
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single-chain derivative of the relaxin hormone is a functionally selective agonist of the G protein-coupled receptor, RXFP1†
Mohammed Akhter
Hossain
 *ab,
Martina
Kocan
c,
Song T.
Yao
a,
Simon G.
Royce
d,
Vinojini B.
Nair
ab,
Christopher
Siwek
c,
Nitin A.
Patil
ab,
Ian P.
Harrison
d,
K. Johan
Rosengren
e,
Stavros
Selemidis
d,
Roger J.
Summers
c,
John D.
Wade
*ab,
Ross A. D.
Bathgate
*af and
Chrishan S.
Samuel
*d
*ab,
Martina
Kocan
c,
Song T.
Yao
a,
Simon G.
Royce
d,
Vinojini B.
Nair
ab,
Christopher
Siwek
c,
Nitin A.
Patil
ab,
Ian P.
Harrison
d,
K. Johan
Rosengren
e,
Stavros
Selemidis
d,
Roger J.
Summers
c,
John D.
Wade
*ab,
Ross A. D.
Bathgate
*af and
Chrishan S.
Samuel
*d
aFlorey Institute of Neuroscience and Mental Health, The University of Melbourne, Victoria 3010, Australia. E-mail: akhter.hossain@florey.edu.au; john.wade@florey.edu.au; bathgate@florey.edu.au
bSchool of Chemistry, The University of Melbourne, Victoria 3010, Australia
cMonash Institute of Pharmaceutical Sciences, Monash University, Victoria, Australia
dCardiovascular Disease Program, Biomedicine Discovery Institute and Department of Pharmacology, Monash University, Victoria, Australia. E-mail: chrishan.samuel@monash.edu
eThe University of Queensland, School of Biomedical Sciences, Brisbane, QLD 4072, Australia
fDepartment of Biochemistry, The University of Melbourne, Victoria 3010, Australia
First published on 26th February 2016
Abstract
Human gene-2 relaxin (H2 relaxin) is a pleiotropic hormone with powerful vasodilatory and anti-fibrotic properties which has led to its clinical evaluation and provisional FDA approval as a treatment for acute heart failure. The diverse effects of H2 relaxin are mediated via its cognate G protein coupled-receptor (GPCR), Relaxin Family Peptide Receptor (RXFP1), leading to stimulation of a combination of cell signalling pathways that includes cyclic adenosine monophosphate (cAMP) and extracellular-signal-regulated kinases (ERK)1/2. However, its complex two-chain (A and B), disulfide-rich insulin-like structure is a limitation to its facile preparation, availability and affordability. Furthermore, its strong activation of cAMP signaling is likely responsible for reported detrimental tumor-promoting actions that may preclude long-term use of this drug for treating human disease. Here we report the design and synthesis of a H2 relaxin B-chain-only analogue, B7-33, which was shown to bind to RXFP1 and preferentially activate the pERK pathway over cAMP in cells that endogenously expressed RXFP1. Thus, B7-33 represents the first functionally selective agonist of the complex GPCR, RXFP1. Importantly, this small peptide agonist prevented or reversed organ fibrosis and dysfunction in three pre-clinical rodent models of heart or lung disease with similar potency to H2 relaxin. The molecular mechanism behind the strong anti-fibrotic actions of B7-33 involved its activation of RXFP1-angiotensin II type 2 receptor heterodimers that induced selective downstream signaling of pERK1/2 and the collagen-degrading enzyme, matrix metalloproteinase (MMP)-2. Furthermore, in contrast to H2 relaxin, B7-33 did not promote prostate tumor growth in vivo. Our results represent the first known example of the minimisation of a two-chain cyclic insulin-like peptide to a single-chain linear peptide that retains potent beneficial agonistic effects.
Introduction
Cardiovascular (CVDs) and inflammatory lung diseases (such as asthma and chronic obstructive pulmonary disease (COPD)) are leading causes of death globally. A central feature of these diseases is fibrosis1 that represents an aberrant wound healing response that contributes to organ dysfunction and failure through excessive build-up of associated extracellular matrix (ECM) components. Despite the availability of various treatments for patients with CVD and inflammatory lung disorders, these therapies fail to effectively abrogate fibrosis, highlighting the need for more direct anti-fibrotic strategies.Human gene-2 relaxin (H2 relaxin) is structurally similar to human insulin (Fig. 1) and is recognized to have several unique organ-protective actions.2–9 It has therefore been evaluated as a potential treatment for patients with various diseases.6–12 In late 2012, recombinant H2 relaxin (serelaxin; RLX030) successfully completed a phase III clinical trial for the treatment of acute heart failure (AHF).13,14 In addition to its immediate vasodilatory13 and cardioprotective15 effects that are likely to benefit patients with AHF, H2 relaxin is also well-known to exert anti-fibrotic actions14 that we have shown to occur independently of etiology.16–19 These pleiotropic effects of H2 relaxin are mediated by the activation of several different cellular signaling pathways including cyclic adenosine monophosphate (cAMP) and extracellular-signal-regulated kinases (ERK)1/2 coupled to its cognate G protein-coupled receptor (GPCR), Relaxin Family Peptide Receptor 1 (RXFP1), initially discovered as LGR7 (Fig. S1A†).20,21
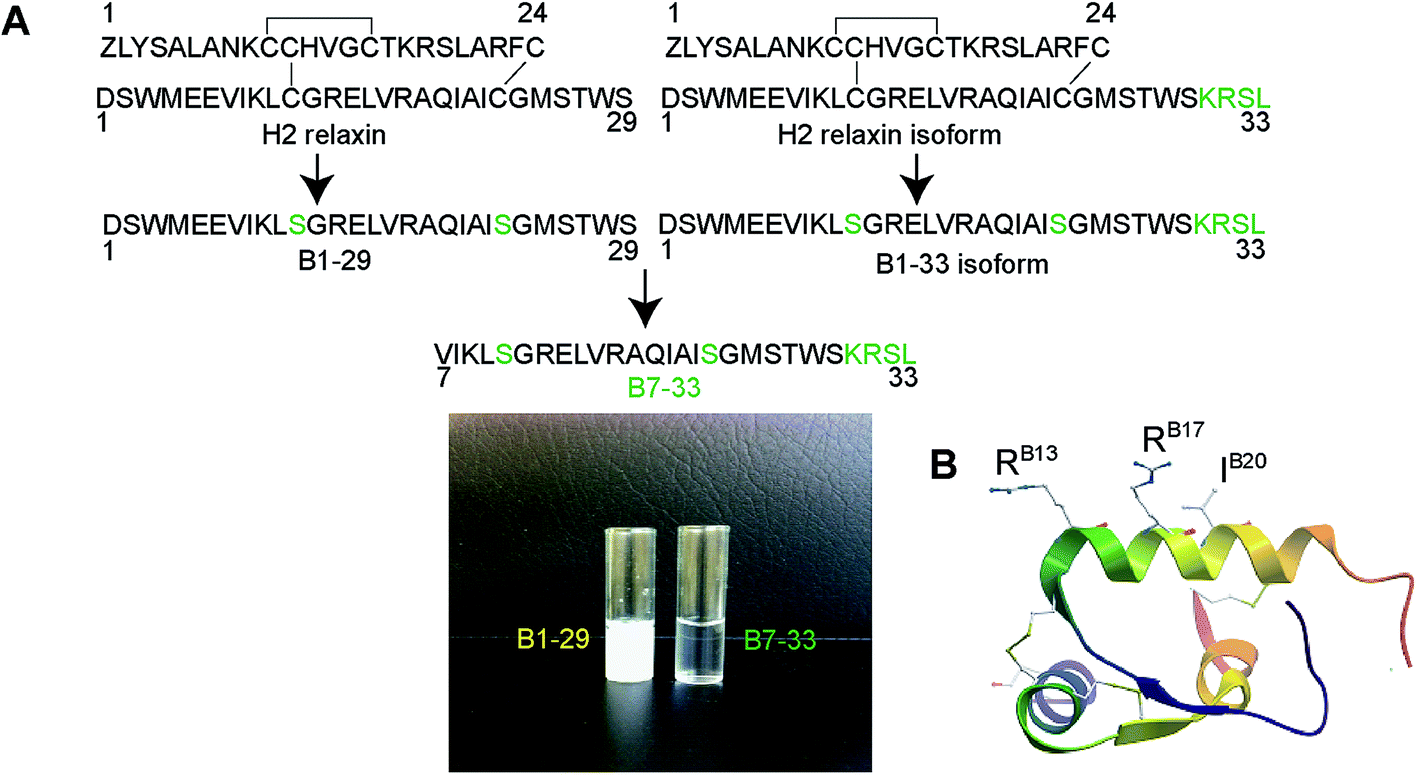 | ||
| Fig. 1 (A) Design of B7-33. Truncation of 6 residues from the N-terminus and elongation of 4 residues (KRSL) at the C-terminus of B1-29 (or truncation of 6 residues from B1-33) resulted in a soluble B-chain derivative of H2 relaxin, B7-33. The native H2 B-chain (B1-29) is insoluble (left vial) whereas the B7-33 is soluble (right vial) at concentration 4 mg mL−1 H2O. (B) X-ray crystal structure of H2 relaxin showing the key RXFP1 binding residues (RB13, RB17 and IB20) in the B-chain. | ||
While a separate phase III trials are in progress, H2 relaxin has been approved for sale in Russia, for human use in clinical settings and is the first new treatment for AHF patients in 20 years.22 However, the current form of the drug (53 amino acids, two chains – A and B connected by 3 disulfide bonds; Fig. 1A and B) is costly and laborious to synthesize, both chemically and recombinantly.23 Additionally, the potent cAMP activation by H2 relaxin has been associated with adverse effects including inotropy and mortality24 and is also involved with the reported ability of H2 relaxin to promote cancer progression particularly in the prostate.25–28 Therefore, the development of a small, easily synthesized, functionally selective RXFP1 agonist that retained the beneficial effects (e.g. anti-fibrotic effects) of H2 relaxin with minimization of its cAMP-activating properties (e.g. cancer promoting effects) would be highly desirable. Such an agonist would be of enormous therapeutic importance as it would be a cost-effective drug with reduced side-effects for the treatment of fibrosis and related disorders. It would also represent an invaluable pharmacological tool to delineate the complex signaling mechanism of RXFP1.
The primary mode of H2 relaxin's interaction with RXFP1 has been extensively studied and well characterized. The receptor-binding cassette (RB13XXX RB17XXIB20) present within the mid-region of the B-chain (Fig. 1B) is responsible for the primary interaction between H2 relaxin and the large extracellular domain (ECD) and, in particular, the leucine-rich repeat (LRR) region of RXFP1 (Fig. S1†).29,30 The corresponding residues in RXFP1 that interact with the B-chain binding motif were later identified.29 Although some reports suggested that there was a secondary interaction involving the A-chain of H2 relaxin and the transmembrane (TM) exoloops of RXFP1,31–34 no single amino acid residue within the A-chain was found to dictate RXFP1 binding and activation.35 This clearly suggested that the B-chain possessed most, if not all, of the residues responsible for high affinity RXFP1 binding, and that rationally designed analogues of the B-chain peptide (Fig. 1A and S2†) could display the characteristic in vitro and in vivo H2 relaxin-like activity.
Thus, we undertook to develop such analogues and herein report for the first time that a chemically synthesized linear H2 relaxin B-chain-only analogue, B7-33 (Fig. 1A), displays potent activity in physiologically-relevant RXFP1-expressing cells and improves heart and airway/lung function by ameliorating fibrosis without exacerbating prostate tumor development. We have also determined how B7-33 interacts with RXFP1 and identified that in fibroblasts, it preferentially signals towards pERK1/2 in agreement with its potent anti-fibrotic effects in vitro and in vivo.
