
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly selective catalytic trans-hydroboration of alkynes mediated by borenium cations and B(C6F5)3†
John S.
McGough
,
Samuel M.
Butler
,
Ian A.
Cade
and
Michael J.
Ingleson
 *
*
University of Manchester, Oxford Road, Manchester M13 9PL., UK. E-mail: michael.ingleson@manchester.ac.uk
First published on 12th February 2016
Abstract
The trans-hydroboration of terminal alkynes mediated by borenium cations [NHC(9-BBN)]+ (NHC = N-heterocyclic carbene, 9-BBN = 9-borabicyclo(3.3.1)nonane) exclusively affords Z-vinylboranes. NHCs and chelating dialkyl substituents on the borenium cation and “non”-basic anions were essential to preclude alternative reactions including dehydroboration. Deuterium labelling studies indicate the mechanism involves addition of the boron electrophile to the alkyne and transfer of hydride to the opposite face of the activated alkyne. trans-Hydroboration proceeds with only catalytic amounts of B(C6F5)3 or [Ph3C][B(C6F5)4] to activate the (NHC)9-BBN(H) precursor with the borenium regenerated in the hydride transfer step. The NHC can be removed from the trans-hydroborated products by the addition of Et2O–BF3 providing access to vinylBBN species effective for Suzuki–Miyaura couplings to generate Z-alkenes. Combinations of catalytic B(C6F5)3 and stoichiometric [HB(C6F5)3]− also lead to trans-hydroboration of terminal alkynes to form Z-isomers of [arylCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHB(C6F5)3]−.
CHB(C6F5)3]−.
Introduction
Vinylboranes and boronate esters are ubiquitous reagents in synthesis widely used in C–C, C–N and C–O bond formation.1 The hydroboration reaction is a powerful transformation for generating vinylboranes which with terminal alkynes yields E-vinylboranes via syn-addition of a B–H moiety.2 In contrast, the trans-hydroboration of terminal alkynes to form Z-vinylboranes is rare with the first example only reported in 2000 using Rh or Ir catalysts.3 Subsequent breakthroughs have been limited excluding four notable exceptions using Ru, Pd and Co based catalysts.4–6 A transition metal free catalytic methodology for trans-hydroboration of alkynes to form Z-vinyl boranes using strong boron electrophiles represents a conceptually new approach not realised to date. In contrast, the transition metal free trans-hydrosilylation of alkynes has been reported using AlCl3 as activator.7 This was subsequently applied to alkene and alkyne trans-hydrosilylation using other main group Lewis acids as catalysts.8,9 In these systems the unusual selectivity is achieved by a stepwise mechanism, with activation of the alkyne by a silicon electrophile followed by subsequent transfer of a hydride to the opposite face in a steric controlled step. An analogous stepwise approach has not been reported for boron electrophiles. This is partly due to the challenge of generating a sufficiently electrophilic borane to activate the alkyne that does not then engage in syn-1,2- or 1,1-elementoboration (eqn (1) and (2)) of alkynes.The trans-hydroboration of alkynes could be achieved by using an appropriate borenium cation10 to activate the alkyne followed by intermolecular hydride transfer from a borane to the less hindered face of the intermediate. Borenium cations have been used in the elementoboration of alkynes but to date only via syn-1,2 or 1,1 addition to the alkyne.11–14 Quenching the borenium activated alkyne with an external donor (essential for trans-selectivity) requires strongly bound “non-migratory” groups on boron to preclude syn-1,2- or 1,1-elementoboration; thus chelating dianionic substituents and a strong Lewis base are essential. The transfer of a hydride intermolecularly from a borane-Lewis adduct would generate further equivalents of the borenium ion and thus render trans-hydroboration catalytic in the activator initially used to form the borenium cation (Fig. 1, bottom). A related catalytic cycle has been reported, albeit for imine hydroboration, using [PinB(DABCO)][HB(C6F5)3] as catalyst.15 Herein we report the trans-hydroboration of terminal alkynes using catalytic B(C6F5)3 as an activator. The reaction exclusively generates Z-vinylboranes in excellent yields that can be used for subsequent Suzuki–Miyaura couplings to form Z-alkenes.
