
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile syntheses of [3]-, [4]- and [6]catenanes templated by orthogonal supramolecular interactions†
Kai
Wang
,
Chi-Chung
Yee
and
Ho Yu
Au-Yeung
 *
*
Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: hoyuay@hku.hk
First published on 15th January 2016
Abstract
A water soluble [6]catenane consisting of two interlocking [3]catenanes was synthesised in 91% yield using readily accessible precursors. The new strategy features the simultaneous use of orthogonal Cu+–phenanthroline and CB[6]–ammonium interactions for preorganising the precursors and the efficient CB[6]-catalysed azide–alkyne cycloaddition as bond forming reactions for ring closing, resulting in high structural complexity and fidelity of the products without compromising interlocking efficiency. A related [4]catenane with three different types of macrocycles was also obtained in good yield.
Introduction
Catenanes have long been attracting research interest because of their unique stereochemistry, structural complexity and mechanical properties that are associated with their non-trivial topology. Despite the rapid development of different assembly strategies towards various interlocked molecular topologies, only the simplest Hopf link (two interlocked macrocycles with one crossing) has been recently considered to be straightforwardly accessible.1,2 While some examples of entwined catenanes with more molecular crossings (e.g. Solomon links3 and Star of David [2]catenane4) have been recently reported, [n]catenanes with multiple numbers of interlocked macrocycles still remain a synthetic challenge. The low number of interlocked macrocycles also limits the available type of topoisomers of different ring connectivity (e.g. linear, branched, radial, circular… etc., Scheme 1) of the [n]catenanes, with the radial [n]catenanes that have n − 1 rings interlocked on one large central macrocycle being the most common. The largest discrete [n]catenane that has been isolated and characterised to date is a [7]catenane, with only a handful of [n]catenanes (n ≥ 5) having been reported.1,5–14 The synthesis of these [n]catenanes usually requires special reaction conditions and/or specific precursors and templates, and the yields are often modest. For example, the first [7]catenane reported by Stoddart and co-workers was assembled using π donor–acceptor templation under an ultrahigh pressure of 12 kbar in 27% yield, along with a series of related [4]-, [5]- and [6]catenanes.5 More recently, Nitschke and Sanders have reported an equilibrating system of tetrahedral metallocages with the six π-deficient edges interlocked with different numbers of complementary π-rich macrocycles, in which the formation of the [7]catenaned cage is favoured by shifting the equilibrium with a large excess of the π-rich macrocycle.6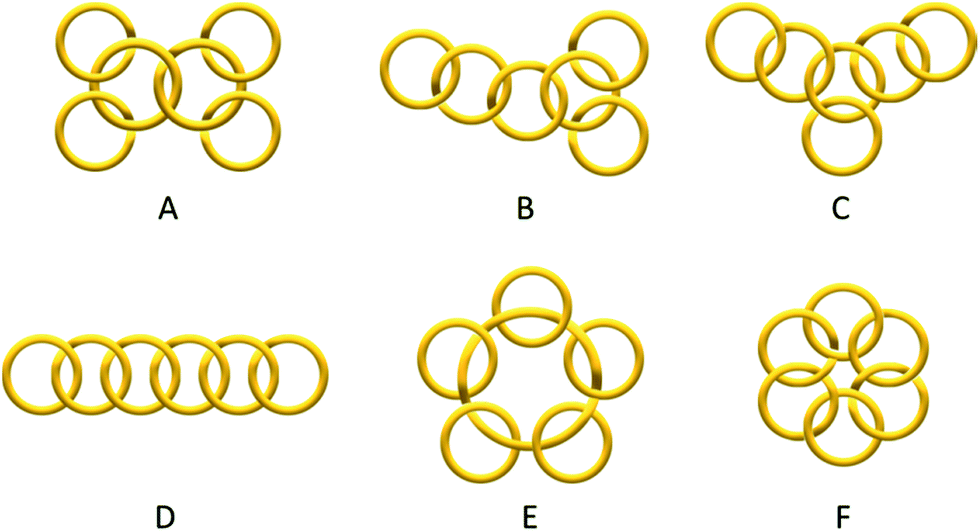 | ||
