased-valence structures
With Heitler–London AO type wavefunctions for 2c–2e bonds – for example a(1)b(2) + b(1)a(2) for the 2c–2e A–B bond – increased-valence structures for electron-rich molecules summarize resonance between two types of Lewis structures, namely the familiar, standard/Kekulé type Lewis octet structures, and “long-bond”/formal bond/singlet diradical/Dewar type Lewis (octet) structures. As indicated already, the latter type of Lewis VB structure is not considered in ref. 1. For the O3 increased-valence structure 2b, these two types of Lewis octet structures (namely structure 3a and structures 3b–3d) are displayed in Scheme 3. None of them is hypervalent, but resonance between them generates hypervalence for the resulting increased-valence structure.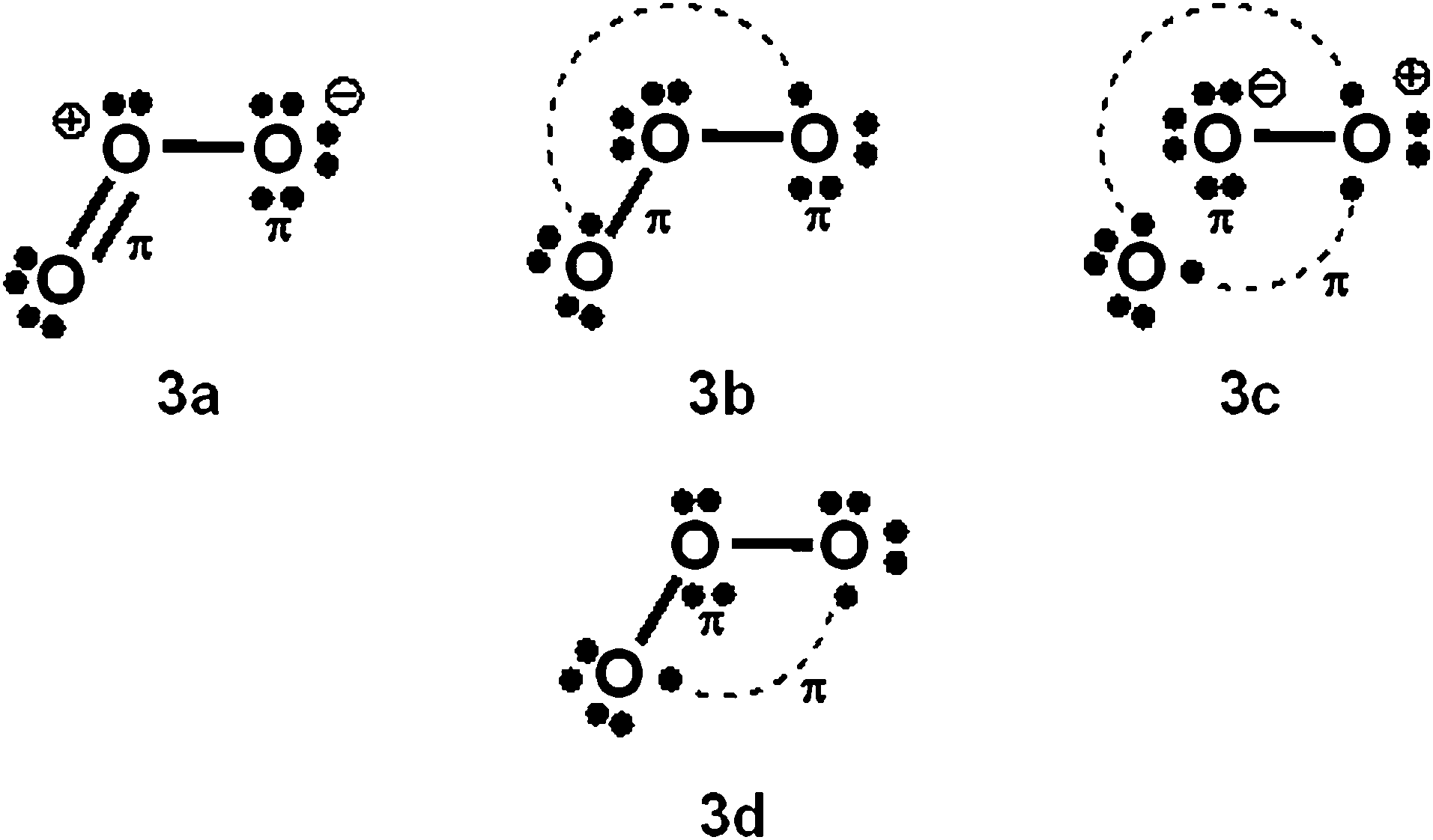 | ||
| Scheme 3 Component Kekulé and Dewar/singlet diradical Lewis structures for increased-valence structure 2b of Scheme 2. | ||
The PCl5 and O3 Dewar structures 1b, 3b and 3d do not carry atomic formal charges, and 1b and 3d in particular were not considered by Durrant.