 | ||
| Fig. 1 Previous work on alkyne elemento-boration (eqn (1) and (2)), and the work described herein (eqn (3)). Bottom, intermolecular hydride transfer for trans-hydroboration (intermediate shown as a vinyl cation a π complex between borenium and alkyne is also feasible). | ||
Results and discussion
Initial studies used 9-BBN(H)(amine) (9-BBN = 9-borabicyclo[3.3.1]nonane) ligated with strongly nucleophilic amines in an attempt to disfavour amine dissociation and alkyne cis-hydroboration.2 However, using 9-BBN(H)(quinuclidine), 1-H, in 1,2-C6H4Cl2 with 50 mol% B(C6F5)3 as activator dehydroboration of 1-pentyne to form 9-BBN(1-pentynyl)(quinuclidine), 2, and cis-hydroboration (from the reaction of free BBN and 1-pentyne) were the major outcomes (the identity of 2 was confirmed by independent synthesis using the frustrated Lewis pair (FLP) of [1][B(C6F5)4]/P(mesityl)3, Scheme 1, top right).16 Presumably dissociation of quinuclidine from boron is occurring at some point during the reaction enabling deprotonation of the borenium activated alkyne and formation of 2.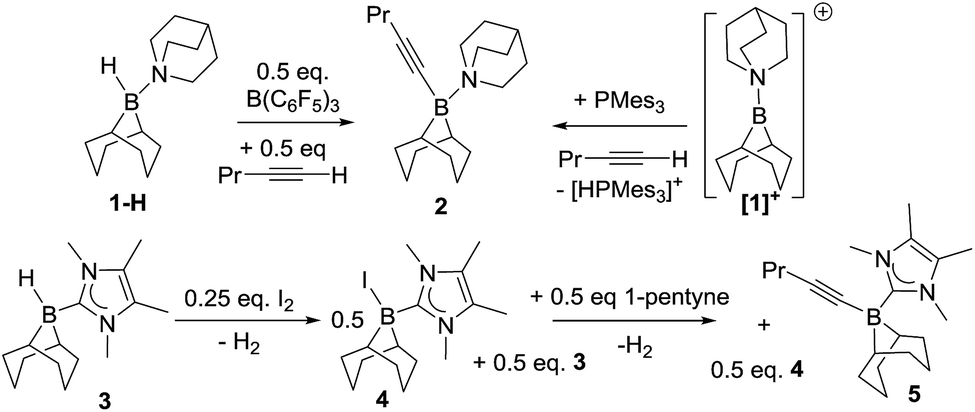 | ||
| Scheme 1 Alkyne dehydroboration with boreniums (or functional equivalents). | ||
N-Heterocyclic carbenes (NHCs) are stronger sigma donors than amines and 1,3,4,5-tetramethylimidazolylidene (IMe4) was selected as its low steric bulk (%Vbur = 26.1%)17 maximises nucleophilicity favouring irreversible binding to boron whilst minimising the kinetic barrier to alkyne and borenium cation interaction. Curran and co-workers have previously used (NHC)BH3 compounds activated with I2 to form (NHC)BH2I (a functional equivalent of a borenium) in alkene syn-hydroboration.12b 9-BBN(H)(IMe4), 3 was readily synthesised and activated with I2 (0.25 equiv.) forming a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 9-BBN(I)(IMe4) (4) and 3in situ. Subsequent addition of 1-pentyne to this mixture led to dehydroboration to form the alkynyl borane, 5, instead of trans-hydroboration. Addition of further equivalents of 3 and 1-pentyne to this reaction demonstrated that it is catalytic (in I2) with further 5 produced along with H2 (presumably from reaction of the HI by-product from dehydroboration with 3 which regenerates 4). Iodide is therefore sufficiently Brønsted basic to deprotonate the borenium activated alkyne; thus “non”-basic anions and NHCs are both required to realise trans-hydroboration. Whilst not-productive for trans-hydroboration the formation of 5 does represent a catalytic (in activator) electrophilic C–H borylation with H2 the only by-product and this topical catalytic conversion18 is being explored in a separate study.
:1 mixture of 9-BBN(I)(IMe4) (4) and 3in situ. Subsequent addition of 1-pentyne to this mixture led to dehydroboration to form the alkynyl borane, 5, instead of trans-hydroboration. Addition of further equivalents of 3 and 1-pentyne to this reaction demonstrated that it is catalytic (in I2) with further 5 produced along with H2 (presumably from reaction of the HI by-product from dehydroboration with 3 which regenerates 4). Iodide is therefore sufficiently Brønsted basic to deprotonate the borenium activated alkyne; thus “non”-basic anions and NHCs are both required to realise trans-hydroboration. Whilst not-productive for trans-hydroboration the formation of 5 does represent a catalytic (in activator) electrophilic C–H borylation with H2 the only by-product and this topical catalytic conversion18 is being explored in a separate study.