| Scheme 1 Some topoisomers of a [6]catenane with different ring connectivity: branched (A–C), linear (D), radial (E) and circular (F) [6]catenanes. In this work, only isomer A was obtained. | ||
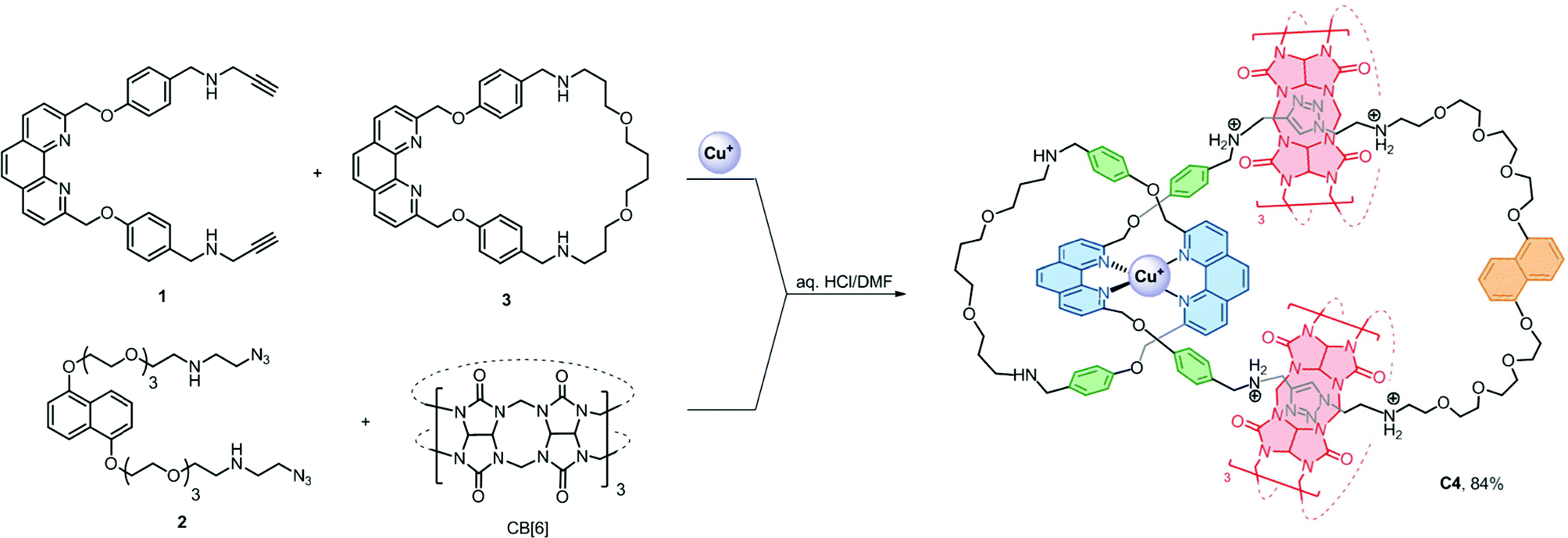
Efficient strategies that are facile, general, controllable and applicable to [n]catenanes that contain more interlocked macrocycles, different interlocking topology and ring connectivity are yet to be developed and will be necessary if the distinct properties of catenanes are to be developed into new molecular machines or incorporated into functional materials.1a,b We anticipated that using more than one type of orthogonal supramolecular interaction as a template,15 in conjunction with highly efficient bond forming reactions, could independently and simultaneously interlock multiple macrocycles without compromising the interlocking efficiency and simplicity in the precursor design. More importantly, products with fewer numbers of interlocking rings and other topological isomers can be minimised and therefore could give the desired [n]catenanes in good yields. Here we describe a new strategy that combines CuI–phenanthroline coordination16 and the ion–dipole and hydrophobic interactions between ammonium and cucurbit[6]uril (CB[6])17 with the CB[6]-catalysed azide–alkyne cycloaddition18 as a ring-closing reaction to obtain a rare [6]catenane and a related [4]catenane with three different types of macrocycles in 91% and 84% yield, respectively.
Results and discussion
The [6]catenane C6, consists of two interlocking [3]catenanes, was synthesised through dropwise addition of 45 ml of a 1 mM solution of the diazide–CB[6] complex [2 ⊂ CB[6]2] in 0.2 M aq. HCl to an equal volume of a 0.5 mM solution of the alkyne-functionalised CuI–phenanthroline complex [Cu(1)2][PF6] in DMF at 60 °C over two hours, followed by stirring of the reaction mixture at the same temperature for two days (Scheme 2). LCMS analysis of the crude product mixture in solution revealed the formation of C6 in 91% yield along with 7% of the [3]catenane C3 as determined using the corresponding peak areas in the chromatogram.19 The latter can also be obtained in 85% yield from a similar reaction of 1 and 2 in the absence of Cu+ (see the ESI† for details). Both C6 and C3 (and C4, vide infra) are water soluble in the form of chlorides, and their water solubility offers good opportunities for studying the unique mechanical motions of catenanes conferred by the aqueous environment which have not yet been extensively studied.20 Also, the branched structure of C6 represents a rare form of ring connectivity for high order [n]catenanes when compared to the more common radial [n]catenanes.7–13,21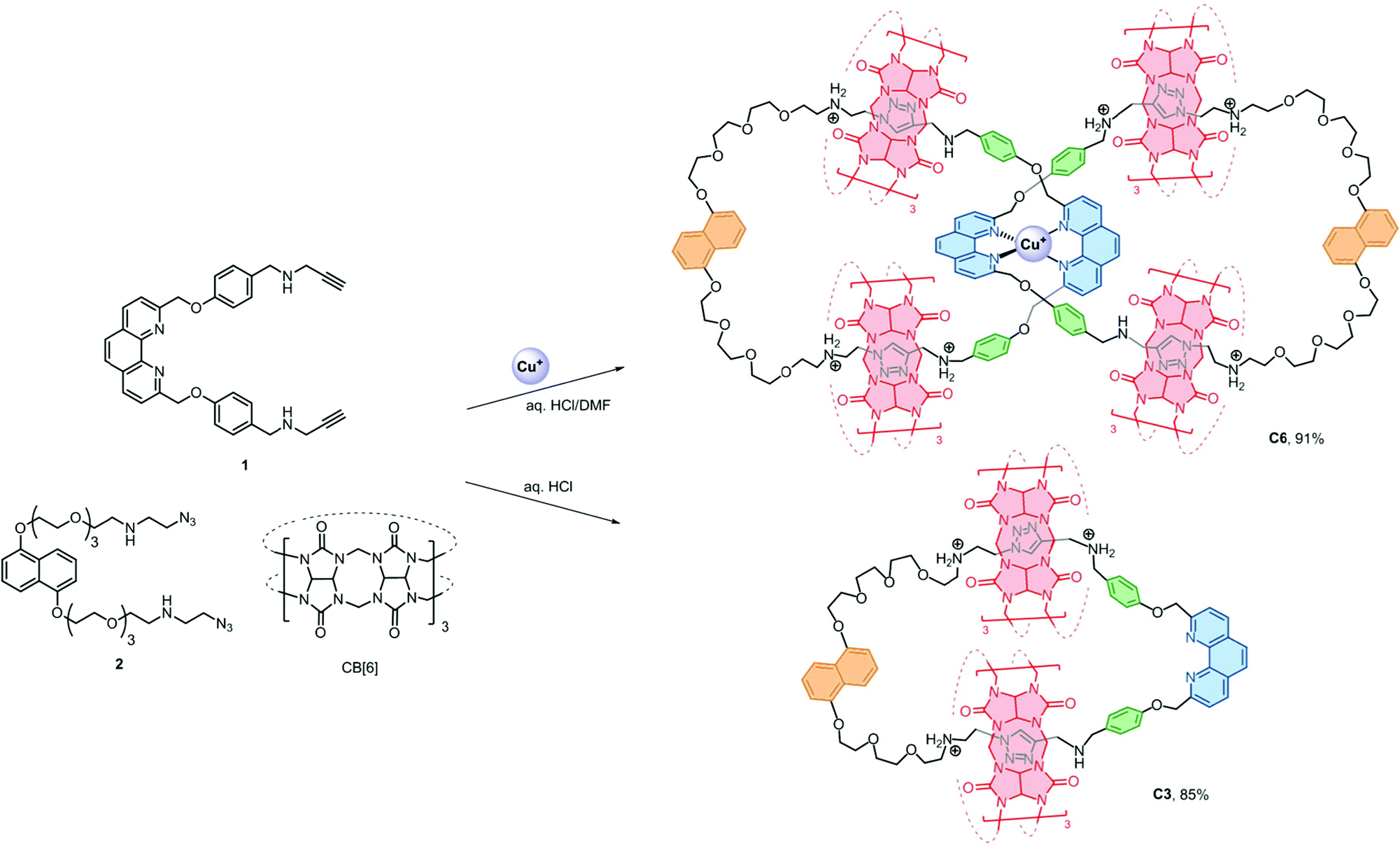 | ||