For an increased-valence structure that does not involve a valence shell expansion to provide an additional AO for bonding, none of the component Lewis structures violates the octet rule, but resonance between them to generate the increased-valence structure leads to (an apparent) violation of the octet rule.6,8,9
Increased-valence structures for SO42−, NO3− and N2O4
In Schemes 4–6, we have generated increased-valence structures for SO42−, NO3− and N2O4 from familiar octet Kekulé type Lewis structures via 2c–1e delocalizations of oxygen O− electrons. In these figures, the hypervalent VB structures with only 2c–2e bonds displayed in ref. 1 are also displayed. We suggest that the increased-valence structures with 1-electron bonds and fractional 2c–2e bonds provide better insight into the possible origin of some molecular properties than do the hypervalent VB structures of ref. 1. For example: | ||
| Scheme 4 SO42−. As well as structure 4b, another type of increased-valence structure can be constructed with four fractional 2c–2e bonds + four 2c–1e bonds and oxygen atom formal charges of −1/2. | ||
 | ||
| Scheme 5 NO3−. As well as 5b, another type of increased-valence structure can be constructed, with two fractional 2c–2e bonds and two oxygen atoms with formal charges of −1/2. | ||
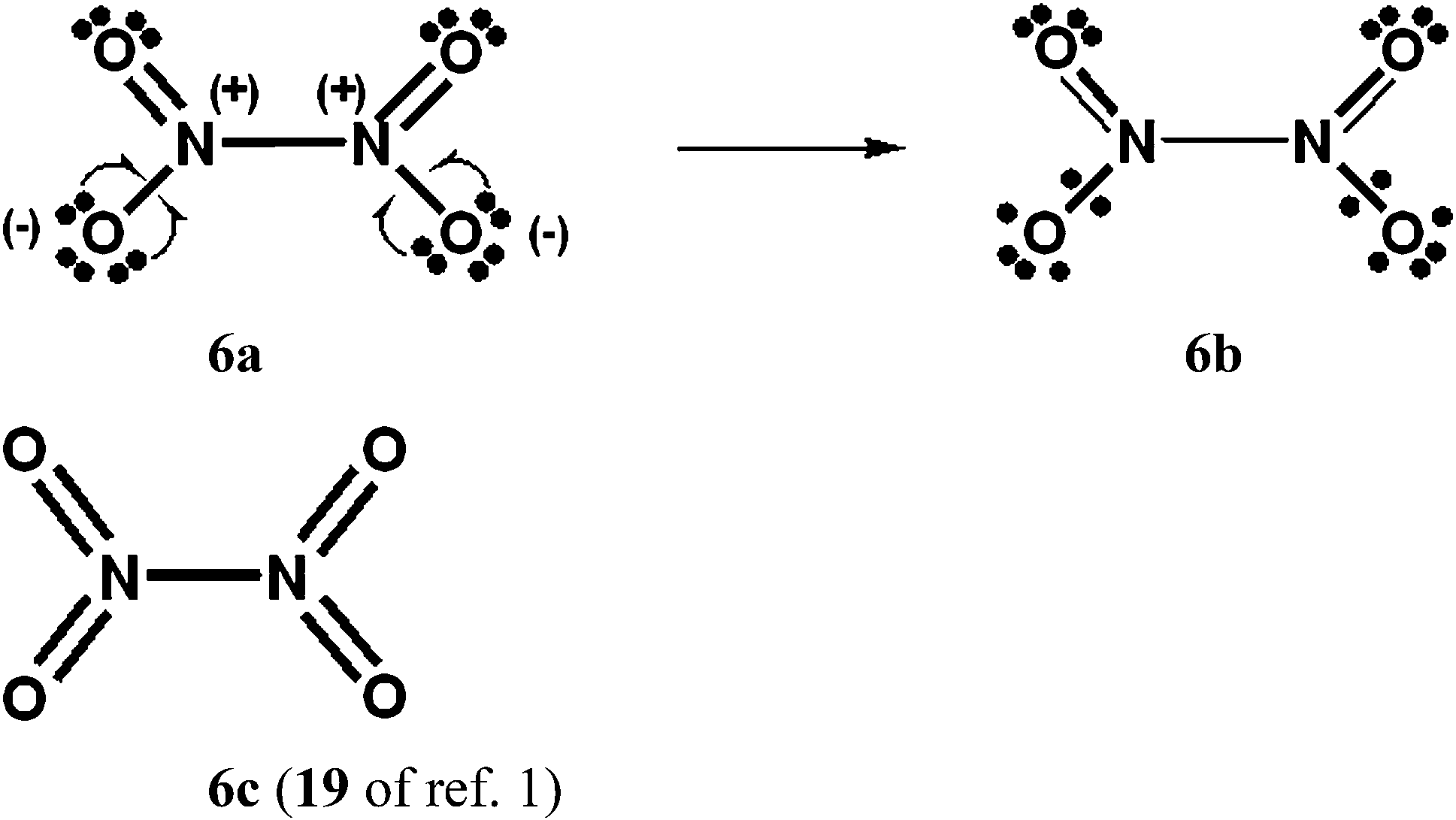 | ||