The reaction of 3 with 1 equivalent of B(C6F5)3 affords the borenium salt [9-BBN(IMe4)][HB(C6F5)3] (6) quantitatively by NMR spectroscopy. 1:1 mixtures of 3 and 6 only display a single broad 11B NMR signal (instead of discrete signals for the adduct 3 and the borenium 6) that shifts depending on the ratio of 3:6. This is attributed to rapid hydride transfer between the boron centres via a hydride bridged intermediate.19,20 Low temperature NMR studies on mixtures of 3 and 6 did not reach the slow exchange regime at −80 °C in CD2Cl2. Nevertheless, a 50:50 mixture of 3 and 6 in DCM reacts with 1-pentyne to generate a new signal in the 11B NMR spectrum at −16.1 ppm, along with 2 vinyl proton signals in a 1:1 ratio at 6.03 ppm (d, 3JHH = 13.3 Hz) and 5.54 ppm (dt, 3JHH = 13.2 and 6.1 Hz). These two signals correspond to the alkene protons of the trans-hydroborated product 9-BBN(IMe4)-1-pentene (7a, Scheme 2) based on 2D NMR experiments, and by comparison with the cis-hydroborated E-alkyne generated by the hydroboration of 1-pentyne by 9-BBN followed by the addition of IMe4. The E-vinylborane (7b) showed vinyl signals at 6.01 ppm (d, 3JHH = 17.4 Hz) and 4.74 ppm (dt, 3JHH = 17.5 and 6.4 Hz). The large 3JHH coupling constant observed between the cis-vinyl protons in 7a is comparable with the 3JHH coupling constants observed for other Z-vinylboranes (which are generally in the range of 13–15 Hz),3,4a,21 further supporting isomer identity. Whilst this provided proof of principle for alkyne trans-hydroboration using boreniums the reaction takes days for significant conversion even with 50 mol% of B(C6F5)3 (relative to 3).
 | ||
| Scheme 2 Hydroboration of 1-pentyne to give 7a and 7b. | ||
Enhancing electrophilicity at boron would potentially increase the rate of reaction. However, attempts using catecholborane/NHC derived species led to rapid ligand scrambling, therefore modification of the NHC was explored. Stephan and co-workers previously used the chlorinated NHC 1,3-dimethyl-4,5-dichloroimidazolylidene (I-DCDM) to increase the rate of hydrogenation of imines catalysed by [9-BBN(NHC)][B(C6F5)4], presumably due to the enhanced electrophilicity at boron.19b The neutral adduct 9-BBN(H)(I-DCDM) (8) was reacted with B(C6F5)3 to generate the borenium species [9-BBN(I-DCDM)][HB(C6F5)3] (9). In DCM a 1:1 mixture of 8 and 9 shows similar 11B NMR spectra to that of 3 and 6 indicating a fast exchange. A 1:1:1 mixture of 8, 9 and 1-pentyne resulted in rapid consumption of the alkyne to produce a number of species with alkene proton signals. The major new product had a 11B NMR signal at −15.9 ppm, and two vinyl protons, a doublet at 5.96 ppm (3JHH = 13.20 Hz), and a pseudo quintet (overlapped d of t, 3JHH = 6.60, 13.20 Hz) at 5.59 ppm consistent with trans-hydroboration of the alkyne (confirmed by 2D and NOE NMR spectroscopy). The minor products that increase with time are attributed to borocation induced isomerisation reactions (analogous to that observed by Curran, Vedejs, Lacôte and co-workers during borenium mediated syn-hydroboration).12a Reducing the loading of B(C6F5)3 reduces the rate of vinylborane-isomerisation; however, neither low loadings of B(C6F5)3 or lower temperatures prevented the formation of the minor products. Furthermore, using 8/10 mol% B(C6F5)3trans-hydroboration does not go to completion, with ca. 30% conversion observed (by 1H NMR spectroscopy). Nevertheless, the observation of ca. 3 turnovers confirmed that catalytic in B(C6F5)3trans-hydroboration is indeed viable.