Results
Design of B-chain analogues
It is known that the B-chain of H2 relaxin can be processed in vivo into three equally active isoforms (B1-29, B1-31 and B1-33).36 The native H2 B-chain with 29 residues (B1-29) and its cyclic derivatives are insoluble in water (Fig. 1A) and functionally inactive.37 The overall net charge of B1-29 is zero at neutral pH (four positively charged and four negatively charged amino acids). We were intrigued to find out if a soluble peptide was able to interact with the receptor. In order to create a soluble peptide, we truncated six residues from the N-terminus of B1-29 as we had previously shown that these residues within H2 relaxin were not functionally important.33,38 Then four residues (KRSL) from the B1-33 isoform were added at the C-terminus to increase overall cationic charges. In other words, we truncated six residues from the N-terminus of the B1-33 isoform (Fig. 1A). The resulting analogue with two cysteine residues had an overall positive charge (+5) and fewer hydrophobic residues. Replacement of two cysteines at positions 11 and 23 with isosteric serine residues prevented peptide dimerization and aggregation. This highly positively charged peptide with increased polar residues, B7-33, was freely water-soluble unlike B1-29 (Fig. 1A). A further five B-chain analogues were designed targeting key binding residues RB13/17 and IB20 (Fig. S2A†) to understand the interaction of B7-33 with RXFP1, as H2 relaxin uses these residues to interact with RXFP1.29,30Solid phase synthesis of B-chain analogues
Peptides were solid phase synthesized as C-terminal amides and purified using RP-HPLC via a preparative column while the final purity of individual synthetic peptides was assessed by analytical RP-HPLC. The molecular masses of all analogues were determined by electrospray ionization mass spectroscopy (ESI MS) – B7-33: m/z 2986.4 [M + H]+, calcd 2986.59; AcB7-33: m/z 3028.2 [M + H]+, calcd 3028.6; R13A (AcB7-33): m/z 2943.1 [M + H]+, calcd 2943.5; R17A (AcB7-33): m/z 2943.1 [M + H]+, calcd 2943.5; I20A (AcB7-33): m/z 2986.6 [M + H]+, calcd 2986.6; R13/17A. I20A (AcB7-33): m/z 2816.1 [M + H]+, calcd 2816.3. The peptide content for each analogue was quantified by Direct Detect® spectrometer, an infrared (IR) – based protein quantitation system. Peptides including B7-33 were also bought from a commercial source (Mimotopes Pty, Melbourne, Australia) and the results described below were reproducible. The purity of each of the analogues (≥94%) was determined by using analytical RP-HPLC peak area integration. Importantly, the purity of B7-33 was found to be 97%. The detailed characterization (purity Fig. S3;† traces of analytical RP-HPLC and ESI-MS Fig. S4†) of all the analogues is provided in the ESI.†From a synthetic point of view, development of the B7-33 peptide represents a dramatic improvement over the currently used H2 relaxin. While chemical or recombinant assembly of the H2 relaxin drug involves multiple purification steps,23 B7-33 requires only one. As illustrated in Fig. 2 and S5,† the cost of production of B7-33 is a fraction of that of H2 relaxin.
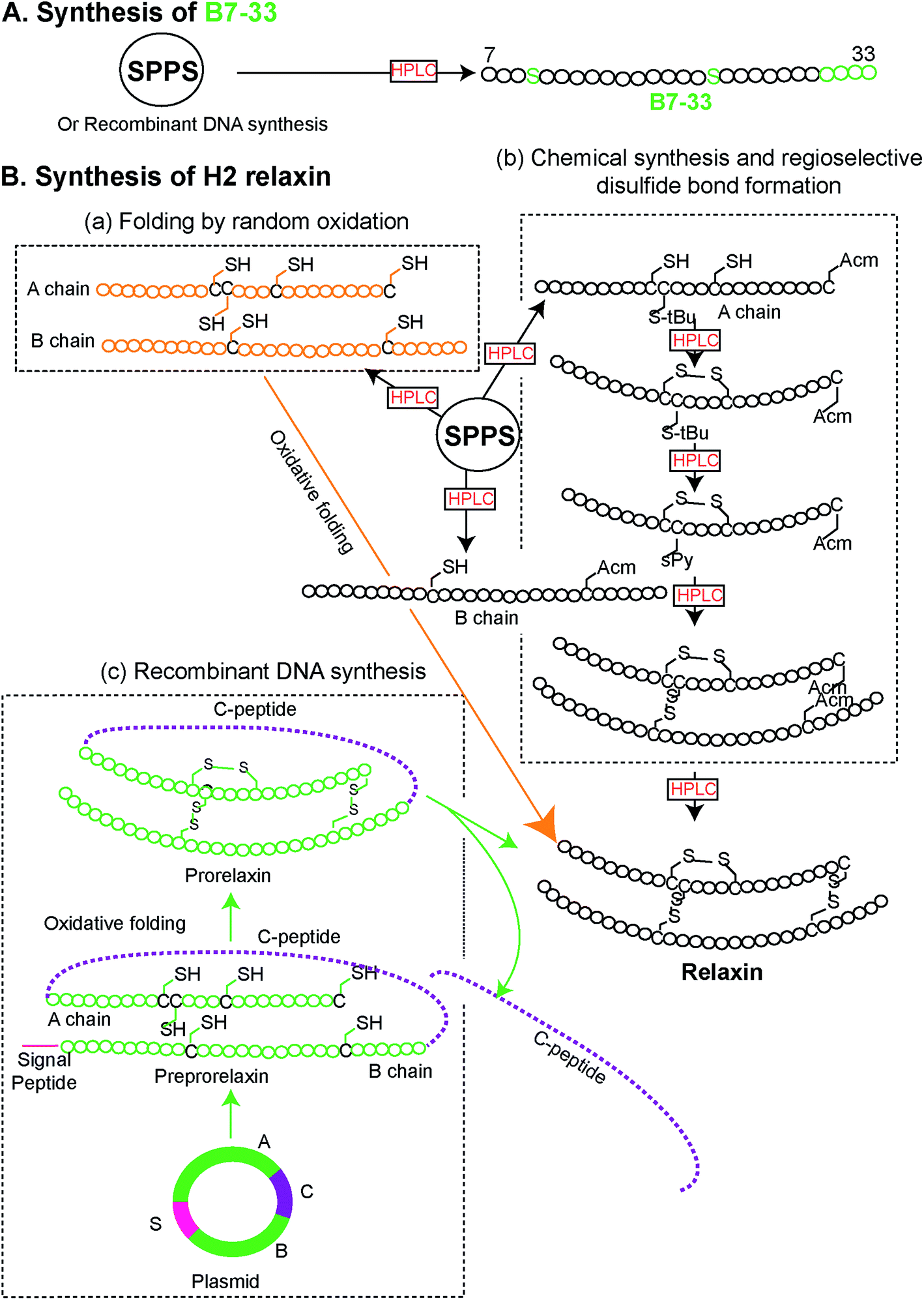 | ||
| Fig. 2 Comparison of synthetic routes (chemical or recombinant) to the (A) B7-33 and (B) H2 relaxin (modified from ref. 23). (A) B7-33 is directly assembled in its final form on resin, requiring a single purification step after cleavage. (B-b) In contrast synthesis of the two chain H2 relaxin requires assembly of two separate chains (A and B chains), four separate reactions for the regioselective formation of its disulfide bonds, and six purification steps, resulting in low overall yields. | ||
Binding affinity and cAMP activity of B7-33 and its analogues
B7-33 was first tested for its ability to bind human RXFP1 receptors stably expressed in HEK-293T cells (HEK-RXFP1, Fig. 3A). Data from competition binding assays showed that B7-33 bound to RXFP1 albeit with significant less affinity (B7-33: pKi = 5.54 ± 0.13, n = 5) compared with H2 relaxin (H2 relaxin: pKi = 8.96 ± 0.03, n = 5) (Fig. 3A and S2B†). This is the first report of binding of a B-chain analogue of H2 relaxin to RXFP1. The B7-33 peptide was then examined in these cells (HEK-RXFP1) for its ability to stimulate cAMP accumulation (Fig. 3B–D). Consistent with the binding data, the potency of B7-33 on human RXFP1 (Fig. 3B) was significantly lower than H2 relaxin (B7-33: pEC50 = 5.12 ± 0.06, n = 3; H2 relaxin: pEC50 = 10.49 ± 0.13, n = 4; Fig. 3B and S2B†). In addition, B7-33 also exhibited weak potency in cAMP activity assays conducted in HEK-293 cells expressing rat (Fig. 3C) or mouse (Fig. 3D) RXFP1 receptors. To assess if B7-33 could stimulate cAMP activity in cells that endogenously express RXFP1, it was tested in the human THP1 monocytic cell line where it also demonstrated weak cAMP potency (Fig. 3E). We also tested B7-33 for its ability to stimulate cAMP in HEK cells stably expressing the related receptor, RXFP2. B7-33 was not found to activate RXFP2 (Fig. S6†) even at micromolar concentrations which were sufficient to activate RXFP1 (Fig. 3).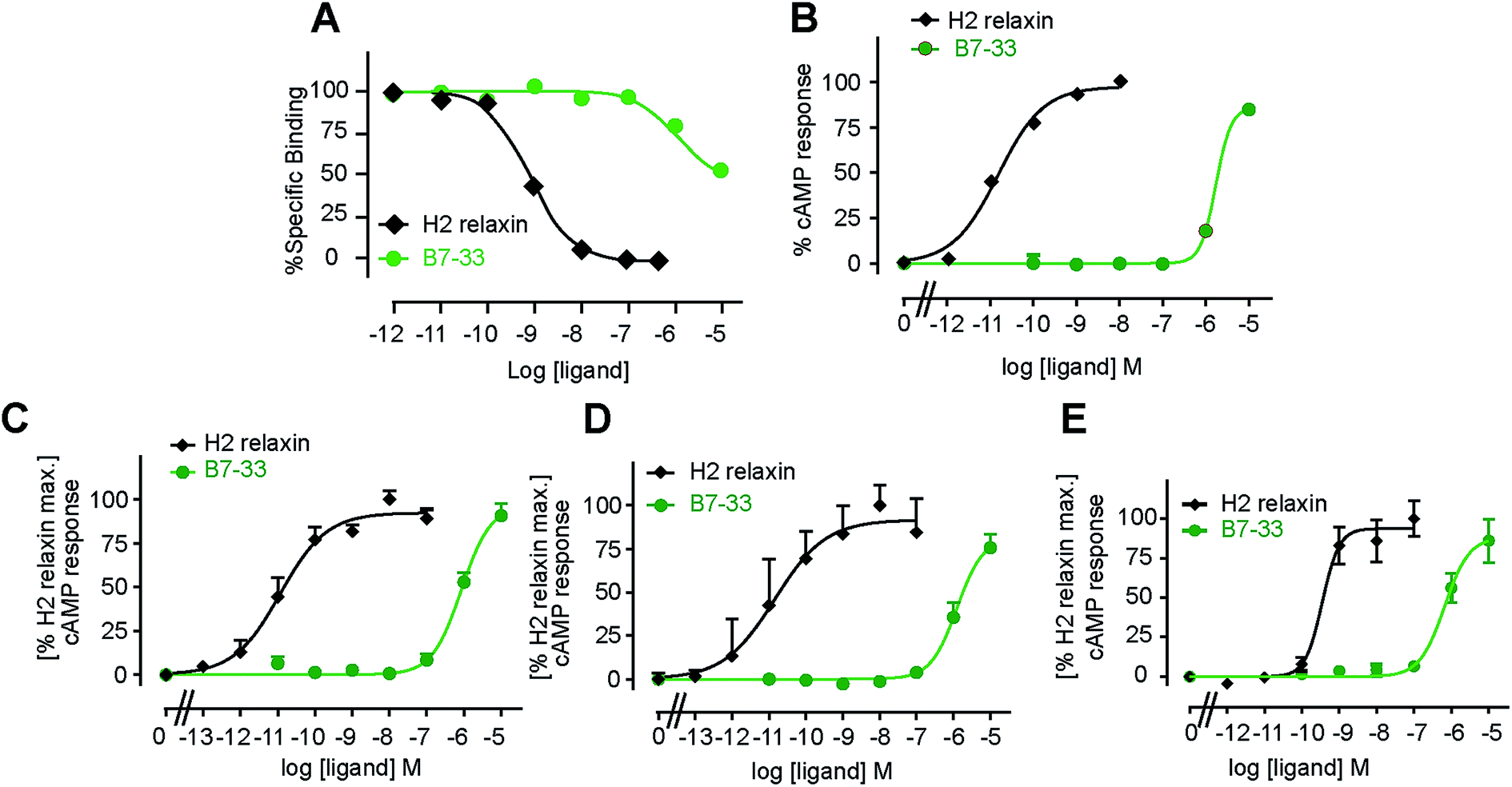 | ||
| Fig. 3 B7-33 weakly bound to and activated cAMP responses in human and rodent RXFP1 cells. (A) Competition binding of B7-33 and H2 relaxin with europium labeled H2 relaxin in human RXFP1 (HEK-293T) cells. B7-33 completed with H2 relaxin for binding to RXFP1 albeit with significantly less affinity compared with H2 relaxin. (B–E) cAMP responses to B7-33 compared with native H2 relaxin in HEK-293T cells that stably express (B) human, (C) rat, and (D) mouse or (E) endogenously express (THP1) RXFP1. Consistent with binding, B7-33 weakly activated cAMP responses in cells over-expressing or naturally expressing RXFP1 across species. The data are expressed as mean ± SEM of n = 3–6 independent experiments. | ||
Acetylated B7-33 (AcB7-33) showed no change in binding affinity and cAMP activity at HEK-RXFP1 (Fig. S2B†). The four acetylated RB13/17 and IB20 mutated analogues, however, completely lost binding and RXFP1 activity in these cells (Fig. S2B†).