| Scheme 6 N2O4 with D2h symmetry. The 16 component Kekulé and Dewar Lewis structures are displayed in ref. 9a and b, as well as an increased-valence structure for an ONONO2 isomer. There are two “cis” type structures for each of 6a and 6b, and two “trans” type structures. | ||
(a) The fractional N–N 2c–2e bond in the increased-valence structure 6b for N2O4 is in accord with the presence of a long, weak N–N single bond.9
(b) The results of VB calculations for O3 and related 1,3-dipolar systems from numerous laboratories – see for example ref. 7d and e and 10 and references therein – indicate that their ground-states possess substantial singlet-diradical character. It arises primarily from the contribution of the Lewis structure 3d of Scheme 3 to the ground-state Lewis structure resonance scheme. In contrast to structure 2c (i.e. structure 5b of ref. 1), increased-valence structure 2b of Scheme 2 reflects the diradical character.
SN2 reactions
With Coulson–Fischer11 type BOs (a + k1b) and (b + k2a) replacing the a and b AOs of the Heitler–London wavefunction for a 2c–2e bond, the course of an SN2 reaction, X− + AY → XA + Y− has been formulated12 as
For it, the increased-valence structures for the (XAY)− reactant-like and product-like complexes, each with an additional bonding electron relative to the AY reactant and XA product, are both hypercoordinate and hypervalent relative to the VB structures for the latter species.