The hydroboration of a range of arylacetylenes was subsequently investigated as these substrates are not amenable to extensive hydride/borane migration derived isomerisation during or post trans-hydroboration. The reaction of 8/10 mol% B(C6F5)3 and 4-ethynyltoluene proceeds rapidly to give a single product with an 11B NMR signal at −14.9 ppm. The in situ1H NMR spectrum in DCM shows two doublets (6.79 ppm, 3JHH = 14.4 Hz and 6.62 ppm, 3JHH = 14.4 Hz) for the cis-vinyl protons. The magnitude of this coupling constant is again fully consistent with other Z-vinyl boranes,3,4a,21 and the stereochemistry was unambiguously assigned via 2D-NOE NMR experiments. It was found that 5 mol% B(C6F5)3 was optimal for complete trans-hydroboration, with the reaction being finished within 5 minutes at room temperature in CH2Cl2. The products are then isolated without column chromatography simply by addition of pentane, filtration (removing minor quantities of ionic by-products) and drying. trans-Hydroboration is tolerant of a range of functional groups (Scheme 3) and in all cases only the Z-vinylborane isomer was formed with no E-isomer or over reduction products (diborylated alkanes) observed. Electron donating groups such as para-methoxy (10c) and para-NMe2 (10d) are compatible as are electron withdrawing groups including p-chloro- (10e), p-fluoro- (10f) p-trifluoromethyl- (10g) and m-chloro (10h) substituted phenylacetylenes. The substrate scope was extended to a hindered alkyne with mesityl acetylene forming 10i in good yield (84%). The compatibility of hetero-aromatic substrates was demonstrated using 2-ethynylthiophene with the formation of 10j in an excellent yield (91%). The reaction is not limited to heteroaryl and arylacetylenes with conjugated enynes also amenable with hydroboration exclusively occurring at the alkyne, affording the borylated dienes 10k and 10l. The trans-hydroboration of alkyl alkynes such as 1-octyne is problematic in terms of isolating the trans-hydroborated products cleanly due to intractable minor products from isomerisation reactions.12 However, the trans-hydroboration products are the major products formed.
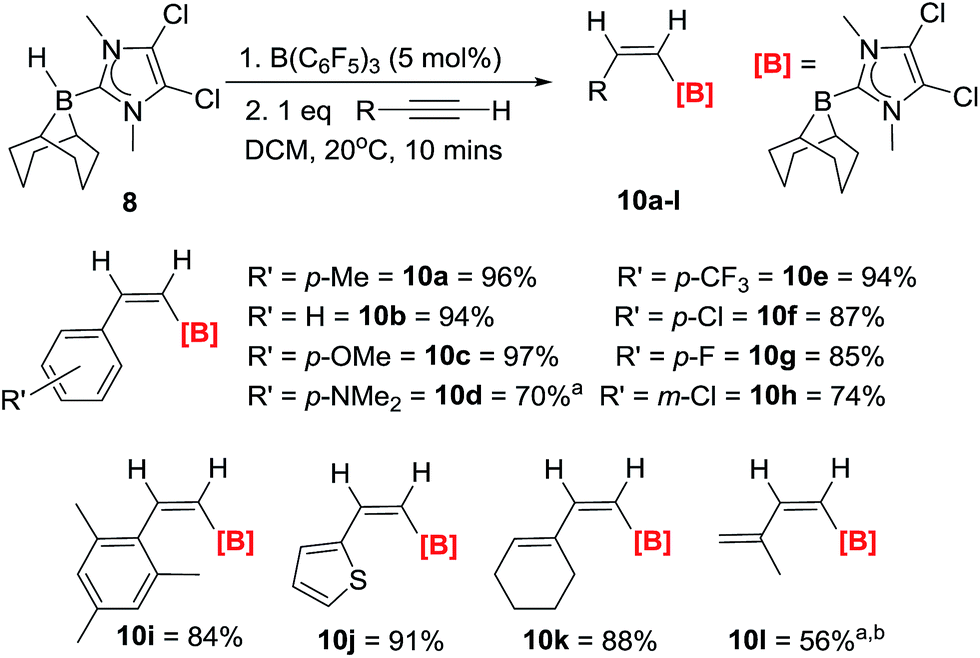 | ||
| Scheme 3 Reaction scope and isolated yields (unless otherwise stated) for the trans-hydroboration of terminal alkynes. a = yield by NMR spectroscopy versus mesitylene as internal standard. b = 10 mol% B(C6F5)3. | ||
The cross coupling of 10a with 4-iodo-fluorobenzene using a range of Pd catalysts/bases principally generated para-methylstyrene from proto-deborylation. It was hypothesized that either NHC dissociation at raised temperatures was poisoning the Pd catalyst or strong NHC binding to boron prevented transmetallation, thus prior removal of the carbene was explored. Heating 10a and one equiv. of BF3:OEt2 in toluene at 60 °C for 1 h led quantitatively to the formation of 11a and the NHC–BF3 adduct (Scheme 4). Importantly, there was no loss of alkene Z-stereochemistry (by NOE spectroscopy). After removal of toluene, the mixture of 11a (or 11c) and NHC–BF3 can be used in Suzuki–Miyaura couplings without any purification. Cross coupling proceeds to give exclusively the cis-stilbenes 12a and 12c in good isolated yields (81 and 73%, respectively), with the NMR spectra of 12c consistent with that previously reported,22 further confirming the trans selectivity of the hydroboration. The NHC adducts 10a–l therefore represent air stable precursors that are readily unmasked for use in Suzuki–Miyaura reactions (and other transformations previously reported using vinylBBN species),23 analogous to recent elegant work using NHCBH2(aryl) compounds as boronic acid precursors.24
 | ||
| Scheme 4 Suzuki Miyaura cross coupling of 10x. (i) 1 equiv. BF3(OEt2), toluene, 60 °C, 1 h. (ii) Dried in vacuo, then Pd(PtBu3)2 (10 mol%), 4-iodo-fluorobenzene (1.1 eq.), THF, KOtBu (2.5 eq.) 20 °C, 18 h. | ||
Mechanistic studies