MMP-promoting effects of B7-33
While HEK cells stably expressing RXFP1 are an excellent tool for screening drug candidates, the pharmacology of the drugs in natively expressing systems may be different. Thus, B7-33 was examined in human cardiac fibroblasts (Fig. 4A and B) and rat renal myofibroblasts (Fig. 4C–F) endogenously expressing RXFP1 for its ability to promote collagen-degrading matrix metalloproteinase (MMP)-2 levels, a well-known response to H2 relaxin.39,40 Excitingly, the B7-33 peptide significantly promoted MMP-2 levels when administered at 30 nM (≃180 ng mL−1) to human cardiac fibroblasts (Fig. 4A and B) or 16.8 nM (≃100 ng mL−1) to rat renal myofibroblasts (Fig. 4C–F) over 72 hours in culture, to a similar extent as H2 relaxin at the equivalent concentrations, as determined by gelatin zymography (Fig. 4A, C and E) and densitometry (Fig. 4B, D and F) (both P < 0.01 vs. untreated cells). Importantly, these MMP-promoting effects of B7-33 in the rat renal myofibroblasts were completely abrogated by an RXFP1 antagonist26,41 which incorporated lysine substitutions for the two arginine residues situated at positions 13 and 17 within the relaxin B-chain (Fig. 4C and D), indicating that the effects of B7-33 were mediated through RXFP1.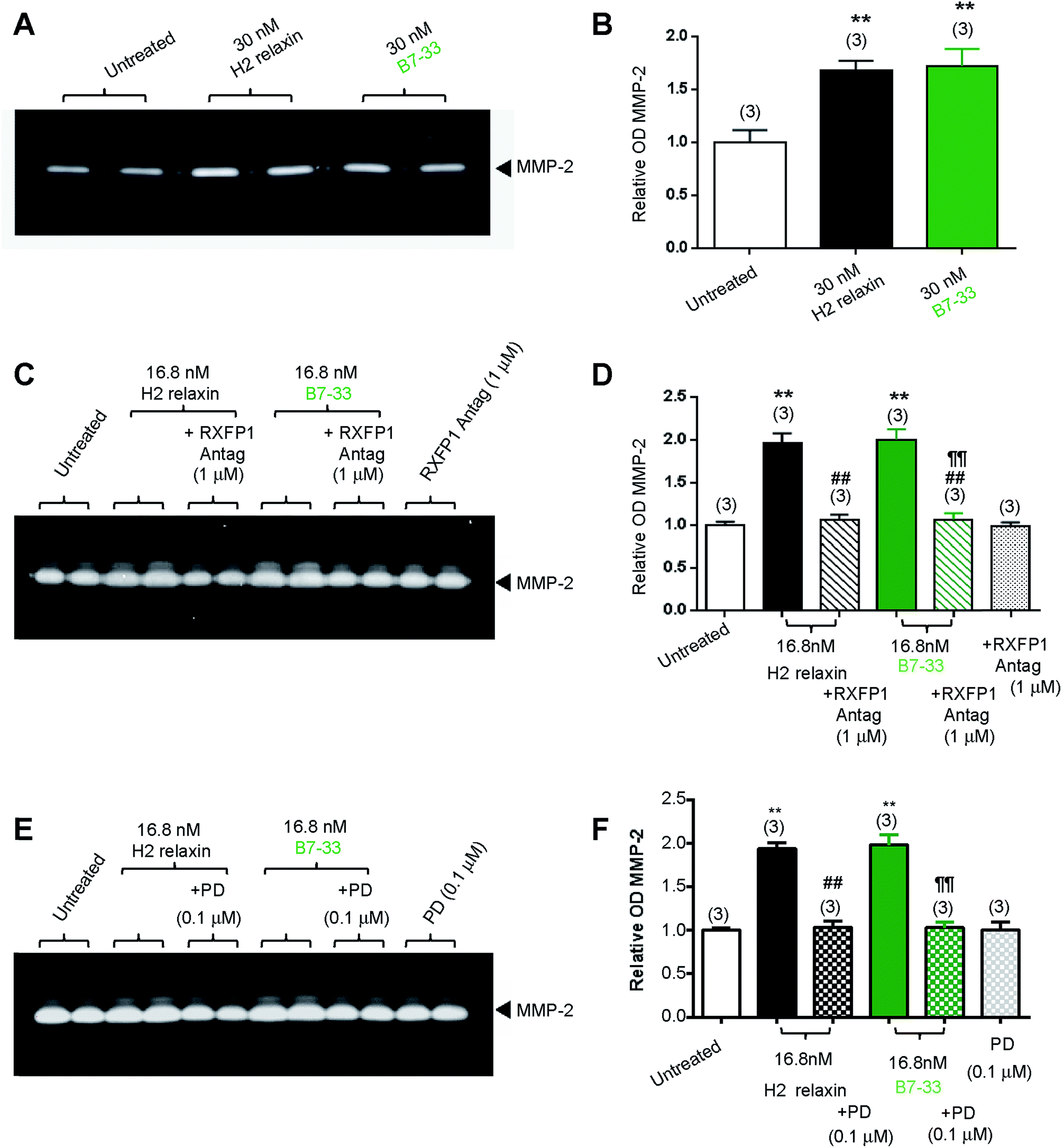 | ||
| Fig. 4 B7-33 promoted matrix metalloproteinase-2 (MMP-2 a collagen-degrading enzyme) with high potency in (myo)fibroblasts endogenously expressing RXFP1. (A) B7-33 stimulated MMP-2 to a similar extent to native H2 relaxin in both (A and B) human cardiac fibroblasts and (C–F) rat renal myofibroblasts (as demonstrated by (A, C and E) representative gelatin zymography and (B, D and F) densitometry). Importantly, the ability of H2 relaxin and B7-33 to promote MMP-2 was equivalently blocked either by (C and D) an RXFP1 antagonist, or (E, F) angiotensin II type 2 (AT2) receptor antagonist (PD = P123319). Data shown (B, D and F) are mean ± SEM (from n = 3 independent experiments conducted in duplicate). **P < 0.01 vs. untreated/control; ##P < 0.01 vs. H2 relaxin; ¶¶P < 0.01 vs. B7-33. | ||
To further explore the molecular mechanisms of its actions, we also investigated the effects of an angiotensin II type 2 (AT2) receptor antagonist (PD12331942) (Fig. 4E and F) on the MMP-promoting effects of B7-33; as we had recently shown that the AT2 receptor (AT2R) was critically required for H2 relaxin's anti-fibrotic actions.43 We demonstrated that H2 relaxin signaled through constitutive RXFP1–AT2R heterodimers to induce downstream functional effects at the level of pERK1/243 which in turn promoted MMP levels. Interestingly, as determined by gelatin zymography (Fig. 4E) and densitometry (Fig. 4F), the MMP-2-promoting effects of B7-33 in the rat renal myofibroblasts were completely blocked by PD123319, suggesting that like H2 relaxin,43 B7-33 was mediating its effects via RXFP1–AT2R heterodimers. On the other hand, neither the RXFP1 nor AT2 receptor antagonists alone affected basal MMP-2 expression (Fig. 4C–F).
pERK1/2-promoting effects of B7-33
Since MMP-2 stimulation by H2 relaxin occurs downstream of ERK1/2 phosphorylation,39,43 pERK1/2 activity following stimulation by B7-33 was then examined in HEK-293T cells stably expressing RXFP1 (Fig. 5A and B). Consistent with the cAMP data, the potency of B7-33 in the pERK assay was found to be modest and significantly lower than H2 relaxin in HEK-RXFP1 cells (Fig. 5A and B). This result did not explain the marked MMP-promoting effects of B7-33 in myofibroblasts as shown in Fig. 4. It also did not match our recent findings that H2 relaxin signaled through RXFP1–AT2R heterodimers to induce downstream functional effects on pERK1/2 to inhibit the pro-fibrotic actions of TGF-β1 at the level of Smad2 (an intracellular protein that promotes TGF-β1 signal transduction).43 Therefore, we examined pERK1/2 activity following stimulation by B7-33 in rat renal myofibroblasts (Fig. 5C and D) that endogenously expressed RXFP1. Interestingly, ERK1/2 activation peaked at 3–5 minutes in rat renal myofibroblasts (Fig. 5C) following administration of either B7-33 (0.1 μM) and H2 relaxin (0.1 μM). In order to compare the potency and efficacy of B7-33 and H2 relaxin, we constructed concentration–response relationships in rat renal myofibroblasts (Fig. 5D). Excitingly, in these myofibroblasts, the concentration–response curves demonstrated that B7-33 increased ERK1/2 activity (5 min treatment) with a similar efficacy and potency to H2 relaxin (Fig. 5D). Given that B7-33 stimulated ERK1/2 activity and increased MMP-2 levels in vitro, we tested its anti-fibrotic potential in three models of disease in vivo.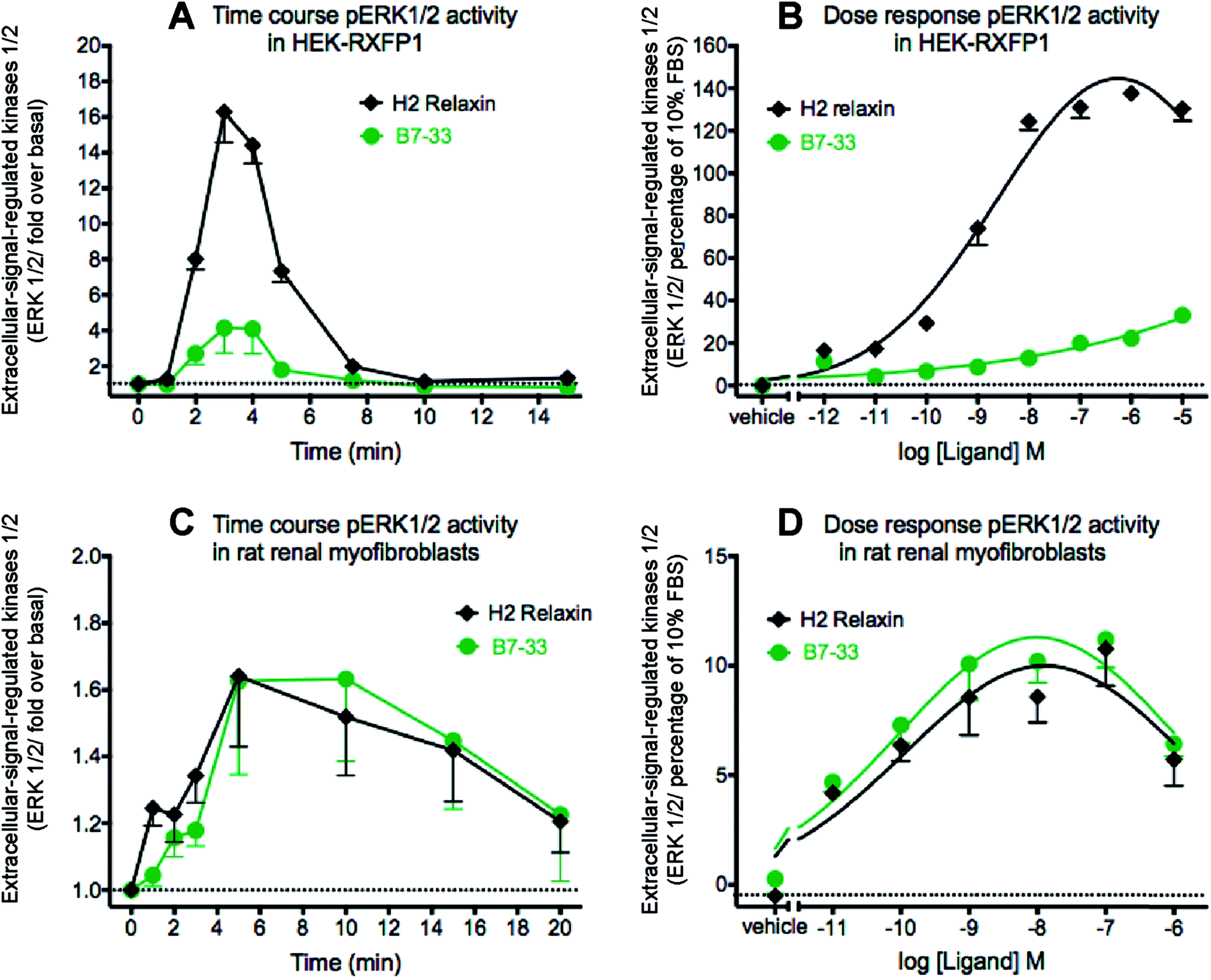 | ||