Further comments and conclusion
When 2-centre Coulson–Fisher type BOs, such as the a + k1b and b + k2a above, are used to accommodate the electrons that form the fractional 2c, 2e bonds in the increased-valence structures, allowance can be made for polarization of these bonds.In ref. 13, the wavefunctions for 3c–4e VB structures of the types X—A—Y (as would occur in the Durrant structures 2c, 5c and 6c for O3, NO3− and N2O4 if expansion of the valence shell does not occur) and •X • A • Y•, and the Rundle-Hach14–Pimentel15 3c–4e MO configuration have been shown to be special cases of wavefunctions for resonance between the increased-valence structures •X • A—Y and X—A • Y•, with 2-centre Coulson–Fischer orbitals, and one variational parameter.16
Increased-valence structures can also be constructed for: (a) systems that involve 3c–3e bonding units,17 with non-reactant and non-product ionic structures replacing the singlet-diradical structures of electron-rich systems, and (b) diatomic molecules.18
Regardless of the method used to construct the wave-functions for increased-valence structures, because they involve the participation of more electrons in bonding than do the Kekulé-type Lewis structures from which they are derived, increased-valence structures are hypervalent relative to these Lewis structures. As indicated above, for electron-rich systems, this is due to the inclusion of singlet diradical structures in the Lewis structure resonance scheme. Also, increased-valence structures involve at least one Pauling three-electron bond as a diatomic component.9
Appendix: valence electron equivalent parameter γ(A) and atomic valencies
In ref. 1, the valence electron equivalent parameter γ(A) for atom A is used to determine whether a molecule exhibits hypervalence.19 Here for the linear, symmetrical, triatomic systems of Table 1, each with one 3c–4e bonding unit, we shall show that the Durrant method1 to construct γ(A) is equivalent to a method that uses a 3c–4e MO configuration with AO overlap integrals omitted.| Molecule | A-atom charge1 | k 2 | γ(A) | A-Atom valence |
|---|---|---|---|---|
| F3− | −0.056 | 2.137 | 8.11 | 1.35 |
| ClF2− | +0.291 | 1.098 | 7.42 | 1.11 |
| Cl3− | −0.019 | 2.077 | 8.04 | 1.34 |
| BrCl2− | +0.108 | 1.160 | 7.78 | 1.27 |
| ICl2− | +0.263 | 1.167 | 7.47 | 1.14 |
| XeF2 | +1.230 | 1.252 | 7.54 | 1.17 |
| KrF2 | +1.003 | 1.988 | 7.99 | 1.06 |
| XeCl2 | +0.763 | 3.242 | 8.47 | 1.37 |
MO wavefunction
As the wavefunction for the 3c–4e electrons, we shall use the Hach-Rundle–Pimentel MO configuration14,15 of eqn (1).| |ψ1αψ1βψ2αψ2β| ∝ |(2x + ka)α(2x + ka)β(2y + ka)α(2y + ka)β| | (1) |
The x, a and y are the overlapping AOs on the three atomic centres, and ψ1 = x + ka + y and ψ2 = x − y are the bonding and non-bonding MOs that can be constructed from them.14,15
The right-hand side of eqn (1) gives the valence-bond structure21 (X—A—Y)q, with fractional 2c–2e bonds that arise from double-occupation of two non-orthogonal BOs, and q = −1 or 0.
To determine the value for k, we equate the charge QA of ref. 1 for atom A to XA − 2k2/(k2 + 2). The XA is the core charge of atom A when the 3c–4e electrons are removed, and 2k2/(k2 + 2) = Paa is the A-atom charge density that arises from the 3c–4e bonding. For the neutral species and anions of Table 1, XA = 2 and 1, respectively.
Durrant's method1 to construct γ(A)
To construct the γ(A) parameter for the systems considered in Table 1, initially we follow Durrant's methodology,1 as described in ref. 1 for CO, and for SCl4 in the ESI for ref. 1. We use the expanded valence-shell (hypervalent) covalent structure X![[thick line, graph caption]](https://www.rsc.org/images/entities/b_char_e117.gif) A(q)Y (with two non-fractional 2c–2e bonds) and the ionic structure X(−)A(q+2)Y(−) (with q = XA − 2).
A(q)Y (with two non-fractional 2c–2e bonds) and the ionic structure X(−)A(q+2)Y(−) (with q = XA − 2).