The mechanism for formation of 10a–l could proceed by 1,2-hydroboration or 1,1-hydroboration. 1,1-Hydroboration would involve a 1,2-hydride shift in the borenium activated intermediate prior to intermolecular hydride transfer; related 1,2-hydride shifts have been reported on combination of B(C6F5)3 with terminal alkynes which ultimately results in 1,1-carboboration of the alkyne.25 The hydroboration of 1-octyne with 8 proceeds to give a single major hydroborated product 10m (by 1H NMR spectroscopy, using 10 mol% B(C6F5)3), with signals at 5.95 ppm (d, 3JHH = 13.2 Hz for C1 bound proton) and 5.61 (dt, 3JHH = 13.2 and 6.5 Hz) for the C2 proton. In contrast, when 1-octyne-d1 is used, 10mD forms with the signal at 5.95 ppm not observed whilst a vinylic signal at 5.62 ppm is observed although now as a triplet (3JHH = 5.6 Hz); furthermore a vinyl deuteron signal (5.37 ppm) is seen in the 2H NMR spectrum (see ESI†). Based on these results, we conclude that the reaction is a 1,2-trans-hydroboration and not 1,1-hydroboration (Scheme 5). | ||
| Scheme 5 Deuterium labelling experiment with 8. | ||
With deuterium labelling studies indicating a 1,2-trans-hydroboration mechanism the trans-hydroboration of internal alkynes is feasible. The reaction with a range of internal alkynes was not successful under a range of catalytic loadings and conditions with minimal activation of the alkyne observed (<5%).26 However, using the nucleophilic internal alkyne 4-(1-propynyl)-N,N-dimethylaniline trans-hydroboration was effective, albeit at a slower rate to that observed with terminal alkynes. After stirring for 3 hours the 11B NMR spectra shows one major new signal at −12.6 ppm, along with a new singlet in the 1H NMR spectrum at 6.68 ppm indicating a single new vinyl borane product is formed in 70% conversion and confirmed as the E isomer by NMR spectroscopy (10n, Scheme 6) thus is formed by trans-1,2-hydroboration. This represents the first highly selective trans-hydroboration of an unsymmetric internal aryl-alkyne, as previous work produced varying ratios of isomeric products.4b
 | ||
| Scheme 6 trans-Hydroboration of an internal alkyne. | ||
The failure to trans-hydroborate the majority of internal alkynes investigated is in part due to decomposition of the mixtures of 8 and 9 over the longer reaction times required (relative to terminal alkynes which react within minutes). A freshly prepared solution of equimolar 8 and 9 shows only a single broadened signal (due to fast exchange) in the 11B NMR spectrum but on standing for 3 h at 20 °C in DCM a new 11B NMR signal grows in at −12.8 ppm, concurrent with the signal for 8 (−17 ppm, 1JBH = 79 Hz). The signal at −12.8 ppm in the 11B NMR spectrum also is observed in the reactions with internal alkynes that do not undergo trans-hydroboration indicating that borenium cation decomposition out-competes the slower trans-hydroboration of most internal alkynes. The new boron species was isolated by layering a DCM solution with hexanes to produce single crystals suitable for X-ray diffraction analysis. This revealed the compound to be the boronium salt [9-BBN(I-DCDM)2][HB(C6F5)3] (13, Fig. 2). Borenium decomposition therefore proceeds by NHC transfer which may occur via the hydride bridged intermediate, with NHC transfer coming within the solvent shell via a concerted process. No activation of the DCM solvent by the NHC is observed, disfavouring an intermolecular pathway (8 is stable in DCM for days at 20 °C, precluding NHC → B dissociation as NHCs react rapidly with DCM).27 The by-product from formation of 13 is 9-BBN and low quantities of alkyne cis-hydroboration products also are observed during attempted catalytic trans-hydroboration of internal alkynes. Attempts to preclude this decomposition pathway by using larger N-substituents on the NHC (e.g., mesityl) was successful in preventing boronium formation, but these more sterically hindered borenium/BBN(H)(NHC) mixtures did not react even with terminal alkynes, presumably due to a larger steric barrier.