| Scheme 2 Syntheses of C3 and C6. Structures of C6 and C3 are shown as the +7 and +3 ions which are the most abundant and stable forms of the catenanes as observed in the ESI-MS studies. Other protonation states of the catenanes are also observed in the MS studies. | ||
ESI-MS analysis of C6 shows a series of peak clusters that are consistent with the molecular formulae of C6 in charge states of +5 to +8 (Fig. 1a). In addition, it is found that the peak at m/z = 915.5, which corresponds to the +7 ion, has the strongest intensity, suggesting that the most abundant and stable form of the Cu+-coordinated [6]catenane under the ESI conditions is the one with six of the eight secondary amines being protonated. Nevertheless, under the acidic conditions used in the synthesis of C6, it is likely that all of the secondary amines are protonated and the ion–dipole interactions between the ammonium and CB[6] are maximised for the CB[6]-catalysed click reactions. HRMS analysis of the peak at m/z = 915.5 (the +7 ion) showed an isotopic pattern that is consistent with the expected molecular formula of the catenane (Fig. 1b). The interlocked structure of C6 was confirmed through MS2 and MS3 experiments. Fragmentation of the peak at m/z = 801.3 resulted in fragments corresponding to C3 (m/z = 793.1) and its smaller fragments. Further fragmentation of the peak at m/z = 793.1 produced a MS3 spectrum that is consistent with the MS2 spectrum of C3, supporting that C6 is composed of two interlocked C3 (Fig. 1c and d). It is noted from both the MS2 and MS3 spectra of C6 (and MS2 of C3, Fig. S39†) that the binding of CB[6] to the ammonium is so strong that the pseudorotaxane fragments are stable enough to be observed under the MSn conditions.22
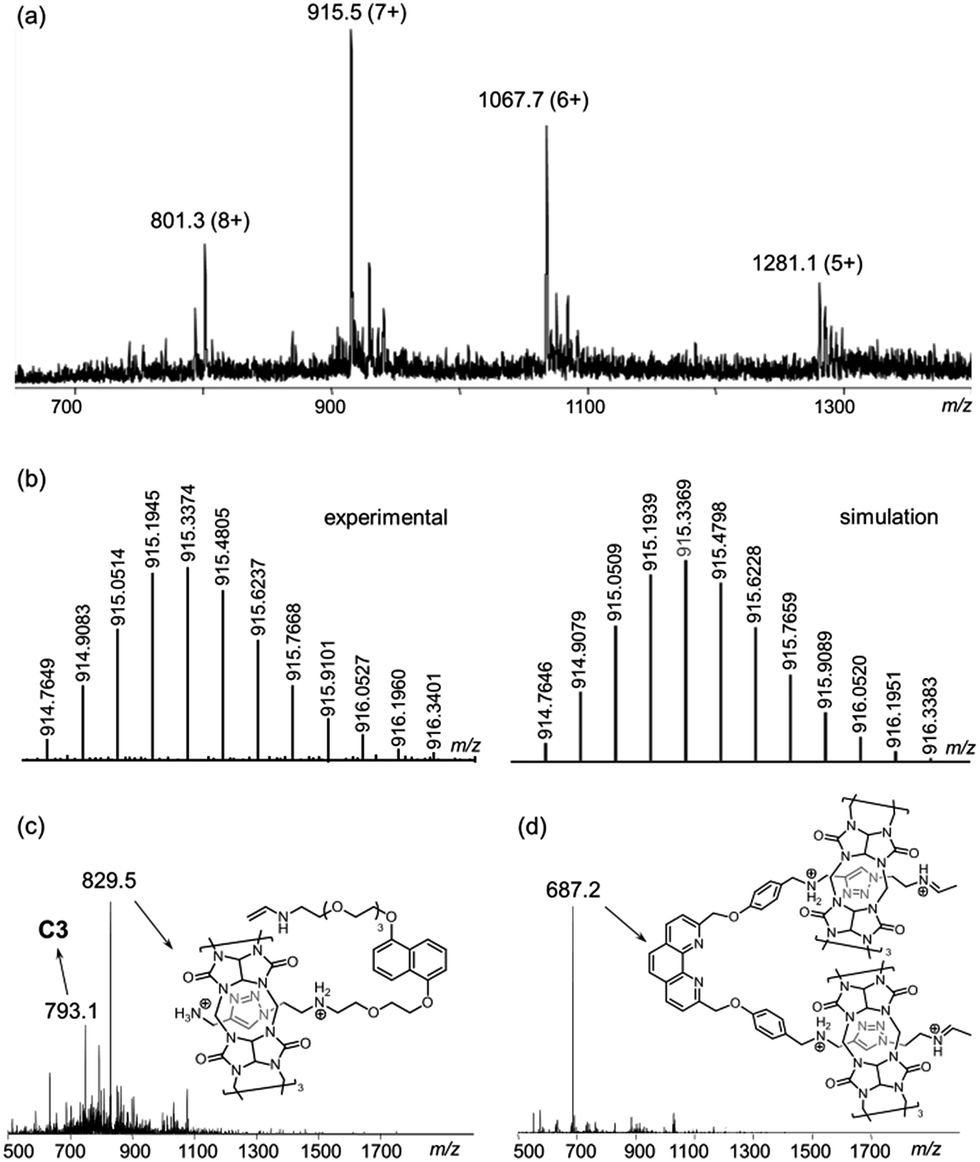 | ||
| Fig. 1 (a) ESI-MS spectrum of C6; (b) HRMS of the peak at m/z = 915.5 (left: experimental; right: simulation); (c) the MS2 and (d) MS3 spectra of C6. | ||
A sample of C6 purified through preparative HPLC was further characterised using NMR spectroscopy (1H, 13C{1H}, COSY, NOESY and DOSY). The 1H spectrum (400 MHz, D2O and 298 K) of C6 shows one set of resonances, indicating a highly symmetrical structure of C6 in aqueous solution. While the 1H resonances from the CB[6] in C6 may be obscured by the resonances of other aliphatic protons in the molecule and the four inequivalent chemical environments of the CB[6] methylene protons can only be vaguely observed in the 1H spectrum, the 13C{1H} spectrum of C6 clearly shows the two different chemical