These structures are weighted according to the value of q = XA − 2 so that the QTAIM charge QA of ref. 1 is reproduced viaeqn (2)
| xq + (1 − x)(q + 2) = QA | (2) |
The γ(A) is then calculated from eqn (4)viaeqn (3),
| γ(A) = 10x + 6(1 − x) | (3) |
| = 6 + 2(XA − QA) | (4) |
MO method to construct γ(A)
Alternatively we can use the 3c–4e MO configuration of eqn (1), and a k-dependent probably density function (P(k)) for the 3c–4e bonding unit. If we choose P(k) = 4k2/(k2 + 2) ≡ 2Paa, with QA = XA − 2k2/(k2 + 2), the resulting γ(A) parameter is given by eqn (6)viaeqn (5).| γ(A) = 6 + 4k2/(k2 + 2) | (5) |
| = 6 + 2(XA − QA) | (6) |
Therefore eqn (3) is identical to eqn (5), thereby showing that the Durrant methodology to determine γ(A) for a symmetric 3c–4e bonding unit corresponds to using a contribution by the A-atom's charge density to a MO formulation of 3c–4e bonding.
Three of the calculated values for γ(A) reported in Table 1 are greater than 8, which indicates hypervalency1 for the associated species. However of course different numerical values and conclusions would be obtained with different types of 3c–4e wavefunctions and P(k) functions.
A-Atom valence
In ref. 22, with AO overlap integrals omitted, it is deduced that the A-atom valence (VA) for the MO configuration of eqn (1) is given by eqn (7),| VA = Vax + Vay = 16k2/{(k2 + 4)(k2 + 2)} | (7) |
In Table 1, the MO estimates of the A atom valence for each species exceeds unity, and therefore its A atom is hypervalent. However, regardless of the values of the A-atom valence and the γ(A), because of the presence of 2c–1e bonds in addition to the 2c–2e bonds, more electrons participate in nearest-neighbour and non-neighbour bonding6,8,9 for each of the increased-valence structures than does occur in any of their component Lewis structures. Therefore all increased-valence structures are electronically hypervalent.
Note added in proof
For PCl5, an increased-valence analogue of structure 1a is obtained via the delocalization of two electrons from the Cl− of the Kekulé structure 1c rather than one (as in structure 1c), to give two 1-electron P–Cl bonds, a fractional equatorial 2c–2e P–Cl bond as well as the fractional axial 2c–2e P–Cl bond of structure 1d.Acknowledgements
We thank Dr Margaret-Jane Crawford for drawing the valence bond structures. We also thank and appreciate the Reply by Durrant to our Comment.Notes and references
- M. C. Durrant, Chem. Sci., 2015, 6, 6614–6623 RSC , see also ref. 2.
- (a) H. Cowley, Chemistry World, 2015, 29 Search PubMed , (i) September 4. (ii) October; (b) I. Miller, MyRSC blogs, 2015, October 26; (c) Anorganische Chemie, Resurrection of Hypervalence 2015, October 12.
- R. F. Bader, Atoms in Molecules. A Quantum Theory, Oxford University Press, Oxford, UK, 2003 Search PubMed.
- Schleyer5 has made the distinction between geometric hypervalence or hypercoordination, and electronic hypervalence. Cowley2a has written: Traditionally, hypervalence describes any atom that breaks the octet rule (or duet rule for hydrogen) by bonding to more neighbouring atoms than its valence electrons should allow. This point of view is adopted here. See for example, ref. 6 and 7 for discussions on hypervalence with6 and without7 increased-valence. Some of the discussions of ref. 7 assume that resonance between all of the ionic structures, such as structure 1c for PCl5, generates hypervalence for hypercoordinate systems.
- P. von R. Schleyer, Chem. Eng. News, 1984, 62, 4 Search PubMed , May 28.