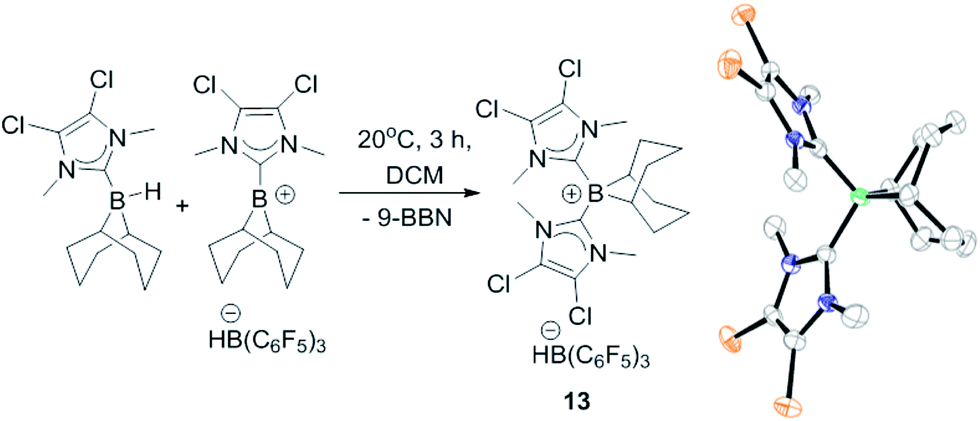 | ||
| Fig. 2 Formation of 13 and structure of 13 (thermal ellipsoids at 50% probability). Hydrogens and [HB(C6F5)3]− omitted for clarity. | ||
trans-Hydroboration is proposed to proceed by activation of the alkyne by 9 to generate a vinyl cation (or π complex), which is then quenched by transfer of a hydride to the activated alkyne to generate 10x; however, this last step could involve hydride transfer from 8 or from [HB(C6F5)3]− (with the latter finding precedence in olefin reduction).28 The former will regenerate an equivalent of 9 directly whilst if hydride transfer is from [HB(C6F5)3]− the subsequent reaction of B(C6F5)3 with 8 will then reform 9. Hydride transfer is feasible directly from 8 as the use of 5 mol% of [Ph3C][B(C6F5)4] as an activator in place of B(C6F5)3 in the trans-hydroboration of ethynyltoluene proceeded with no loss of activity (complete in <5 minutes at 20 °C) to generate 10a in an excellent yield (97%). The reaction of stoichiometric 9 with 4-ethynyltoluene also led to trans-hydroboration and formation of 10a and B(C6F5)3, (as observed by in situ NMR spectroscopy), indicating that [HB(C6F5)3]− is able to transfer hydride to the activated alkyne as in the absence of 8 the anion is the only hydride source. The extremely rapid reaction at 5 mol% loading precludes rate comparison between [B(C6F5)4]− and [HB(C6F5)3]−, whilst attempts with lower activator loadings led to irreproducible results due to the sensitivity of borenium cations to varying quantities of trace protic impurities.
Analysis of the low quantity of ionic by-products formed in the catalytic trans-hydroboration provided further insight into the minor processes operating alongside borenium mediated trans-hydroboration. Along with boronium salt 13 an additional C6F5 containing species was observed (by 19F NMR spectroscopy) in the pentane insoluble fraction post trans-hydroboration of terminal alkynes. The second ionic by-product displayed 19F signals at −132.5, −164.5 and −167.3 ppm consistent with a [(C6F5)3B-vinyl]− formulation.29 Recrystallization of the ionic by-products from borenium mediated trans-hydroboration of 4-ethynyltoluene unambiguously identified this species as [(C6F5)3B-Z-(4-methylstyrene)]− [14a]− (Fig. 3). In this structure the anion is partnered with [1,3-dimethyl-4,5-dichloro imidazolium]+ presumably formed from trace adventitious moisture. Examination (by 11B and 19F NMR spectroscopy) of the crude trans-hydroboration reaction mixtures pre-workup revealed anion [14a]− was present as a minor product during the borenium mediated trans-hydroboration of 10a–10d and 10h–10k.