environments of the carbonyl (at 156.3 and 156.5 ppm) and methylene (at 51.4 and 51.7 ppm) carbons, which are the result of the interlocking of CB[6] on the unsymmetrical triazole (Fig. S34†). In addition, all the 13C resonances of the CB[6] carbons (156.5, 156.3, 70.4, 51.7 and 51.4 ppm) in C6 are upfield shifted by ca. 4 ppm when compared to that of the guest-free CB[6] (160.0, 74.2 and 55.3 ppm),23 further confirming the interlocking of the macrocycle on the [6]catenane. Comparing the 1H spectra of C6 and C3, the phenylene protons of C6 are upfield shifted by 0.59 and 1.14 ppm, while the phenanthroline protons are downfield shifted by 0.26, 0.24 and 0.29 ppm, suggesting close proximity of the phenylene and phenanthroline units with an edge to face orientation (Fig. 2). These chemical shift changes are comparable to those observed between the phenanthroline ligand 1 and the CuI complex [Cu(1)2][(PF6)] (Fig. S37†), showing the presence of the same CuI–phenanthroline coordination motif in C6. On the other hand, the triazole protons of C6 and C3 at 6.31 and 6.23 ppm, respectively, are significantly upfield shifted when compared to that of the reference compound which lacks the CB[6] binding at 7.72 ppm (Fig. S20†), consistent with the inclusion of the triazole in the cavity of CB[6] in both C6 and C3.18 The close proximity between the phenylene and phenanthroline units in C6, and that between the triazole and CB[6] is also supported by the corresponding NOE cross peaks in the 2D NOESY spectrum (Fig. S36†). Careful analysis of the 1H spectrum of C6 revealed the presence of a minor species. Further LCMS analysis of the isolated C6 sample from preparative HPLC showed a minor portion of the Cu+ in C6 was decomplexed from the [6]catenane, suggesting that the set of minor resonances is due to the copper-free C6 (Fig. S5a†). Diffusion ordered spectroscopy (DOSY) experiments showed that both sets of resonances have the same diffusion coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D = −9.93), further supporting the assignment of the minor component to the metal free form of the [6]catenane, as both the Cu+-complexed and Cu+-free forms of C6 are expected to have similar sizes and hydrodynamic volumes (Fig. 2c). Attempts to obtain a pure sample of the copper-free C6 through treating the Cu+-containing C6 with common demetallation reagents (e.g. CN−) were unsuccessful. The required alkaline medium is incompatible with the solubility of C6 so that the latter precipitated from the aqueous solution and the demetallation reaction could not be performed.24
D = −9.93), further supporting the assignment of the minor component to the metal free form of the [6]catenane, as both the Cu+-complexed and Cu+-free forms of C6 are expected to have similar sizes and hydrodynamic volumes (Fig. 2c). Attempts to obtain a pure sample of the copper-free C6 through treating the Cu+-containing C6 with common demetallation reagents (e.g. CN−) were unsuccessful. The required alkaline medium is incompatible with the solubility of C6 so that the latter precipitated from the aqueous solution and the demetallation reaction could not be performed.24
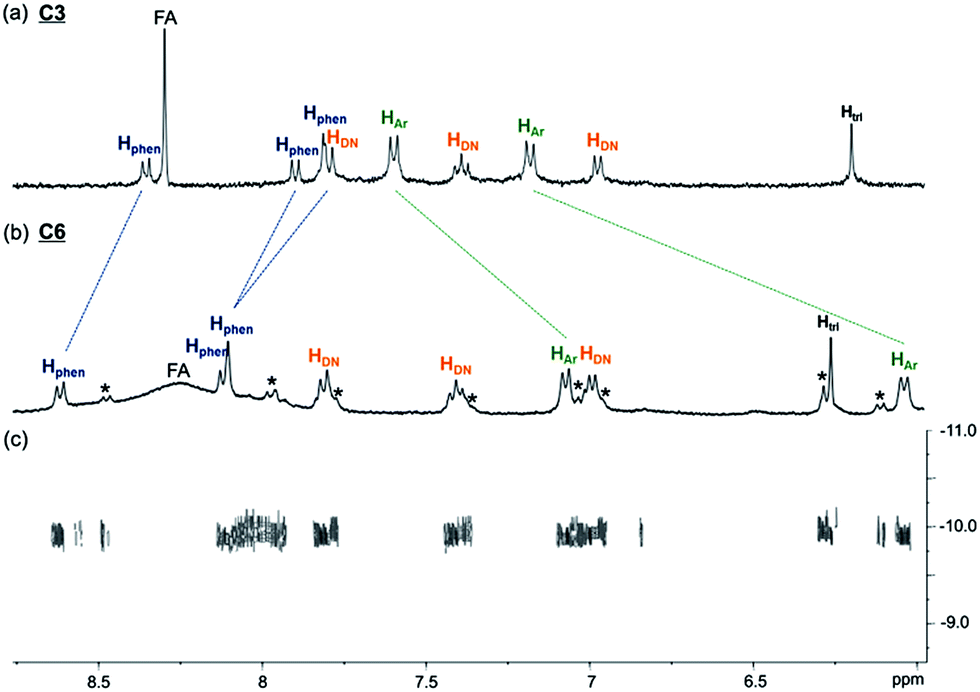 | ||
| Fig. 2 Partial 1H NMR (400 MHz, D2O and 298 K) of (a) C3 and (b) C6. 1H resonances from the phenanthroline, naphthalene, phenylene and triazole units are labelled with Hphen, HDN, HAr and Htri respectively. Resonances from the copper-free C6 are labelled with *. Signals at ca. 8.3 ppm are assigned as the residual formate (FA) from preparative HPLC (see the ESI† for details); (c) partial 2D DOSY (500 MHz, D2O and 298 K) of C6. | ||
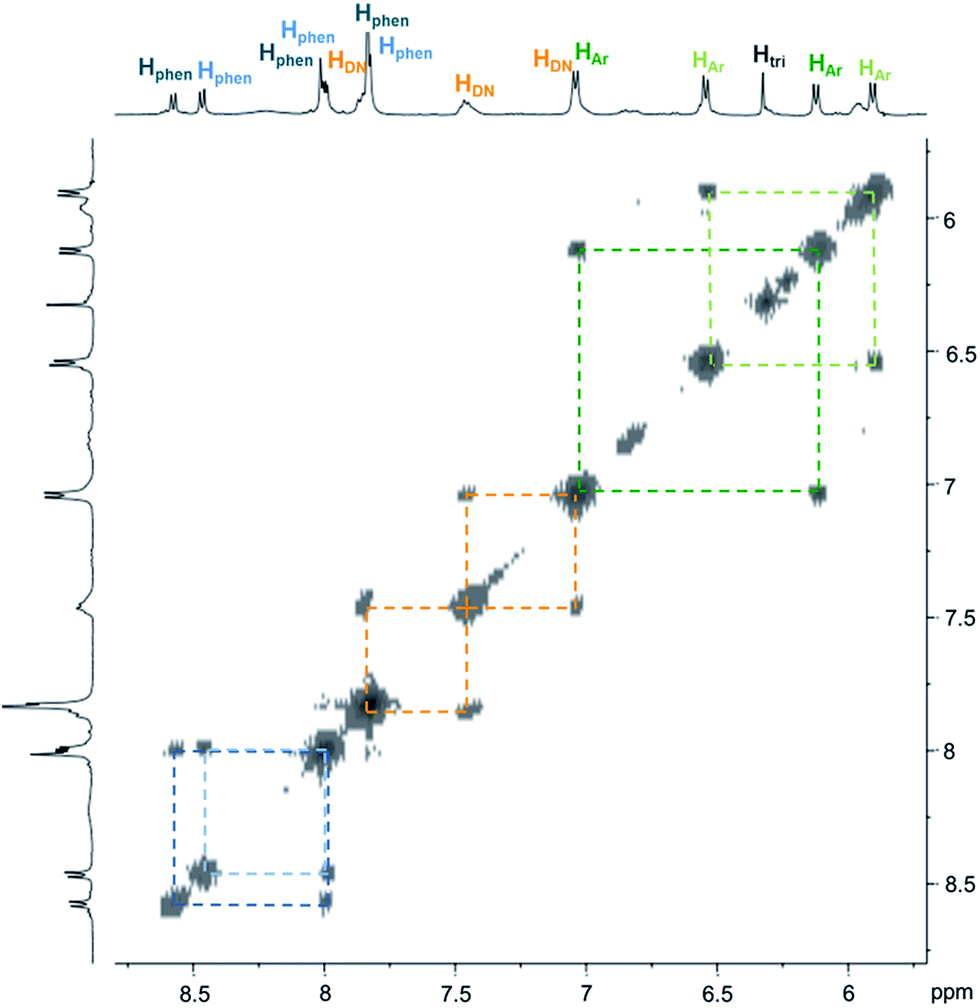 | ||
| Fig. 3 Partial COSY (500 MHz, D2O and 298 K) of C4. Correlations within the two inequivalent phenanthrolines and phenylenes are highlighted by dash lines. | ||
The formation of topological isomers, including those with different numbers of interlocking macrocycles or different interlocking patterns, connectivities or topologies, other than the target [n]catenane that leads to low yield and difficult purification processes is one main reason for the low efficiency of [n]catenane assembly. Notably, the use of CB[6]-catalysed azide–alkyne cycloaddition to ring-close the precursors in our strategy not only resulted in a good yield of C6 due to its high efficiency in bond formation and ring-closure, but the prerequisite CB[6] binding for ring-closure also simultaneously ensures that the macrocycle will be interlocked and therefore minimises the formation of other catenanes with less interlocked macrocycles. While the CB[6]-catalysed reaction has been employed to obtain rotaxanes, its application for cyclising and synthesising catenanes has not yet been demonstrated as far as we are concerned.18 In addition, the use of different orthogonal interactions as templates also directs the macrocycles to their designated interlocking site with good fidelity. The formation of [6]catenanes other than C6 with different topologies or connectivity patterns is minimised. Only the branched structure with one cross-point for each interlocking pair of the macrocycles was identified. Moreover, the flexibility of the diazide 2, in conjunction with the dilute conditions used for the macrocyclisation reaction, also favours only the [1 + 1] cyclisation that leads to C6 but not other higher order [n]catenanes with larger macrocycles derived from other cyclic oligomers (Fig. S3†). Taken together, all of these effects help to suppress the formation of undesired topological isomers other than C6 and highlight the effectiveness of our strategy in its synthesis.
The presence of the π rich dioxynaphthalene units in C6 (and C3) offers an opportunity to further interlock a π deficient macrocycle to give a higher order [n]catenane. Introduction of the π-deficient cyclobisparaquat(p-phenylene) (CBPQT4+) macrocycle to the CB[6]-catalysed click reaction between 1 and 2, however, did not result in any higher order [n]catenane, only C3 and the free CBPQT4+ macrocycle (Fig. S4†). The failure to incorporate CBPQT4+ into any cyclised product is probably due to the repulsive Columbic interaction between CBPQT4+ and 2 under the acidic reaction conditions which prohibits the formation of the charge transfer complex (see the ESI† for details). On the other hand, the compatibility of the CuI–phenanthroline coordination and CB[6]–ammonium binding as demonstrated by the successful synthesis of C6 prompted us to study and diversify the use of other phenanthroline building blocks to synthesise other interlocked structures. Following a similar procedure, a CB[6]-catalysed click reaction between 2 and the heteroleptic complex [Cu(1)(3)][(PF6)] afforded the [4]catenane C4 in 84% yield as demonstrated through LCMS analysis of the crude product mixture (Scheme 3). Data from MS, MSn and NMR studies (Fig. 3, S28–32 and S40†) on C4 are all consistent with the expected [4]catenane structure with three different types of macrocycles interlocked in a radial fashion. The two inequivalent chemical environments for the phenanthroline and phenylene units due to the unsymmetrical coordination environment can be clearly observed and identified from the 2D COSY spectrum (Fig. 3). [n]Catenanes with three or more types of macrocycle are not common.5 Correctly positioning the different macrocycles through using orthogonal templates is one of the keys to the successful synthesis of the [4]catenane, otherwise other topological and/or positional isomers will result. Incorporation of different types of macrocycle into a single [n]catenane will facilitate further functionalisation with good selectivity and/or integration of the interlocked compound into different materials. The successful syntheses of C4 and C6 demonstrate that the present strategy is effective and modular. By proper design of the precursor building blocks and control of the reaction conditions, two different high order [n]catenanes (C4 and C6) can be facilely and selectively obtained. The strategy is controllable and could be easily extended to other interlocked structures with different numbers of interlocking macrocycles and connectivity.
 | ||
| Scheme 3 Synthesis of C4. Its structure is shown as the +5 ion which is the most abundant and stable form as observed in the ESI-MS study. | ||
Conclusions
In summary, a new strategy that employs orthogonal supramolecular interactions (metal–ligand coordination, ion–dipole and hydrophobic interactions) and an efficient bond forming reaction (CB[6]-catalysed click) to preorganise and construct multi-macrocyclic [n]catenanes has been successfully demonstrated through the efficient syntheses of the [3]-, [4]- and [6]catenane products, C3, C4 and C6. Their syntheses are highly facile and the yields are good (>80%). This modular approach to [n]catenane will serve not only as a starting point to a general synthetic method that can be extended to higher order [n]catenanes with more interlocked macrocycles and different topological patterns of the interlocking macrocycles, but the new interlocked molecules could also be novel candidates for the development of new functional molecular materials that are based on the unique mechanical properties and structural complexities of the [n]catenanes. Further studies to extend our strategy to other high order [n]catenanes with more interlocked rings and/or different interlocking topologies and connectivity patterns in a predictable and controllable way, and subsequent studies on their molecular motions are currently under way.Acknowledgements
The work described in this paper was supported by a grant from the Research Grants Council of the Hong Kong Special Administration Region, China (Early Career Scheme, Project No. HKU 27300014) and the Croucher Foundation. KW and CCY acknowledge the receipt of the Postgraduate Scholarship from The University of Hong Kong. We also thank Dr Eva Y. M. Fung and Prof. C. M. Che for their help in the HRMS experiments, and Ms Bonnie Yan for her technical assistance in the NMR experiments.Notes and references
- (a) G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef PubMed; (b) N. H. Evans and P. D. Beer, Chem. Soc. Rev., 2014, 43, 4658–4683 RSC; (c) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; (d) L. Fang, M. A. Olson, D. Benítez, E. Tkatchouk, W. A. Goddard and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17–29 RSC; (e) K. D. Hännia and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC; (f) Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS PubMed.
- (a) Y. Ye, S.-P. Wang, B. Zhu, T. R. Cook, J. Wu, S. Li and P. J. Stang, Org. Lett., 2015, 17, 2804–2807 CrossRef CAS PubMed; (b) S. Li, J. Huang, T. R. Cook, J. B. Pollock, H. Kim, K.-W. Chi and P. J. Stang, J. Am. Chem. Soc., 2013, 135, 2084–2087 CrossRef CAS PubMed; (c) S. Li, M. Liu, B. Zheng, K. Zhu, F. Wang, N. Li, X.-L. Zhao and F. Huang, Org. Lett., 2009, 11, 3350–3353 CrossRef CAS PubMed.
- (a) J. E. Beves, J. J. Danon, D. A. Leigh, J.-F. Lemonnier and I. J. Vitorica-Yrezabal, Angew. Chem., Int. Ed., 2015, 54, 7555–7559 CrossRef CAS PubMed; (b) C. Schouwey, J. J. Holstein, R. Scopelliti, K. O. Zhurov, K. O. Nagornov, Y. O. Tsybin, O. S. Smart, G. Bricogne and K. Severin, Angew. Chem., Int. Ed., 2014, 53, 11261–11265 CrossRef CAS PubMed; (c) J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Angew. Chem., Int. Ed., 2014, 53, 7823–7827 CrossRef CAS PubMed; (d) N. Ponnuswamy, F. B. L. Cougnon, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2014, 136, 8243–8251 CrossRef CAS PubMed.
- D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978–982 CrossRef CAS PubMed.
- D. B. Amabilino, P. R. Ashton, V. Balzani, S. E. Boyd, A. Credi, J. Y. Lee, S. Menzer, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 4295–4307 CrossRef CAS.
- S. P. Black, A. R. Stefankiewicz, M. M. J. Smulders, D. Sattler, C. A. Schalley, J. R. Nitschke and J. K. M. Sanders, Angew. Chem., Int. Ed., 2013, 52, 5749–5752 CrossRef CAS PubMed.
- M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897–1901 RSC.
- S. Li, J. Huang, F. Zhou, T. R. Cook, X. Yan, Y. Ye, B. Zhu, B. Zheng and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 5908–5911 CrossRef CAS PubMed.
- C.-F. Chang, C.-J. Chuang, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S. H. Chiu, Angew. Chem., Int. Ed., 2012, 51, 10094–10098 CrossRef CAS PubMed.
- S. Dasgupta and J. Wu, Org. Biomol. Chem., 2011, 9, 3504–3515 CAS.
- K.-M. Park, S.-Y. Kim, J. Heo, D. Whang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2002, 124, 2140–2147 CrossRef CAS PubMed.
- S.-G. Roh, K.-M. Park, G.-J. Park, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 1999, 38, 637–641 CrossRef.
- F. Bitsch, C. O. Dietrich-Buchecker, A. K. Khémiss, J.-P. Sauvage and A. Vandorsselaer, J. Am. Chem. Soc., 1991, 113, 4023–4025 CrossRef CAS.
- H. Iwamoto, S. Tafuku, Y. Sato, W. Takizawa, W. Katagiri, E. Tayama, E. Hasegawa, Y. Fukazawab and T. Haino, Chem. Commun., 2016, 52, 319–322 RSC.
- (a) P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC; (b) X.-Y. Hu, T. Xiao, C. Lin, F. Huang and L. Wang, Acc. Chem. Res., 2014, 47, 2041–2051 CrossRef CAS PubMed; (c) M. L. Saha, S. De, S. Pramanik and M. Schmittel, Chem. Soc. Rev., 2013, 42, 6860–6909 RSC; (d) C.-H. Wong and S. C. Zimmerman, Chem. Commun., 2013, 49, 1679–1695 RSC.
- (a) J.-P. Sauvage, Acc. Chem. Res., 1990, 23, 319–327 CrossRef CAS; (b) C. O. Dietrich-Buchecker and J.-P. Sauvage, Chem. Rev., 1987, 87, 795–810 CrossRef CAS.
- (a) W. L. Mock and N.-Y. Shih, J. Org. Chem., 1983, 48, 3619–3620 CrossRef CAS; (b) W. L. Mock, T. A. Irra, J. P. Wepsiec and M. Adhya, J. Org. Chem., 1989, 54, 5302–5308 CrossRef CAS.
- (a) M. K. Sinha, O. Reany, M. Yefet, M. Botoshansk and E. Keinan, Chem.–Eur. J., 2012, 18, 5589–5605 CrossRef CAS PubMed; (b) G. Celtek, M. Artar, O. A. Scherman and D. Tuncel, Chem.–Eur. J., 2009, 15, 10360–10363 CrossRef CAS PubMed; (c) D. Tuncel and M. Katterle, Chem.–Eur. J., 2008, 14, 4110–4116 CrossRef CAS PubMed; (d) D. Tuncel, Ö. Özsar, H. B. Tiftika and B. Salih, Chem. Commun., 2007, 1369–1371 RSC; (e) D. Tuncel and J. H. G. Steinke, Macromolecules, 2004, 37, 288–302 CrossRef CAS; (f) K. Kim, Chem. Soc. Rev., 2002, 31, 96–107 RSC; (g) D. Tuncel and J. H. G. Steinke, Chem. Commun., 1999, 1509–1510 RSC.
- Yields of C3, C4 and C6 syntheses were calculated based on the relative peak areas in the HPLC chromatograms. Relative absorbances of different chromophores were determined using independent UV-Vis measurements. The [n]catenanes were also isolated from preparative HPLC and the isolated yields of C3, C4 and C6 were determined to be 66%, 54%, and 69% respectively. See ESI† for details.
- (a) F. B. L. Cougnon, N. Ponnuswamy, G. D. Pantoş and J. K. M. Sanders, Org. Biomol. Chem., 2015, 13, 2927–2930 RSC; (b) M. J. Langton and P. D. Beer, Chem. Commun., 2014, 50, 8124–8127 RSC; (c) S. Grunder, P. L. McGrier, A. C. Whalley, M. M. Boyle, C. Stern and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 17691–17694 CrossRef CAS PubMed; (d) R. S. Forgan, J. J. Gassensmith, D. B. Cordes, M. M. Boyle, K. J. Hartlieb, D. C. Friedman, A. M. Z. Slawin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 17007–17010 CrossRef CAS PubMed.
- D. Whang, K.-M. Park, J. Heo and K. Kim, J. Am. Chem. Soc., 1998, 120, 4899–4900 CrossRef CAS.
- (a) S. W. Heo, T. S. Choi, K. M. Park, Y. H. Ko, S. B. Kim, K. Kim and H. I. Kim, Anal. Chem., 2011, 83, 7916–7923 CrossRef CAS PubMed; (b) D. V. Dearden, T. A. Ferrell, M. C. Asplund, L. W. Zilch, R. R. Julian and M. F. Jarrold, J. Phys. Chem. A, 2009, 113, 989–996 CrossRef CAS PubMed; (c) H. Zhang, E. S. Paulsen, K. A. Walker, K. E. Krakowia and D. V. Dearden, J. Am. Chem. Soc., 2003, 125, 9284–9285 CrossRef CAS PubMed.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- Preliminary LCMS analysis of a purified aqueous sample of C6 after prolonged standing at 4 °C for 4 months could give the copper free, demetalated [6]catenane in about 50% yield (Fig. S5†). Presumably, the CuI was slowly oxidised under air to CuII, which has a weaker binding to the phenanthroline coordination pocket in the catenane.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR, MS, HPLC and UV-Vis data. See DOI: 10.1039/c5sc04774a |
| This journal is © The Royal Society of Chemistry 2016 |