| Fig. 5 B7-33 strongly activated pERK1/2 in cells endogenously expressing RXFP1 (rat renal myofibroblasts) but weakly activated pERK1/2 in stably expressing RXFP1 (HEK-293T) cells as shown by (A and C) time course and (B and D) concentration–response curves for activation of ERK1/2 by B7-33. B7-33 stimulated pERK peaking at (A) 3–5 minutes in HEK-293T cells and (C) at 5–10 minutes in rat renal myofibroblasts. (B) B7-33 activated pERK1/2 (3 min treatment) with lower potency and efficacy compared with H2 relaxin. The data are expressed as mean ± SEM of n = 3–4 independent experiments. (D) B7-33 promoted ERK1/2 activity (5 min treatment) in rat renal myofibroblasts endogenously expressing RXFP1 with similar potency and efficacy to H2 relaxin. The data are expressed as mean ± SEM of n = 3–5 independent experiments. | ||
B7-33 significantly reduced cardiac fibrosis and improved heart function
Systemic administration of B7-33 or H2 relaxin (each at 0.5 mg kg−1 d−1), a dose of H2 relaxin that had been used previously to successfully demonstrate anti-fibrotic actions,18,44 and produce circulating levels of 20–40 ng mL−1,45 from weeks 8–12 post-myocardial infarction (MI) injury significantly reduced collagen deposition in the left ventricle (LV) of rat compared to the injured/vehicle-treated group at 12 weeks post-MI (both P < 0.01 vs. saline-treated MI group; Fig. 6A and B). Left ventricular end-diastolic pressure (LVEDP) was significantly reduced in both the B7-33 (P < 0.05 vs. saline-treated MI group) and H2 relaxin (P < 0.05 vs. saline-treated MI group) treated rats (Fig. 6C) suggesting that treatment with B7-33 was as effective as H2 relaxin for improving LV function in the MI-induced rat model of heart failure.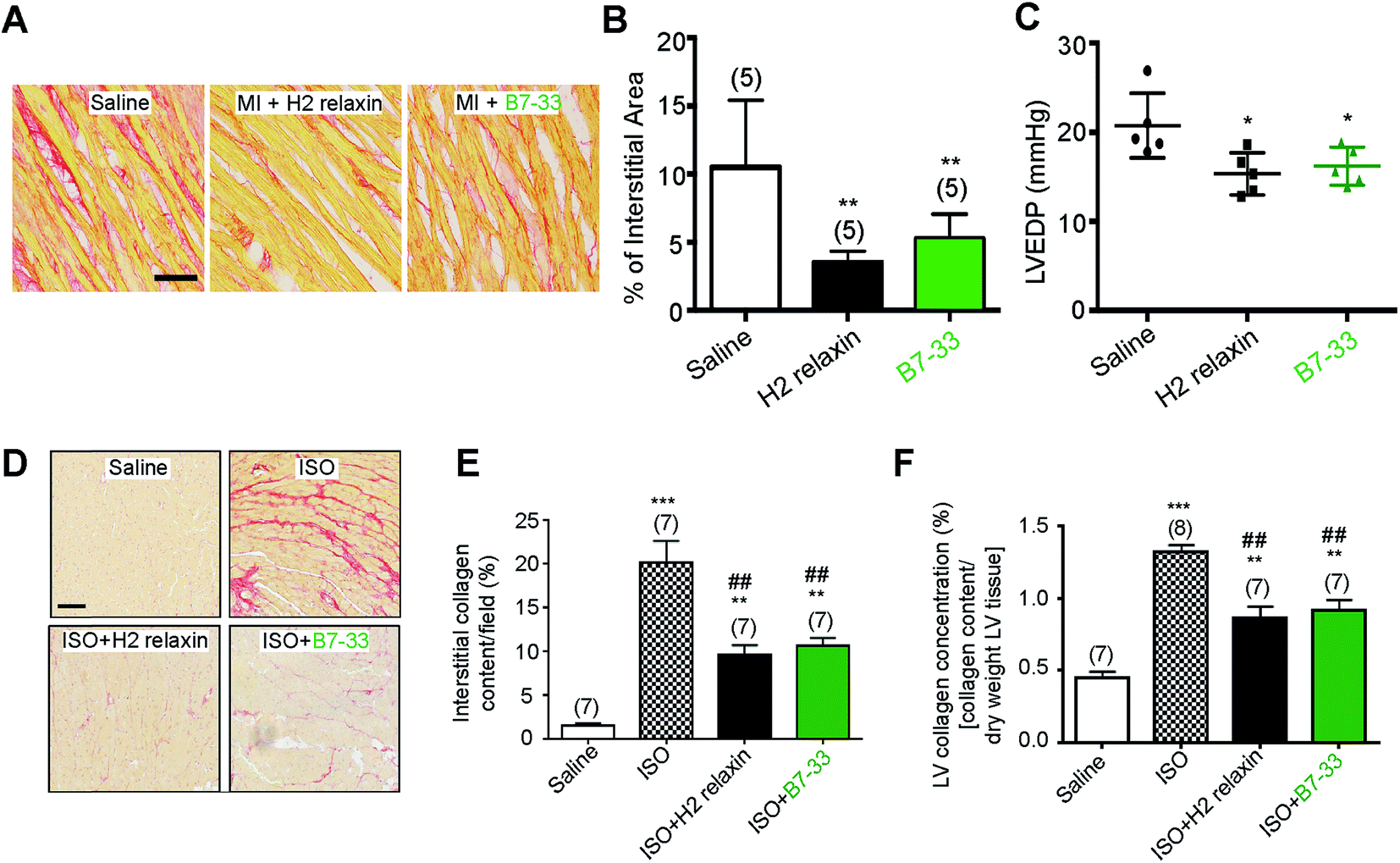 | ||
| Fig. 6 (A–C) B7-33 significantly reduced myocardial infraction (MI)-induced fibrosis and improved heart function. (A) Representative photomicrographs of collagen stained with picrosirius red in LVs of the heart from vehicle (saline), H2 relaxin and B7-33 treated rats with MI. (B) H2 relaxin and B7-33 significantly reduced left ventricular fibrosis post-MI compared with saline treated rats. (C) Animals treated with H2 relaxin or B7-33 were also associated with reduced left ventricular end-diastolic pressure (LVEDP) compared to saline-treated animals demonstrating peptide-induced improvement of LV function 12 weeks after MI. Numbers in parentheses represent the number of animals used per group. *P < 0.05, **P < 0.01 vs. saline group. Scale bar 100 μm. (D–F) B7-33 prevented isoproterenol (ISO)-induced cardiac fibrosis. (D) Collagen was stained with picrosirius red to determine (E) % interstitial LV collagen and assessed by hydroxyproline to determine (F) total LV collagen concentration. B7-33 (0.25 mg kg−1 d−1) significantly prevented LV fibrosis to a similar extent as native H2 relaxin (0.5 mg kg−1 d−1) by day 14 post-ISO-induced injury. Numbers in parentheses represent the number of animals used per group. **P < 0.01, **P < 0.001 vs. saline (control) group; ##P < 0.01 vs. ISO group. Scale bar 50 μm. | ||
We then used equimolar amounts of B7-33 and H2 relaxin in a mouse model of isoproterenol (ISO)-induced cardiomyopathy and related fibrosis (Fig. 6D–F). When the effects of B7-33 (0.25 mg kg−1 d−1) were directly compared with those of H2 relaxin (0.5 mg kg−1 d−1; equivalent dose to B7-33 when corrected for molecular weight) in preventing the effects of ISO-induced cardiac fibrosis, B7-33 significantly reduced interstitial collagen staining (by ∼51%; P < 0.01 versus ISO alone; Fig. 6D and E) to a similar extent to H2 relaxin (which decreased ISO-induced interstitial collagen by ∼57%; P < 0.01 versus ISO alone). Likewise, B7-33 significantly reduced ISO-induced increases in total LV collagen concentration (by ∼47%; P < 0.01 versus ISO alone; Fig. 6F) as did H2 relaxin (which decreased ISO-induced total LV collagen concentration by ∼53%; P < 0.01 versus ISO alone; Fig. 6F). However, these collagen-inhibitory effects of B7-33 and H2 relaxin were independent of their ability to regulate systolic blood pressure (SBP) in the ISO model (basal SBP/end SBP (mmHg) for ISO alone: 115 ± 5/110 ± 6; ISO + B7-33: 112 ± 5/110 ± 3; and for ISO + H2 relaxin: 117 ± 5/112 ± 4; n = 7 mice/group).
B7-33 significantly reduced airway/lung fibrosis and improved airway function
Mice with ovalbumin (OVA)-induced chronic allergic airways disease (AAD) presented with significantly increased epithelial thickening (a measure of airway remodeling; Fig. 7A and B), sub-epithelial collagen (blue) staining (Fig. 7A) and total lung collagen concentration (measures of airway fibrosis; Fig. 7C), as well as airway hyperresponsiveness (AHR; a measure of lung dysfunction; Fig. 7D) (all P < 0.001 vs. saline-treated controls). In this model, daily intranasal (i.n.) administration of the B7-33 peptide (0.25 mg kg−1 d−1) or H2 relaxin (0.5 mg kg−1 d−1)46 over a 2 week treatment period totally reversed the increased epithelial thickness associated with chronic AAD (both P < 0.001 vs. OVA alone) (Fig. 7A and B). Total lung collagen concentration was also normalized after 2 weeks of B7-33 (P < 0.05 vs. OVA alone; no different to saline alone) or H2 relaxin treatment over the same time period (P < 0.01 versus OVA alone; no different to saline alone) (Fig. 7C). In addition, AHR was partially but significantly reversed in B7-33-treated mice and to a similar extent as in H2 relaxin-treated mice (at the three highest doses of methacholine tested; all P < 0.05 vs. OVA alone; Fig. 7D). However, airway reactivity in B7-33 or H2 relaxin-treated mice remained significantly higher than that in saline-treated controls (at the four highest doses of methacholine tested; all P < 0.05 vs. saline alone; Fig. 7D) again demonstrating that B7-33 could improve lung dysfunction through its anti-remodeling and anti-fibrotic effects.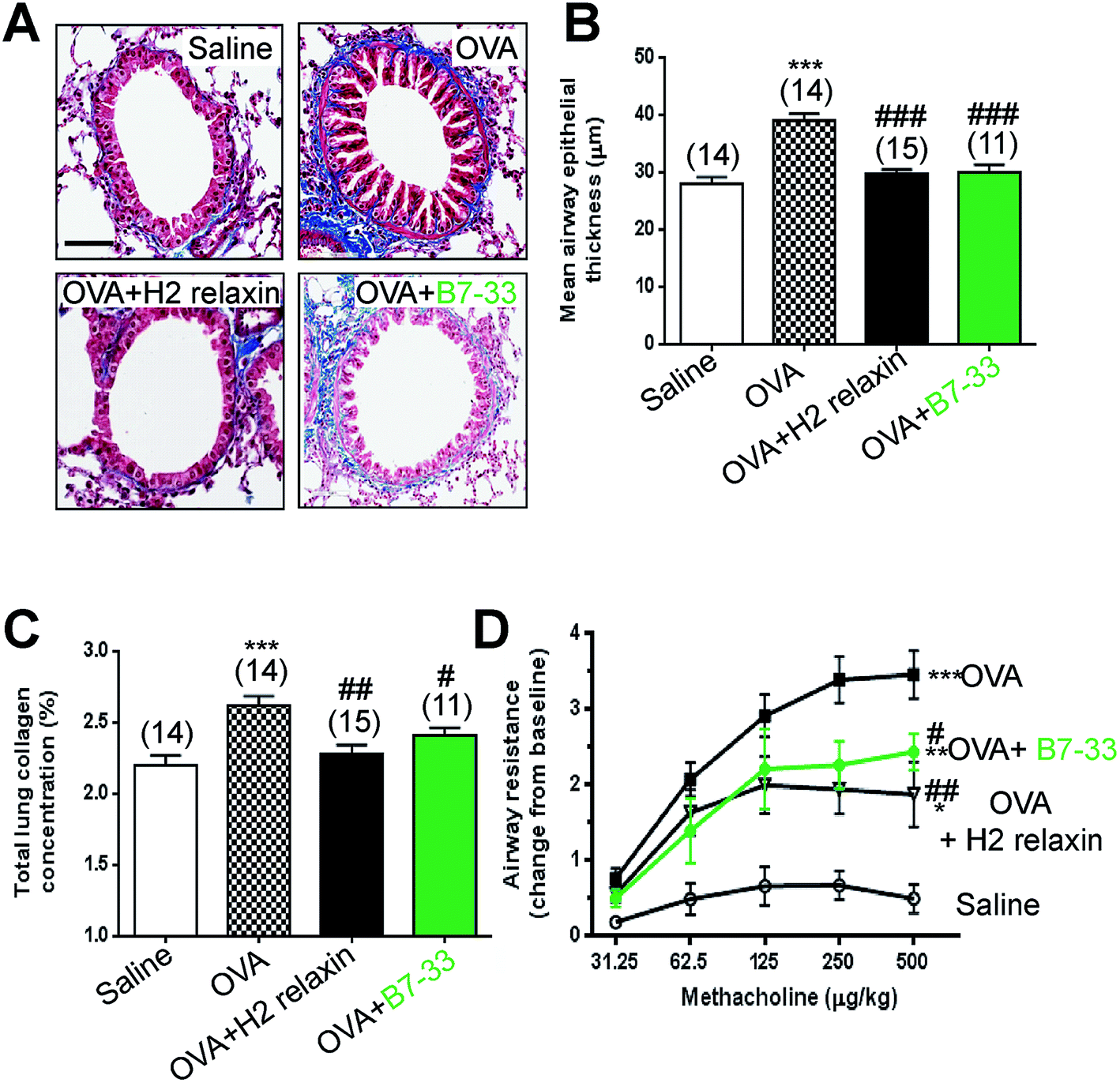 | ||
| Fig. 7 B7-33 reduced ovalbumin (OVA)-induced airway/lung fibrosis and improved airway function. In (A) representative photomicrographs of Masson’s trichrome-stained airway/lung sections of saline, OVA, OVA + H2 relaxin and OVA + B7-33 mice. Scale bar = 100 μm; In (B) subepithelial collagen thickness (mm) in the lamina reticularis as mean ± SEM. In (C) total lung collagen (hydroxyproline) content (a measure of fibrosis) from each of the groups studied. In (D) airway resistance (airway hyperresponsiveness) (AHR); change from baseline a measure of lung dysfunction in saline, OVA, OVA + H2 relaxin mice and OVA + B7-33 (n = 11–15 mice/group), in response to increasing concentrations of the bronchoconstrictor, methacholine, by invasive plethysmography as mean ± SEM; numbers in parantheses represent number of mice analysed per group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. saline group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. OVA group. | ||
B7-33 did not exacerbate tumor development in a mouse model of prostate tumor growth in vivo
RM1 prostate cancer cells (5 × 103 cells) were orthotopically implanted in the prostates of wild-type mice and after 10 days a significant tumor developed (Fig. 8). B7-33 and H2 relaxin were then evaluated for their ability to promote prostate tumor growth, which H2 relaxin had previously been demonstrated to do.25–28 In this study, H2 relaxin (0.15 mg kg−1 d−1; which produces circulating levels of 5–10 ng mL−1, that promoted prostate tumor growth in mice25) increased prostate size by more than 150% (P < 0.001 vs. untreated mice with prostate tumors) when administered from 2–10 days post-RM1 cell-induction of prostate tumors (Fig. 8A and B). However, administration of an equivalent (0.075 mg kg−1 d−1) or higher dose (0.25 mg kg−1 d−1; the dose used to demonstrate its anti-fibrotic effects in the ISO model) of B7-33 failed to exacerbate mouse prostate tumor size in vivo when administered over the same period of time.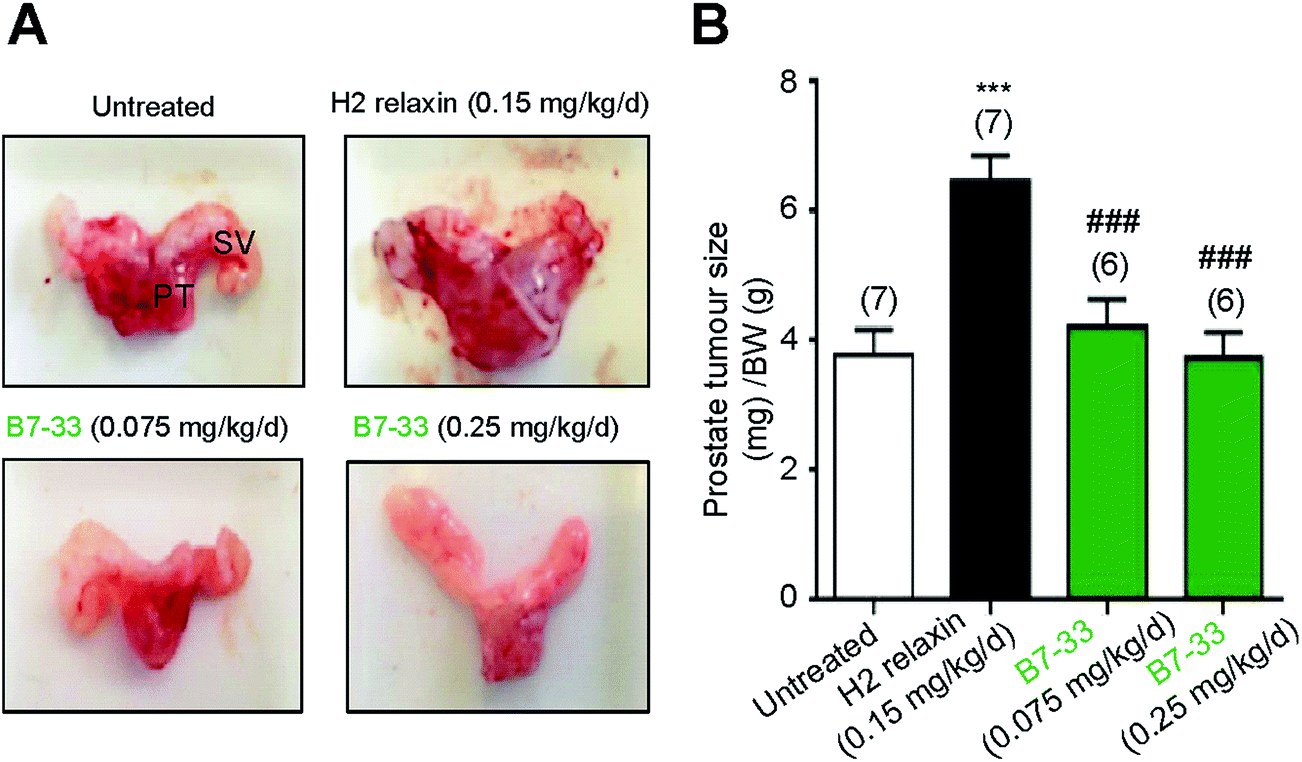 | ||
| Fig. 8 B7-33 did not promote tumor growth. In (A), representative prostate tumors of male C57B6J mice 10 days after their prostates were injected with RM1 cells. PT: prostate tumor; SV: seminal vesicle. In (B) the mean ± SEM prostrate tumor size/BW ratio is shown from untreated, B7-33-treated mice. Tumor development was enhanced by H2 relaxin but not B7-33 (A and B). ***P < 0.001 vs. untreated group; ###P < 0.001 vs. H2 relaxin treated group. | ||
Structural analysis of B7-33 by NMR spectroscopy
To investigate whether the single-chain B7-33 could adopt a native H2 B-chain-like conformation in solution without structural support from the A-chain, we examined its structural features using two-dimensional NMR spectroscopy. NOESY and TOCSY spectra were recorded at 600 MHz and the spectra were of good quality in terms of line width and signal-to-noise but had poor signal dispersion, consistent with an unstructured peptide lacking a well-defined structural core. Resonance assignments were achieved by sequential assignment strategies.47 Secondary Hα-chemical shifts, that is, differences between observed Hα-chemical shifts and shifts from random coil peptides, are good indicators of the presence of secondary structure. A comparison of the secondary chemical shifts of B7-33 and H2 relaxin are shown in Fig. 9. Stretches of negative values are indicative of helical character, positive values of extended region and values between 0.1 and −0.1 are characteristic of random coil. The profile of native H2 relaxin was dominated by positive values between residues 8–11 and negative values between residues 13 and 22, consistent with a short extended region and a helical segment, respectively.48 In contrast, the values of B7-33 were close to random coil throughout, confirming that in solution the structural features of the H2 B-chain had been lost.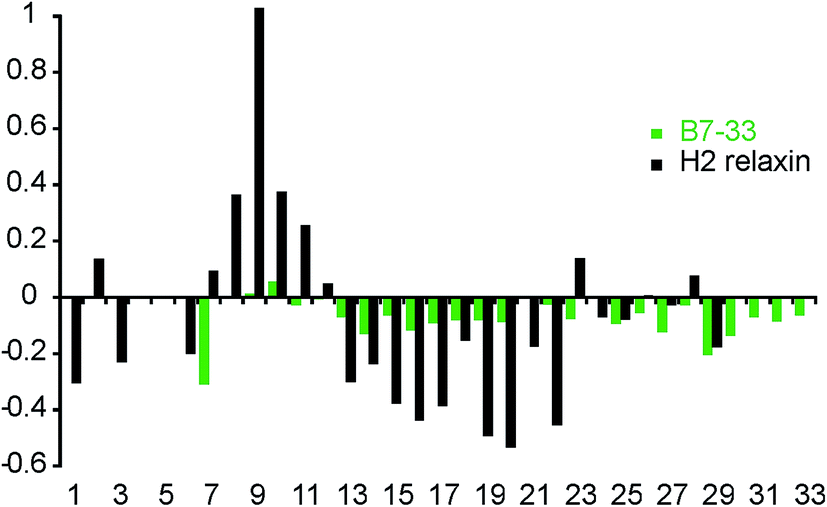 | ||
| Fig. 9 Secondary Hα-shift analysis for B7-33 and H2 relaxin. Stretches of negative values indicate α-helix and positive values indicate β-sheet. B7-33 displayed only small secondary shifts, lacking the stretch of large negative values seen in the helical region of native H2 relaxin, suggesting that B7-33 mainly adopted a random coil structure. | ||
Discussion
Since its discovery in 1926,49 H2 relaxin has been the subject of intense endocrinological and pharmacological research. The cognate receptor for H2 relaxin, RXFP1, was discovered only 13 years ago.20 Since then, significant progress has been made towards understanding the H2 relaxin–RXFP1 interaction.50 In addition to its classical 7-transmembrane spanning region (TM), RXFP1 has a large extracellular domain containing 10 leucine rich repeats (LRRs) and a NH2-terminal low-density lipoprotein type A (LDLa) module (Fig. S1†) that is only found in RXFP1 and the closely related RXFP2.20 The activation of RXFP1 is complex and requires interactions involving the LRR domain,29 the extracellular loops,31,32 and the N-terminal LDLa module.51–54 Although this complexity presents considerable challenges for the development of domain-minimized analogues as does the notoriously insolubility of the single chains,37 we have engineered an active single-B-chain derivative of H2 relaxin, B7-33, with an increased cationic charge and aqueous solubility. This is in fact the first report of any domain-minimized single chain insulin- or relaxin-related peptide displaying agonist activity.This peptide acted as a full agonist of cAMP activation but with modest to poor affinity and potency in HEK-293T cells stably expressing RXFP1, and THP1 cells endogenously expressing RXFP1. In order to better understand the interaction between B7-33 and RXFP1, we synthesized B7-33 analogues targeting the key residues: RB13, RB17 and IB20 in the B-chain of H2 relaxin that are known as the primary drivers for RXFP1 activity55 and that interact with corresponding residues in the LRR region of the RXFP1 receptor.29 The loss of binding affinity and cAMP activity of key-residue mutated analogues for RXFP1 indicated that B7-33 used the same residues as H2 relaxin to interact with the LRRs of RXFP1. In addition to its cognate receptor RXFP1, H2 relaxin is also known to activate RXFP2, which is cognate receptor for the related INSL3 peptide.35,50 However B7-33 did not activate cAMP activity in HEK-RXFP2 cells.
Like H2 relaxin,39,40 B7-33 exhibited high potency in increasing the activity of the collagen-degrading enzyme MMP-2 in human cardiac fibroblasts and rat renal myofibroblasts (key matrix-producing cells that contribute to fibrosis). These effects were demonstrated to be RXFP1-specific as they were blocked by an RXFP1-specific antagonist. The MMP results contrasted with its modest ability to stimulate cAMP in stably- and endogenously-expressing RXFP1 cells and the limited ability of H2 relaxin to stimulate cAMP in (myo)fibroblast cultures.40,44,56 Since the increased MMP-2 activity in response to H2 relaxin is associated with increased ERK1/2 activity39 (Fig. S1†), we examined MAP kinase responses to B7-33. In HEK-RXFP1 cells, B7-33 poorly induced pERK1/2 phosphorylation. However, in (myo)fibroblasts, B7-33 potently activated pERK1/2 with high efficacy. The fact that pERK1/2 and MMP-2 have previously been shown to be associated with the anti-fibrotic actions of H2 relaxin39,43,56 strongly suggested that B7-33 was a functionally selective agonist of RXFP1. Furthermore, the fact that the MMP-2-promoting effects of both H2 relaxin and B7-33 were equivalently blocked by either a RXFP1 or AT2R antagonist suggested that, like H2 relaxin, B7-33 exerted its biological activity through RXFP1 and likely through RXFP1–AT2R heterodimers, as both these GPCRs are expressed on the renal myofibroblasts investigated.43,57
It is known that the same GPCR can couple to several different signaling pathways in different cells and that the binding of the same ligand in different cells can demonstrate pathway-dependent pharmacology.58 The fact that B7-33 was a potent and high efficacy agonist for activating pERK1/2 in fibroblasts but not in recombinant systems expressing RXFP1 may be explained by highly efficient coupling of ERK1/2 to RXFP1 in fibroblasts. It has been previously demonstrated that the actions of H2 relaxin through RXFP1 in fibroblasts are associated with strong pERK activation but virtually no cAMP production40,44,56 We therefore postulated that B7-33 would be a potent anti-fibrotic agent when utilized in vivo with the added benefit of inactivity or limited activity at cAMP pathways associated with the tumor-promoting effects of H2 relaxin (Fig. S1†).59 In accord with this, we showed that, in contrast to H2 relaxin, B7-33 did not exacerbate RM1 cell-induced prostate tumor growth in mice in vivo. Additionally, as the activation of cAMP has been associated with several other adverse effects including inotropy and mortality,24 it is possible that B7-33 may not induce the same level of H2 relaxin-mediated inotropy,60,61 but may improve end-stage mortality by having limited ability to activate cAMP and/or cAMP-dependent protein kinase A. The latter may have important implications in the context of the phase III trials that are currently assessing the ability of H2 relaxin (serelaxin) to improve end-stage mortality in AHF patients.
Based on these findings in fibroblasts and particularly its high potency in activating pERK1/2 in these cells, the efficacy of B7-33 in a pathophysiologically relevant rat model of MI-induced heart failure was examined. Systemic administration of B7-33 or H2 relaxin, after 8 weeks of MI-induced injury, over a 4 week treatment period, significantly reduced the build-up of MI-induced collagen confirming that B7-33 had similar anti-fibrotic activity to H2 relaxin in experimental heart failure. Furthermore, we also observed improved LV function evidenced by a significant reduction in LVEDP. We then investigated the efficacy of systemic B7-33 administration (throughout the onset of injury; from days 1–14) in a mouse model of cardiomyopathy-related cardiac fibrosis and showed that B7-33 had similar efficacy to H2 relaxin in preventing ISO-induced cardiac fibrosis, confirming again its potential as a drug candidate for the treatment of fibrosis and related dysfunction associated with heart failure.
Finally we assessed the anti-remodeling and anti-fibrotic effects of B7-33 given by intranasal administration in a 9 week OVA-induced murine model of chronic AAD in vivo. When administered daily from weeks 9–11, B7-33 significantly diminished airway epithelial thickening, total lung collagen concentration (a measure of fibrosis) and airway hyperresponsiveness (a measure of lung dysfunction) to a similar extent to native H2 relaxin, confirming that it also had therapeutic potential for chronic inflammatory lung diseases. We have previously shown that upon binding to RXFP1, H2 relaxin mediated its anti-fibrotic actions through a pERK1/2-neuronal nitric oxide (NO) synthase (nNOS)-NO-cGMP-dependent pathway in myofibroblasts to disrupt TGF-β1 signal transduction, at the level of Smad2 phosphorylation18,40,43,46,56 in several organs including the heart, lung and kidney. This in turn inhibited TGF-β1-induced myofibroblast differentiation and aberrant ECM/collagen deposition in these organs. Additionally, H2 relaxin is also well known to stimulate MMPs associated with collagen degradation16–18,39,40,43,62 as demonstrated for B7-33. We have now also demonstrated that like H2 relaxin, B7-33 mediates its anti-fibrotic actions by signaling through constitutive RXFP1–AT2R heterodimers. Hence, it is likely that the potent anti-fibrotic effects of B7-33 are mediated through similar mechanisms.
The B7-33 was structurally characterized by solution NMR spectroscopy. Despite its high potency in native cells and in vivo animal models, it was mainly unstructured in solution. This indicated that B7-33 will only adopt the correct conformation for binding upon interaction with RXFP1 consistent with previous studies where an unstructured single-chain relaxin 3 peptide was a potent antagonist of RXFP3.63
Conclusion
In summary, we report for the first time the development of a domain-minimized H2 relaxin single-chain peptide, B7-33, as a functionally selective agonist of RXFP1. This single-chain peptide displayed equivalent efficacy to the natural H2 relaxin in several functional assays that included stimulation of pERK1/2 activity and MMP-2 levels in fibroblasts, reduction of fibrosis in three animal models of disease and improvement of heart and lung function in rodent models of heart failure and chronic AAD, respectively. It is likely that the molecular mechanism behind the strong anti-fibrotic actions of B7-33, as per those of H2 relaxin, involved its activation of RXFP1–AT2R heterodimers. We additionally showed that B7-33 bound using the binding cassette “RXXXRXXI” for receptor activation demonstrating that it uses a similar method of binding and activation of RXFP1 as H2 relaxin. Our findings also demonstrated that B7-33 stimulated pERK1/2 activity to a similar extent to H2 relaxin in fibroblasts and that this correlated with its ability to ameliorate fibrosis regardless of etiology with similar efficacy to H2 relaxin. B7-33 did not promote prostate cancer progression due to its limited ability to activate cAMP signaling and it is likely that this poor coupling to cAMP signaling may benefit the therapeutic potential of the single chain peptide. B7-33 is far easier and cheaper to manufacture by recombinant or chemical means and thus more cost-effective as a drug. It will also be an attractive lead for further modification to improve stability, potency and in vivo efficacy. While B7-33 is an attractive anti-fibrotic lead molecule it is also a valuable tool to determine the mechanism of action of relaxin in animal models of AHF.Materials and methods
Solid phase peptide synthesis
Peptides were solid phase synthesized as C-terminal amides on PAL-PEG-PS resin on one of the following instruments: CEM Liberty™ microwave peptide synthesizer (Ai Scientific, Queensland, Australia) or Protein Technologies Tribute batch-wise peptide synthesizer (Tucson, AZ, USA).Ligand binding assays at RXFP1
Human embryonic kidney (HEK-293T) cells stably transfected with RXFP1 were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 100 μg mL−1 penicillin, 100 μg mL−1 streptomycin and 2 mM L-glutamine and plated into 96 well plates pre-coated with poly-L-lysine for whole cell binding assays. Competition binding experiments Eu3+-labelled H2 relaxin in the absence or presence of increasing concentrations of unlabelled H2 relaxin B-chain derivatives were conducted as previously described.64 All data are presented as the mean ± S.E. of the % specific binding of triplicate wells, repeated in at least three separate experiments, and curves were fitted using one-site binding curves in GraphPad Prism 6 (GraphPad Inc, San Diego, CA). Statistical differences in pIC50 values were analyzed using one-way analysis of variance coupled to Newman Keul's multiple comparison test for multiple group comparisons in GraphPad Prism 6.cAMP activity assays
The ability of the H2 relaxin B-chain peptide derivatives to stimulate cAMP activity in HEK-293T cells transfected with human, rat or mouse RXFP1 was evaluated using a cAMP reporter gene assay as previously described.51 Cells were co-transfected with a pCRE β-galactosidase reporter plasmid plated in 96-well plates and after 24 hours, the co-transfected cells were incubated with increasing concentrations of peptides in parallel with H2 relaxin. cAMP signaling in RXFP1-expressing THP1 cells65 was assessed as described previously66 using direct cAMP measurement (cAMP dynamic 2 HTRF kit, cisbio) after ligand stimulation for 30 minutes in the presence of 50 μM of the phosphodiesterase inhibitor IBMX. Ligand-induced cAMP stimulation was expressed as a % of maximal response to H2 relaxin. Each data point was measured in triplicate and each experiment conducted independently at least three separate times. Statistical differences in pEC50 values were analyzed using one-way ANOVA coupled to Newman Keul's multiple comparison test for multiple group comparisons in GraphPad Prism 6.ERK1/2 phosphorylation surefire® assay
HEK-RXFP1 cells,51 rat renal myofibroblasts or human cardiac fibroblasts43,56 were plated into 96-well plates or 48-well plates, respectively (4 × 104 cells per well) and grown overnight in DMEM medium containing 10% (v/v) FBS at 37 °C, 5% CO2. Cells were serum starved in 0.5% (v/v) DMEM medium for 4 hours before addition of ligands, 0.5% (v/v) DMEM medium plus 0.01% w/v BSA (vehicle control) or 10% FBS (positive control) at 37 °C. After experimentation, cells were lysed with lysis buffer and frozen at −20 °C. ERK1/2 was detected as described previously.67 Briefly, cell lysate was transferred to 384-well microplates and combined with AlphaScreen detection reagents according to the manufacturer's instructions. Samples were counted in 384-well microplates on an EnVision multilabel plate reader (PerkinElmer Life and Analytical Sciences) (excitation 680 nm; emission 520 to 620 nm).Gelatin zymography
Media samples from renal myofibroblasts isolated from injured rat kidneys, 3 days post-unilateral ureteric obstruction (kindly provided by Associate Professor Tim Hewitson, Department of Nephrology, Royal Melbourne Hospital, Parkville, Victoria, Australia39,43,56); or fetal human cardiac fibroblasts (ScienCell Research Laboratories, San Diego, CA, USA)40 (1 × 105 cells per 12-well plate well) were analysed for MMP-2 expression by gelatin zymography, after 72 hours in culture as described before.68 Additionally, rat renal myofibroblasts treated with H2 relaxin or B7-33 were further treated with our without an RXFP1 antagonist41 or AT2R antagonist (PD123319);43 to confirm that both peptides were acting through RXFP1 and possibly RXFP1–AT2R heterodimers. To avoid saturation of the MMP-2 bands, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5–1:10 diluted media samples were analyzed from 3 separate experiments conducted in duplicate.
:5–1:10 diluted media samples were analyzed from 3 separate experiments conducted in duplicate.
Animals
All animals were group housed under a constant temperature of 22 ± 1 °C and a relative humidity of 50–60% under a controlled 12 hour light:dark cycle. Animals were given access to standard laboratory chow and drinking water ad libitum and at least 5 days to acclimatize before any experimentation was conducted on them.
All experiments were conducted with approval from a Florey Institute's or Monash University's Animal Ethics Committee, which adhere to the Australian Code of Conduct for care and use of laboratory animals for scientific purposes.
Induction of MI-induced heart failure
Adult male Sprague-Dawley rats were subjected to MI-induced heart failure, as previously described.69 Briefly, rats were anaesthetised with an intramuscular injection of ketamine (60 mg kg−1) and medetomidine hydrochloride (250 mg kg−1). A left sided thoracotomy through an opening between the fourth and fifth rib was performed, the heart was exteriorised and the left anterior descending coronary artery was ligated. Anaesthesia was reversed with atipamezole hydrochloride (1 mg kg−1). Penicillin (1000 U) and buprenorphine (0.05 mg kg−1) was administered to aid post-operative recovery. Animals were left to recover from the surgery under a heating source. Rats were individually housed after the surgery.Eight weeks after MI surgery, rats were randomly assigned to 3 groups (vehicle, H2 relaxin or B7-33-treated), re-anaesthetised (2–3% isoflurane) and an osmotic mini-pump (model 2ML4, Alzet, Cupertino, CA) implanted intraperitoneally. Vehicle (saline), H2 relaxin (0.5 mg kg−1 d−1; a dose that had been used previously to successfully demonstrate its anti-fibrotic actions18,44 and produce circulating levels of 20–40 ng mL−1 (ref. 45)) or B7-33 (0.5 mg kg−1 d−1) was continuously administered for 28 days. At the conclusion of treatment, rats were anaesthetized (sodium pentobarbitone, 60 mg kg−1 i.p.) and the left ventricular end-diastolic pressure determined by an investigator blinded to the treatment prior to decapitation and removal of the heart for histological analysis.
Induction of ISO-induced heart failure
Male 129SV mice were subcutaneously injected with isoprenaline hydrochloride (25 mg kg−1; Sigma-Aldrich) once daily for 5 consecutive days and then left for a further 9 days for fibrosis progression to occur. Subgroups of animals (n = 7–8/group) received no treatment (injury alone control), recombinant H2 relaxin (0.5 mg kg−1 d−1) or an equivalent dose of B7-33 (0.25 mg mg−1 d−1 corrected for MW) via subcutaneously implanted osmotic minipumps (model 2002; Alzet). A separate subgroup of mice (n = 7) that were not subjected to isoproterenol or peptide treatment were used as untreated controls. Nine days after the fifth ISO injection (at day 14), all mice were weighed and then sacrificed for heart and LV collection. A similar portion of the LV from each animal was then used for the determination of interstitial collagen staining and morphometric analysis of interstitial collagen density18 or hydroxyproline content, as described before44 which was performed in a randomized and blinded fashion. Basal and end systolic blood pressure was also measured from all mice studied using tail cuff plethysmography (MC4000 Blood Pressure Analysis Systems; Hatteras Instruments Inc., NC, USA); where 15–20 measurements per time point were pooled to obtain a mean for each animal.70Induction of chronic AAD
A chronic model of OVA-induced AAD71 was established in female Balb/c mice (n = 40). Mice were sensitized i.p. on day 0 and 14 with 10 μg Grade V chicken egg ovalbumin (Sigma-Aldrich Corp., St. Louis, MO, USA) and 0.4 mg aluminium potassium sulphate (alum) in 0.5 mL saline, then challenged by whole body inhalation exposure to aerosolized 2.5% OVA (weight/volume of saline) three times a week from days 21–63 (30 min per session) using an ultrasonic nebulizer.72 Control mice (n = 14) were sensitized and challenged with saline instead of OVA.Histopathology
The mid-zone of the LV from male rats and 129SV mice, and largest lung lobe from female Balb/c mice were fixed in 10% neutral buffered formalin for 24–48 h before being processed and embedded routinely in paraffin wax. Representative sections of tissue, 3–5 μm each, were taken and stained with either picrosirius red18 for the detection of LV interstitial collagen or Masson's trichrome62 for the detection of airway/lung subepithelial basement membrane collagen deposition.Hydroxyproline analysis
The apical region of the heart or second largest lung lobe from each mouse were treated as described previously45,62 for the determination of hydroxyproline content. Hydroxyproline values were estimated based on a standard curve constructed with serial dilutions of a 0.1 mg mL−1 stock of trans-4-hydroxyproline-L-proline (Sigma-Aldrich). Hydroxyproline values were then converted to collagen content as detailed previously44 and, in turn, divided by the dry weight of each corresponding left ventricular or lung tissue assessed to yield collagen concentration (a measure of fibrosis).Induction of prostate tumor growth
Male C57BL/6 mice (n = 26) were injected with 5000 RM1 (mouse prostate tumor) cells into their prostates to induce tumor growth. One sub-group of mice (n = 7) was left untreated until day 10 post-RM1 cell administration (a time-point which had previously been determined these mice get small but significantly increased prostate tumors). Additional sub-groups of mice were subcutaneously implanted with osmotic mini-pumps (model 1007D; Alzet) containing H2 relaxin alone (0.15 mg kg−1 d−1; n = 7) or B7-33 (0.075 mg kg−1 d−1; corrected for MW (n = 6) or 0.25 mg kg−1 d−1 (n = 6)) on day 2 post-RM1 cell administration and maintained until day 10 post-cell administration. Each pump had a reservoir that allowed it to continuously infuse these peptides for 8 days. At day 10, prostate weight to body weight (BW) ratio was determined by an investigator blinded to the treatment groups studied.Nuclear magnetic resonance spectroscopy
All NMR data were recorded on a Bruker Avance 600 MHz spectrometer equipped with a cryoprobe on a sample containing 0.5 mg peptide dissolved in 0.5 mL of 90% H2O/10% D2O at 298 K and pH ∼ 4. Recorded data sets included two-dimensional homonuclear TOCSY and NOESY (with mixing times of 80 ms and 200 ms, respectively). The data were recorded with 4k data points in the F2 dimension and 512 increments in the F1 dimension, which was subsequently zero-filled to 1k data points prior to transformation. Data were collected and processed using Topspin (Bruker) and analysed using XEASY.73 Spectra were referenced to the water signal at 4.768 ppm at 298 K.Statistics
All data were expressed as the mean ± SEM and analyzed using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA). The results were analyzed by one-way ANOVA, using the Newman–Keuls post hoc test for multiple comparisons between treatments groups in all experiments performed except for the analysis of the lung function (AHR) data, which was assessed by a two-way ANOVA with Bonferroni's post hoc test. P < 0.05 was considered to be statistically significant.Author contributions
M. A. H., J. D. W., R. A. D. B., and C. S. S. designed the research, and wrote the manuscript. R. J. S. advised on the research and edited the manuscript. M. A. H., M. K., J. K. R., S. S., and C. S. S. performed experiments and edited the manuscript. S. T. Y., S. G. R., V. B. N., C. S., N. A. P., and I. P. H. performed experiments.Acknowledgements
This work was supported by National Health & Medical Research Council (NHMRC) of Australia Grants (GNT1023321, GNT1023078, and GNT1065481) awarded to M. A. H., J. D. W., K. J. R., and R. A. D. B., (GNT1050268 and GNT1079680) awarded to S. T. Y., a NHMRC Program Grant GNT1055134 and ARC Linkage grant LP110100288 to R. J. S., a Florey Foundation Fellowship and Melbourne Research Grant Support Scheme (RGSS) awarded to M. A. H., a University of Melbourne Postgraduate Scholarship to V. B. N. and N. A. P., an Australian Postgraduate Award to C. S., Australian Future Fellowships to K. J. R. (FT130100890) and S. S. (FT120100876), an NHMRC Principal Research Fellowship to J. D. W. (GNT5018148) and NHMRC Senior Research Fellowships to R. A. D. B. (GNT1042650) and C. S. S. (GNT1041766). Research at The Florey Institute of Neuroscience and Mental Health is supported by the Victorian Government Operational Infrastructure Support Program. We sincerely thank Mr Sharon Layfield, Ms Tania Ferraro and Ms Kazi Jannatul Ferdous for expert technical assistance; Associate Professor Tim Hewitson (Royal Melbourne Hospital, Melbourne, Australia) for providing the rat renal myofibroblasts; and Professor Robert Widdop (Monash University, Melbourne, Australia) for providing the PD123319.References
- T. A. Wynn and T. R. Ramalingam, Nat. Med., 2012, 18, 1028–1040 CrossRef CAS PubMed.
- L. A. Danielson, O. D. Sherwood and K. P. Conrad, J. Clin. Invest., 1999, 103, 525–533 CrossRef CAS PubMed.
- J. Novak, L. A. Danielson, L. J. Kerchner, O. D. Sherwood, R. J. Ramirez, P. A. Moalli and K. P. Conrad, J. Clin. Invest., 2001, 107, 1469–1475 CrossRef CAS PubMed.
- L. A. Danielson and K. P. Conrad, J. Clin. Invest., 1995, 96, 482–490 CrossRef CAS PubMed.
- G. D. Bryant-Greenwood, Endocr. Rev., 1982, 3, 62–90 CrossRef CAS PubMed.
- E. T. Van Der Westhuizen, R. J. Summers, M. L. Halls, R. A. Bathgate and P. M. Sexton, Curr. Drug Targets, 2007, 8, 91–104 CrossRef CAS PubMed.
- E. Unemori, B. Sibai and S. L. Teichman, Ann. N. Y. Acad. Sci., 2009, 1160, 381–384 CrossRef CAS PubMed.
- C. A. Tozzi, G. J. Poiani, N. A. McHugh, M. P. Shakarjian, B. H. Grove, C. S. Samuel, E. N. Unemori and D. J. Riley, Pulm. Pharmacol. Ther., 2005, 18, 346–353 CrossRef CAS PubMed.
- T. Yoshida, H. Kumagai, A. Suzuki, N. Kobayashi, S. Ohkawa, M. Odamaki, T. Kohsaka, T. Yamamoto and N. Ikegaya, Nephrol., Dial., Transplant., 2012, 27, 2190–2197 CrossRef CAS PubMed.
- E. T. van der Westhuizen, M. L. Halls, C. S. Samuel, R. A. Bathgate, E. N. Unemori, S. W. Sutton and R. J. Summers, Drug Discovery Today, 2008, 13, 640–651 CrossRef CAS PubMed.
- E. Masini, D. Bani, M. Bigazzi, P. F. Mannaioni and T. Bani-Sacchi, J. Clin. Invest., 1994, 94, 1974–1980 CrossRef CAS PubMed.
- D. Bani and M. Bigazzi, Curr. Drug Saf., 2011, 6, 324–328 CrossRef CAS PubMed.
- J. R. Teerlink, G. Cotter, B. A. Davison, G. M. Felker, G. Filippatos, B. H. Greenberg, P. Ponikowski, E. Unemori, A. A. Voors, K. F. Adams Jr, M. I. Dorobantu, L. R. Grinfeld, G. Jondeau, A. Marmor, J. Masip, P. S. Pang, K. Werdan, S. L. Teichman, A. Trapani, C. A. Bush, R. Saini, C. Schumacher, T. M. Severin and M. Metra, Lancet, 2013, 381, 29–39 CrossRef CAS.
- S. L. Teichman, E. Unemori, J. R. Teerlink, G. Cotter and M. Metra, Curr. Heart Failure Rep., 2010, 7, 75–82 CrossRef CAS PubMed.
- C. S. Samuel, T. D. Hewitson, E. N. Unemori and M. L. Tang, Cell. Mol. Life Sci., 2007, 64, 1539–1557 CrossRef CAS PubMed.
- E. D. Lekgabe, H. Kiriazis, C. Zhao, Q. Xu, X. L. Moore, Y. Su, R. A. Bathgate, X. J. Du and C. S. Samuel, Hypertension, 2005, 46, 412–418 CrossRef CAS PubMed.
- C. S. Samuel, T. D. Hewitson, Y. Zhang and D. J. Kelly, Endocrinology, 2008, 149, 3286–3293 CrossRef CAS PubMed.
- C. S. Samuel, S. Cendrawan, X. M. Gao, Z. Ming, C. Zhao, H. Kiriazis, Q. Xu, G. W. Tregear, R. A. Bathgate and X. J. Du, Lab. Invest., 2011, 91, 675–690 CrossRef CAS PubMed.
- E. N. Unemori, L. B. Pickford, A. L. Salles, C. E. Piercy, B. H. Grove, M. E. Erikson and E. P. Amento, J. Clin. Invest., 1996, 98, 2739–2745 CrossRef CAS PubMed.
- S. Y. Hsu, K. Nakabayashi, S. Nishi, J. Kumagai, M. Kudo, O. D. Sherwood and A. J. Hsueh, Science, 2002, 295, 671–674 CrossRef CAS PubMed.
- R. A. Bathgate, R. Ivell, B. M. Sanborn, O. D. Sherwood and R. J. Summers, Pharmacol. Rev., 2006, 58, 7–31 CrossRef CAS PubMed.
- G. M. Felker, P. S. Pang, K. F. Adams, J. G. Cleland, G. Cotter, K. Dickstein, G. S. Filippatos, G. C. Fonarow, B. H. Greenberg, A. F. Hernandez, S. Khan, M. Komajda, M. A. Konstam, P. P. Liu, A. P. Maggioni, B. M. Massie, J. J. McMurray, M. Mehra, M. Metra, J. O'Connell, C. M. O'Connor, I. L. Pina, P. Ponikowski, H. N. Sabbah, J. R. Teerlink, J. E. Udelson, C. W. Yancy, F. Zannad and M. Gheorghiade, Circ.: Heart Failure, 2010, 3, 314–325 CrossRef PubMed.
- M. A. Hossain and J. D. Wade, Curr. Opin. Chem. Biol., 2014, 22, 47–55 CrossRef CAS PubMed.
- J. R. Teerlink, Heart Failure Rev., 2009, 14, 289–298 CrossRef CAS PubMed.
- J. D. Silvertown, J. Ng, T. Sato, A. J. Summerlee and J. A. Medin, J. Int. Cancer, 2006, 118, 62–73 CrossRef CAS PubMed.
- J. D. Silvertown, J. C. Symes, A. Neschadim, T. Nonaka, J. C. Kao, A. J. Summerlee and J. A. Medin, FASEB J., 2007, 21, 754–765 CrossRef CAS PubMed.
- S. Feng, I. U. Agoulnik, N. V. Bogatcheva, A. A. Kamat, B. Kwabi-Addo, R. Li, G. Ayala, M. M. Ittmann and A. I. Agoulnik, Clin. Cancer Res., 2007, 13, 1695–1702 CrossRef CAS PubMed.
- S. Feng, I. U. Agoulnik, Z. Li, H. D. Han, G. Lopez-Berestein, A. Sood, M. M. Ittmann and A. I. Agoulnik, Ann. N. Y. Acad. Sci., 2009, 1160, 379–380 CrossRef CAS PubMed.
- E. E. Bullesbach and C. Schwabe, J. Biol. Chem., 2005, 280, 14051–14056 CrossRef PubMed.
- D. J. Scott, G. W. Tregear and R. A. Bathgate, Ann. N. Y. Acad. Sci., 2009, 1160, 74–77 CrossRef CAS PubMed.
- S. Sudo, J. Kumagai, S. Nishi, S. Layfield, T. Ferraro, R. A. Bathgate and A. J. Hsueh, J. Biol. Chem., 2003, 278, 7855–7862 CrossRef CAS PubMed.
- N. A. Diepenhorst, E. J. Petrie, C. Z. Chen, A. Wang, M. A. Hossain, R. A. Bathgate and P. R. Gooley, J. Biol. Chem., 2014, 289, 34938–34952 CrossRef PubMed.
- M. A. Hossain, K. J. Rosengren, L. M. Haugaard-Jonsson, S. Zhang, S. Layfield, T. Ferraro, N. L. Daly, G. W. Tregear, J. D. Wade and R. A. Bathgate, J. Biol. Chem., 2008, 283, 17287–17297 CrossRef CAS PubMed.
- M. A. Hossain, J. D. Wade and R. A. Bathgate, Peptides, 2012, 35, 102–106 CrossRef CAS PubMed.
- L. J. Chan, K. J. Rosengren, S. L. Layfield, R. A. Bathgate, F. Separovic, C. S. Samuel, M. A. Hossain and J. D. Wade, J. Biol. Chem., 2012, 287, 41152–41164 CrossRef CAS PubMed.
- H. D. Niall, R. A. Bradshaw and G. D. Bryant-Greenwood, Prog. Clin. Biol. Res., 1979, 31, 651–658 CAS.
- M. P. Del Borgo, R. A. Hughes and J. D. Wade, J. Pept. Sci., 2005, 11, 564–571 CrossRef CAS PubMed.
- M. A. Hossain, K. J. Rosengren, C. S. Samuel, F. Shabanpoor, L. J. Chan, R. A. Bathgate and J. D. Wade, J. Biol. Chem., 2011, 286, 37555–37565 CrossRef CAS PubMed.
- B. S. Chow, E. G. Chew, C. Zhao, R. A. Bathgate, T. D. Hewitson and C. S. Samuel, PLoS One, 2012, 7, e42714 CAS.
- M. Sarwar, C. S. Samuel, R. A. Bathgate, D. R. Stewart and R. J. Summers, Br. J. Pharmacol., 2015, 172, 1005–1019 CrossRef CAS PubMed.
- M. A. Hossain, C. S. Samuel, C. Binder, T. D. Hewitson, G. W. Tregear, J. D. Wade and R. A. Bathgate, Amino Acids, 2010, 39, 409–416 CrossRef CAS PubMed.
- Y. Ozawa, Y. Suzuki, K. Murakami and H. Miyazaki, Biochem. Biophys. Res. Commun., 1996, 228, 328–333 CrossRef CAS PubMed.
- B. S. Chow, M. Kocan, S. Bosnyak, M. Sarwar, B. Wigg, E. S. Jones, R. E. Widdop, R. J. Summers, R. A. Bathgate, T. D. Hewitson and C. S. Samuel, Kidney Int., 2014, 86, 75–85 CrossRef CAS PubMed.
- C. S. Samuel, E. N. Unemori, I. Mookerjee, R. A. Bathgate, S. L. Layfield, J. Mak, G. W. Tregear and X. J. Du, Endocrinology, 2004, 145, 4125–4133 CrossRef CAS PubMed.
- C. S. Samuel, C. Zhao, R. A. Bathgate, C. P. Bond, M. D. Burton, L. J. Parry, R. J. Summers, M. L. Tang, E. P. Amento and G. W. Tregear, FASEB J., 2003, 17, 121–123 CAS.
- S. G. Royce, C. X. Lim, K. P. Patel, B. Wang, C. S. Samuel and M. L. Tang, Clin. Exp. Allergy, 2014, 44, 1399–1408 CrossRef CAS PubMed.
- K. Wüthrich, NMR of proteins and nucleic acids, Wiley-Interscience, New York, 1986 Search PubMed.
- L. M. Haugaard-Kedstrom, M. A. Hossain, N. L. Daly, R. A. Bathgate, E. Rinderknecht, J. D. Wade, D. J. Craik and K. J. Rosengren, ACS Chem. Biol., 2015, 10, 891–900 CrossRef CAS PubMed.
- F. L. Hisaw, Proc. Soc. Exp. Biol. Med., 1926, 23, 661 CrossRef.
- R. A. Bathgate, M. L. Halls, E. T. van der Westhuizen, G. E. Callander, M. Kocan and R. J. Summers, Physiol. Rev., 2013, 93, 405–480 CrossRef CAS PubMed.
- D. J. Scott, S. Layfield, Y. Yan, S. Sudo, A. J. Hsueh, G. W. Tregear and R. A. Bathgate, J. Biol. Chem., 2006, 281, 34942–34954 CrossRef CAS PubMed.
- E. J. Hopkins, S. Layfield, T. Ferraro, R. A. Bathgate and P. R. Gooley, J. Biol. Chem., 2007, 282, 4172–4184 CrossRef CAS PubMed.
- R. C. Kong, E. J. Petrie, B. Mohanty, J. Ling, J. C. Lee, P. R. Gooley and R. A. Bathgate, J. Biol. Chem., 2013, 288, 28138–28151 CrossRef CAS PubMed.
- A. M. Svendsen, A. Zalesko, J. Konig, M. Vrecl, A. Heding, J. B. Kristensen, J. D. Wade, R. A. Bathgate, P. De Meyts and J. Nohr, Mol. Cell. Endocrinol., 2008, 296, 10–17 CrossRef CAS PubMed.
- E. E. Bullesbach and C. Schwabe, J. Biol. Chem., 2000, 275, 35276–35280 CrossRef CAS PubMed.
- I. Mookerjee, T. D. Hewitson, M. L. Halls, R. J. Summers, M. L. Mathai, R. A. Bathgate, G. W. Tregear and C. S. Samuel, FASEB J., 2009, 23, 1219–1229 CrossRef CAS PubMed.
- R. Masterson, T. D. Hewitson, K. Kelynack, M. Martic, L. Parry, R. Bathgate, I. Darby and G. Becker, Nephrol., Dial., Transplant., 2004, 19, 544–552 CrossRef CAS.
- J. G. Baker and S. J. Hill, Trends Pharmacol. Sci., 2007, 28, 374–381 CrossRef CAS PubMed.
- V. B. Nair, C. S. Samuel, F. Separovic, M. A. Hossain and J. D. Wade, Amino acids, 2012, 43, 1131–1140 CrossRef CAS PubMed.
- A. R. Kompa, C. S. Samuel and R. J. Summers, Br. J. Pharmacol., 2002, 137, 710–718 CrossRef CAS PubMed.
- T. Dschietzig, K. Alexiou, H. T. Kinkel, G. Baumann, K. Matschke and K. Stangl, J. Card. Failure, 2011, 17, 158–166 CrossRef CAS PubMed.
- S. G. Royce, Y. R. Miao, M. Lee, C. S. Samuel, G. W. Tregear and M. L. Tang, Endocrinology, 2009, 150, 2692–2699 CrossRef CAS PubMed.
- L. M. Haugaard-Kedstrom, F. Shabanpoor, M. A. Hossain, R. J. Clark, P. J. Ryan, D. J. Craik, A. L. Gundlach, J. D. Wade, R. A. Bathgate and K. J. Rosengren, J. Am. Chem. Soc., 2011, 133, 4965–4974 CrossRef CAS PubMed.
- F. Shabanpoor, R. A. Bathgate, A. Belgi, L. J. Chan, V. B. Nair, J. D. Wade and M. A. Hossain, Biochem. Biophys. Res. Commun., 2012, 420, 253–256 CrossRef CAS PubMed.
- A. A. Claasz, C. P. Bond, R. A. Bathgate, L. Otvos, N. F. Dawson, R. J. Summers, G. W. Tregear and J. D. Wade, Eur. J. Biochem., 2002, 269, 6287–6293 CrossRef CAS PubMed.
- R. A. Bathgate, C. S. Samuel, T. C. Burazin, S. Layfield, A. A. Claasz, I. G. Reytomas, N. F. Dawson, C. Zhao, C. Bond, R. J. Summers, L. J. Parry, J. D. Wade and G. W. Tregear, J. Biol. Chem., 2002, 277, 1148–1157 CrossRef CAS PubMed.
- M. Kocan, M. Sarwar, M. A. Hossain, J. D. Wade and R. J. Summers, Br. J. Pharmacol., 2014, 171, 2827–2841 CrossRef CAS PubMed.
- J. F. Woessner Jr, Methods Enzymol., 1995, 248, 510–528 CAS.
- P. J. Ruchaya, V. R. Antunes, J. F. Paton, D. Murphy and S. T. Yao, Exp. Physiol., 2014, 99, 111–122 CrossRef CAS PubMed.
- C. S. Samuel, H. Bodaragama, J. Y. Chew, R. E. Widdop, S. G. Royce and T. D. Hewitson, Hypertension, 2014, 64, 315–322 CrossRef CAS PubMed.
- J. Temelkovski, S. P. Hogan, D. P. Shepherd, P. S. Foster and R. K. Kumar, Thorax, 1998, 53, 849–856 CrossRef CAS PubMed.
- N. R. Locke, S. G. Royce, J. S. Wainewright, C. S. Samuel and M. L. Tang, Am. J. Respir. Cell Mol. Biol., 2007, 36, 625–632 CrossRef CAS PubMed.
- C. Bartels, T. H. Xia, M. Billeter, P. Guntert and K. Wuthrich, J. Biomol. NMR, 1995, 6, 1–10 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04754d |
| This journal is © The Royal Society of Chemistry 2016 |