 | ||
| Fig. 3 ORTEP depiction of [14a]− (thermal ellipsoids at 50%). | ||
It was hypothesised that [14a]− forms during borenium mediated trans-hydroboration when B(C6F5)3 (present after transfer of hydride from [HB(C6F5)3]−) activates the alkyne instead of reacting with 8. The B(C6F5)3 activated alkyne is then quenched by subsequent hydride transfer from 8 or [HB(C6F5)3]− to generate [14a]−. This is related to the reactivity of B(C6F5)3/Lewis base FLPs which undergo 1,2-addition reactions with alkynes;16,30 in this case the nucleophile is [HB(C6F5)3]− or 8. 1,1-Carboboration products can be formed from the reaction of B(C6F5)3 and terminal alkynes but no resonances consistent with these products are observed.31 This indicates quenching the B(C6F5)3 activated alkyne by intermolecular hydride transfer is more rapid than intramolecular hydride migration and 1,1-carboboration. To confirm this hypothesis [NBu4][HB(C6F5)3] was reacted with 4-ethynyltoluene in the presence of 5 mol% B(C6F5)3 to give [14a][NBu4] which could be isolated in 96% yield. The product shows a single 11B signal at −16.8 ppm in the NMR spectrum, along with 2 new vinyl resonances at 6.70 (d, 3JHH = 14.55 Hz) and 6.65 (d, 3JHH = 14.92 Hz) in the 1H NMR spectrum fully consistent with a cis-vinylborane. The scope of B(C6F5)3 catalysed trans-hydroboration was explored with terminal alkynes such as phenylacetylene and 4-ethynylanisole reacting rapidly to give [14b]− and [14c]−, respectively (Scheme 7). However, less nucleophilic terminal alkynes such as 4-(trifluoromethyl)-phenylacetylene and internal alkynes such as 3-hexyne did not undergo trans-hydroboration (even after heating to 100 °C for extended periods). Nevertheless, the use of an inexpensive borane/borohydride combination to achieve transition metal free trans-hydroboration is unprecedented to the best of our knowledge.
 | ||
| Scheme 7 Alkyne trans-hydroboration with B(C6F5)3/[HB(C6F5)3]−. | ||
Conclusions
In conclusion, highly Lewis acidic boranes containing low migratory aptitude substituents (chelated alkyls or C6F5 groups) enable the transition metal-free trans-hydroboration of alkynes in the presence of an appropriate hydroborane or borohydride. Using a borenium cation trans-hydroboration is applicable to a range of aryl (containing electron donating and withdrawing groups), heteroaryl and vinyl substituted terminal alkynes, exclusively generating Z-vinylborane isomers in excellent yields. Mechanistically the reaction does not involve intramolecular hydride transfer steps and is thus applicable to internal alkynes (albeit with limited scope currently). In both borenium cation and B(C6F5)3 mediated trans-hydroboration the Z-vinylborane products are readily isolated as air stable solids and for the former the utility of these products in cross coupling has been demonstrated. Work is ongoing to expand the scope of the trans-elementoboration methodology by rational borocation modification, as well as to probe the range of neutral boranes capable of alkyne activation and trans-elementoboration.Acknowledgements
This work was made possible by financial support from the European Research council (FP7 Grant Agreement 305868), the EPSRC (EP/J000973/1 and EP/K03099X/1), and the Royal Society (for the award of a University Research Fellowship).Notes and references
- (a) Boronic Acids, ed. D. G. Hall, Wiley-VCH, 2011 Search PubMed. For a review on the uses of vinylboranes see: (b) M. Vaulter and G. Alcaraz, Science of Synthesis, Vinylboranes, Georg Thieme Verlag KG, 2014 Search PubMed.
- H. C. Brown and M. Zaidlewicz, Organic Syntheses via Boranes, Aldrich Chemical, Milwaukee, WI, 2001, vol. 2 Search PubMed.
- T. Ohmura, Y. Yamamoto and N. Miyaura, J. Am. Chem. Soc., 2000, 122, 4990 CrossRef CAS.
- For Ru see: (a) C. Gunanathan, M. Holscher, F. Pan and W. Leitner, J. Am. Chem. Soc., 2012, 134, 14349 CrossRef CAS PubMed; (b) B. Sundararaju and A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 14050 CrossRef CAS PubMed.
- For Pd see: S. Xu, F. Haeffner, B. Li, L. V. Zakharov and S.-Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 6795 CrossRef CAS PubMed.
- For Co see: J. V. Obligacion, J. M. Neely, A. N. Yazdani, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2015, 137, 5855 CrossRef CAS PubMed.
- N. Asao, Y. Sudo and Y. Yamamoto, J. Org. Chem., 1996, 61, 7654 CrossRef CAS PubMed.
- For trans-hydrosilylation with catalytic B(C6F5)3 see: (a) M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936 CrossRef CAS PubMed; (b) L. D. Curless and M. J. Ingleson, Organometallics, 2014, 33, 7241 CrossRef CAS.
- M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308 CrossRef PubMed.
- For recent reviews on borocations see: (a) W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016 CrossRef CAS PubMed; (b) T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 116, 4246 CrossRef PubMed; (c) M. J. Ingleson, Top. Organomet. Chem., 2015, 49, 39 CrossRef.
- For syn-haloboration see: J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon and M. J. Ingleson, Angew. Chem., Int. Ed., 2013, 52, 7518 CrossRef CAS PubMed.
- For syn-hydroboration see: (a) A. Prokofjevs, A. Boussonniere, L. Li, H. Bonin, E. Lacote, D. P. Curran and E. Vedejs, J. Am. Chem. Soc., 2012, 134, 12281 CrossRef CAS PubMed; (b) X. Pan, A. Boussonniere and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 14433 CrossRef CAS PubMed; (c) X. Pan, A. Boussonniere, S. J. Geib and D. P. Curran, Organometallics, 2013, 32, 7445 CrossRef.
- For syn-carboboration see: (a) I. A. Cade and M. J. Ingleson, Chem.–Eur. J., 2014, 20, 12874 CrossRef CAS PubMed; (b) M. Devillard, R. Brousses, K. Miqueu, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5722 CrossRef CAS PubMed.
- For 1,1-carboboration with borocations: J. R. Lawson, V. Fasano, J. Cid, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2015 10.1039/c5dt03003j.
- P. Eisenberger, A. Bailey and C. M. Crudden, J. Am. Chem. Soc., 2012, 134, 17384 CrossRef CAS PubMed.
- For the use of FLPs to dehydroborate alkynes see: M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396 CrossRef CAS PubMed.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC.
- M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513 CrossRef PubMed.
- Similar [(NHC)9-BBN]+ cations have been reported: (a) J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728 CrossRef CAS PubMed; (b) J. M. Farrell, R. T. Posaratnanathan and D. W. Stephan, Chem. Sci., 2015, 6, 2010 RSC; (c) P. Eisenberger, B. P. Bestvater, E. C. Keske and C. M. Crudden, Angew. Chem., Int. Ed., 2015, 54, 2467 CrossRef CAS PubMed.
- Hydride bridged borocations have been previously characterised see: (a) T. S. De Vries and E. Vedejs, Organometallics, 2007, 26, 3079 CrossRef CAS PubMed; (b) A. Prokofjevs, J. W. Kampf, A. Solovyev, D. P. Curran and E. Vedejs, J. Am. Chem. Soc., 2013, 135, 15686 CrossRef CAS PubMed.
- M. Hoshi, K. Takahata and A. Arase, Tetrahedron Lett., 1997, 38, 453 CrossRef CAS.
- M. Shi and B. Xu, J. Org. Chem., 2002, 67, 294 CrossRef CAS PubMed.
- For a recent use of vinylBBN in synthesis see: K. Nagao, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2014, 136, 10605 CrossRef CAS PubMed.
- (a) S. Nerkar and D. P. Curran, Org. Lett., 2015, 17, 3394 CrossRef CAS PubMed; (b) For other references on the use of C(sp2)–BBN species in cross coupling see: A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412 RSC and references therein.
- G. Kehr and G. Erker, Chem. Commun., 2012, 48, 1839 RSC.
- Unsuccessful internal alkynes tested include: 3-hexyne, 1-phenyl-1-propyne, bis(p-methoxyphenyl)acetylene, diphenylacetylene, (p-dimethylaminophenyl)-(p-methoxyphenyl)acetylene, (bromo)(para-tolyl)acetylene, (para-tolyl)(2-thiophenyl)acetylene.
- A. J. Arduengo, F. Davidson, H. V. R. Dias, J. R. Goerlich, D. Khasnis, W. J. Marshall and T. K. Prakasha, J. Am. Chem. Soc., 1997, 119, 12742 CrossRef CAS.
- X. Zhao and D. W. Stephan, Chem. Sci., 2012, 3, 2123 RSC.
- C. Jiang, O. Blaque and H. Berke, Organometallics, 2010, 29, 125 CrossRef CAS.
- M. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6594 CrossRef CAS.
- For the 1,1-carboboration of terminal alkynes with B(C6F5)3 see: (a) C. Chen, F. Eweiner, B. Wibbeling, R. Fröhlich, S. Sends, Y. Ohki, K. Tatsumi, S. Grimme, G. Kehr and G. Erker, Chem.–Asian J., 2010, 5, 2199 CrossRef CAS PubMed. For a recent perspective article on carboboration with RB(C6F5)2, see: (b) G. Kehr and G. Erker, Chem. Sci., 2016, 7, 56–65 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details, NMR spectra and crystallographic data in .cif format. CCDC 1429287–1429289 and 1440939. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04798f |
| This journal is © The Royal Society of Chemistry 2016 |