- (a) R. D. Harcourt, Chem. Eng. News, 1985, 63, 3,58 Search PubMed , Jan. 21; (b) R. D. Harcourt, Int. J. Quantum Chem., 1996, 60, 553–566 CrossRef CAS; (c) R. D. Harcourt, J. Phys. Chem. A, 2011, 115, 6610–6616 CrossRef CAS PubMed , 8180 and ref. 6–8, 11 and 12 therein.
- (a) See refs. 15–25 of ref. 6c; (b) A. Kalemos and A. Mavridis, J. Phys. Chem. A, 2011, 115, 2378–2384 CrossRef CAS PubMed; (c) F. R. Knight, K. S. A. Arachchige, R. A. M. Randall, M. Bühl, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2012, 41, 3154–3165 RSC; (d) B. Braida and P. C. Hiberty, Nat. Chem., 2013, 5, 417–422 CrossRef CAS PubMed; (e) B. Braida, T. Ribeyre and P. C. Hiberty, Chem.–Eur. J., 2014, 20, 9643–9650 CrossRef CAS PubMed.
- R. D. Harcourt, J. Chem. Educ., 1968, 45, 779–786 CrossRef CAS ; corrections 1969, 46, 856.
- (a) R. D. Harcourt, Qualitative Valence-Bond Descriptions of Electron-Rich Molecules; Pauling “3-Electron Bonds” and Increased-Valence Theory, Lect. Notes Chem., 1982, 30 Search PubMed; (b) R. D. Harcourt, 2nd Edition: Bonding in Electron-Rich Molecules: Qualitative Valence Bond Approach via Increased-Valence Structures, Lect. Notes Chem., 2016, 90 Search PubMed.
- See for example: (a) P. C. Hiberty, Isr. J. Chem., 1983, 23, 10–20 CrossRef CAS; (b) B. Braida, C. Walter, B. Engels and P. C. Hiberty, J. Am. Chem. Soc., 2010, 132, 7631–7637 CrossRef CAS PubMed; (c) R. D. Harcourt and T. M. Klapötke, (i) J. Fluorine Chem. 2006, 127, 712–718 for PF5 calculations. (ii) Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 935–943.
- C. A. Coulson and I. Fischer, Philos. Mag., 1949, 40, 386–393 CrossRef CAS.
- R. D. Harcourt, ref. 9b, pp. 277–280 and ref. 14a–l and 15 therein. Missing fishhook arrows in the SN2 mechanisms on pages 277 and 279 are included in Fig. 21-4 on page 278. On pages 85 and 202, change y + b + kb and y + b − k*b to y + b + ka and y + b − k*a, and change The Vab of Eqn. (28) to The Vab of Eqn. (30).
- R. D. Harcourt and A. Harcourt, J. Chem. Soc., Faraday Trans. 2, 1974, 70, 743–752 RSC.
- R. J. Hach and R. E. Rundle, J. Am. Chem. Soc., 1951, 73, 4321–4324 CrossRef CAS.
- G. C. Pimentel, J. Chem. Phys., 1951, 19, 446–448 CrossRef CAS.
- For the increased-valence structure •X • A—Y, the orbitals are13x, x + 1/2ka, y + 1/2ka and ka + y. For the increased-valence structure X—A • Y•, the orbitals are13ka + x, x + 1/2ka, y + 1/2ka and y.
- R. D. Harcourt, ref. 9b, 318–319.
- R. D. Harcourt, J. Mol. Struct., 1991, 229, 39–62 CrossRef.
- The γ parameter of ref. 1 provides ‘the formal shared electron count at a given atom, obtained by any combination of ionic and covalent resonance forms20 that reproduces the observed charge distribution’1.
- Whether or not an O, S or N atom expands its valence shell expansion to form the additional 2c–2e bond in each of the covalent structures 2c, 4c, 5c and 6c will determine the nature of the covalent structure (for example XAY and X—A—Y without and with fractional 2c–2e bonds) that may be used to calculate γ(A). Here we use XAY, as has Durrant1.
- C. A. Coulson, J. Chem. Soc., 1964, 1442–1456 RSC.
- R. D. Harcourt, Aust. J. Chem., 2007, 60, 691–695 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |