DOI:
10.1039/C5SC04748J
(Edge Article)
Chem. Sci., 2016,
7, 4059-4066
Regioselective phenylene-fusion reactions of Ni(II)-porphyrins controlled by an electron-withdrawing meso-substituent†
Received
9th December 2015
, Accepted 1st March 2016
First published on 1st March 2016
Abstract
Oxidation of 10,15,20-triaryl Ni(II)-porphyrins bearing an electron-withdrawing substituent at the 5-position with DDQ and FeCl3 gave 10,12- and 18,20-doubly phenylene-fused Ni(II)-porphyrins regioselectively. A doubly phenylene-fused meso-chloro porphyrin thus prepared was reductively coupled to give a meso–meso linked dimer, which was further converted to a quadruply phenylene-fused meso–meso, β–β, β–β triply linked Zn(II)–diporphyrin via inner-metal exchange followed by oxidation with DDQ and Sc(OTf)3. As compared to the usual meso–meso, β–β, β–β triply linked Zn(II)-diporphyrin, this π-extended porphyrin dyad exhibits a smaller HOMO–LUMO gap and a larger two-photon absorption cross-section.
Introduction
Porphyrins having extended π-electronic networks have been attracting considerable attention because of their applications such as organic semiconductors, near-infrared dyes, and nonlinear optical materials.1 Among such π-extended porphyrins, 7,8-dehydropurpurins and their families, which have a double bond across the meso- and β-positions and thus contain a peri-fused five-membered ring directly onto the periphery, display largely altered absorption spectra, small HOMO–LUMO gaps, and weakened aromaticity.2–4 These intriguing properties can be ascribed to their π-extended electronic networks involving a pseudo 20π-antiaromatic electronic circuit.
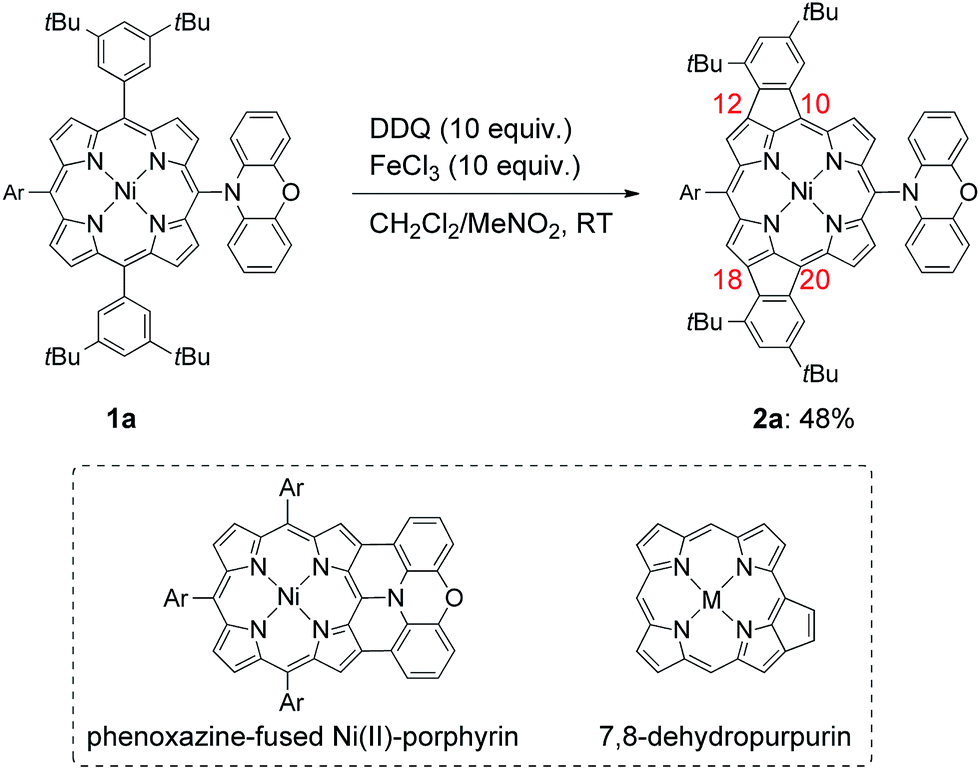
Recently, we reported that the oxidation of meso-phenoxazine-substituted Ni(II)-porphyrin 1a with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) and FeCl3 unexpectedly gave doubly 10,12- and 18,20-phenylene-fused Ni(II)-porphyrin 2a instead of phenoxazine-fused Ni(II)-porphyrin (Scheme 1).5 Doubly phenylene-fused porphyrins such as 2a are attractive pigments in view of altered optical and electronic properties as compared with mono phenylene-fused 7,8-dehydropurpurin derivatives. In addition, their symmetric structures are suitable for further oligomerizations and functionalizations. Despite these potentials, the mechanism and scope of this double fusion reaction is not well understood.
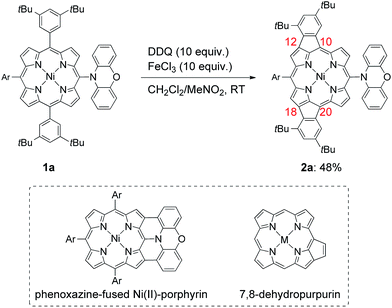 |
| | Scheme 1 Synthesis of 10,12- and 18,20-phenylene-fused meso-phenoxazino Ni(II)-porphyrin 2a, and structures of phenoxazine-fused porphyrin and 7,8-dehydropurpurin. Ar = 3,5-di-tert-butylphenyl. | |
Phenylene-fused 7,8-dehydropurpurins have been often prepared by Pd-catalyzed cyclizations via C–H activations of meso-(2-haloaryl)-substituted porphyrins or β-bromo-meso-aryl-substituted porphyrins.2 In contrast, syntheses of phenylene-fused 7,8-dehydropurpurins upon oxidation have remained rather rare.4 In this sense, the formation of 2a is interesting and thus worth understanding further. In addition, the observed regioselectivity looks curious, considering that seven regioisomers are possible for doubly fusion products (ESI, Scheme S1†). In the oxidation of 1a, the first oxidation at 0.34 V (vs. Fc/Fc+) should occur at the meso-phenoxazine group, since it is easier to oxidize than the Ni(II)-porphyrin core.5 We thought that the one-electron oxidized phenoxazine moiety might serve just as an electron-withdrawing group to the Ni(II)-porphyrin, helping the regioselective fusion reactions at the 12- and 18-positions. In order to check this postulate, we examined the oxidative fusion reactions of Ni(II)-porphyrins bearing an electron-withdrawing substituent at the 5-position.
Results and discussion
Oxidative fusion reactions of meso-functionalized porphyrins
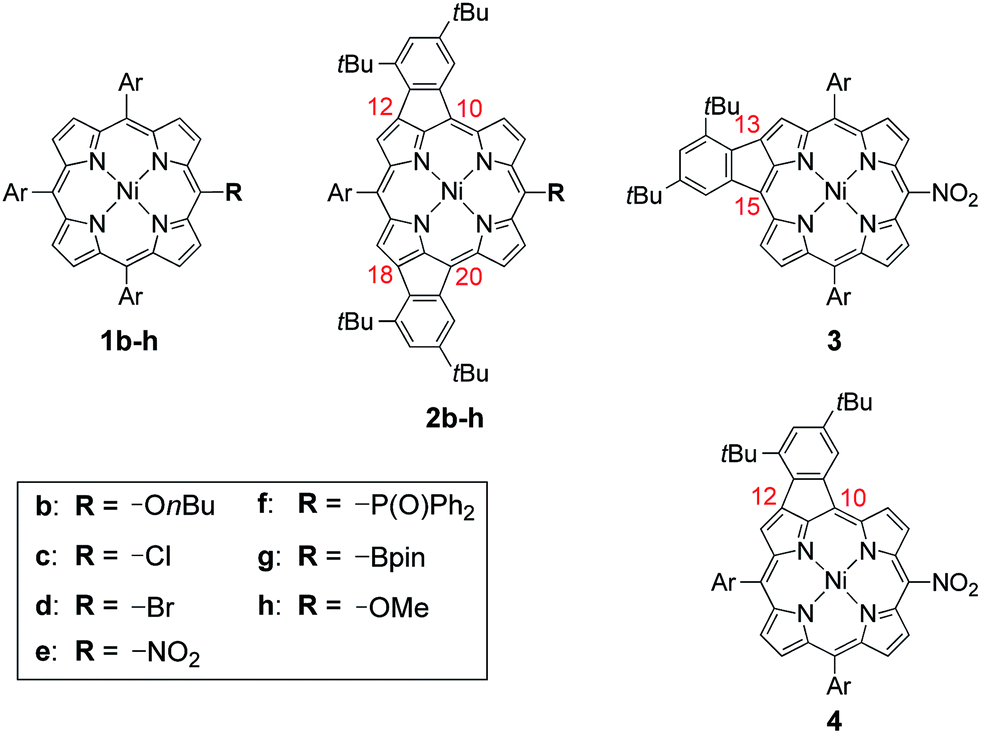
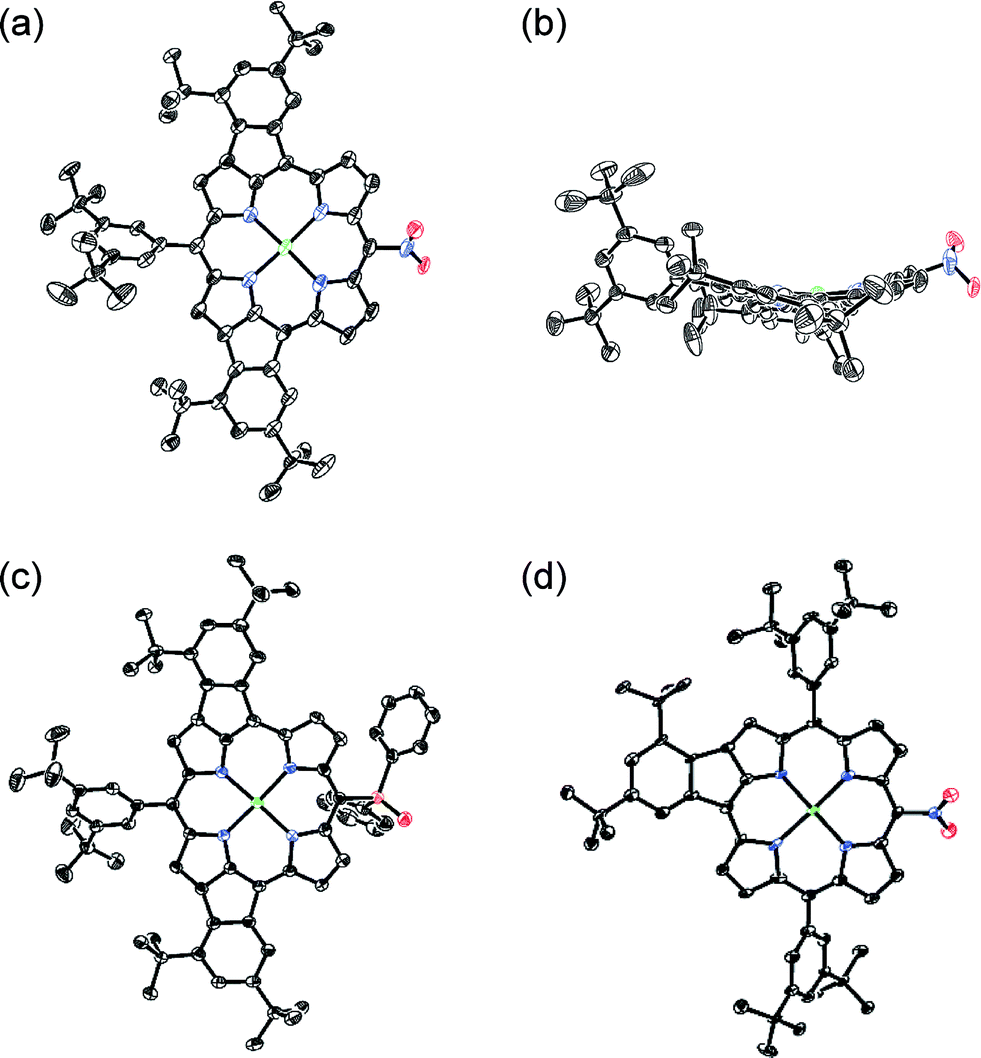
Oxidative fusion reactions of porphyrins 1b–f were examined with 10 equivalents of DDQ and FeCl3 in a mixture of CH2Cl2 and MeNO2 (Scheme 2). Electron-rich substrate 1b6 was readily decomposed under these conditions. Weakly electron-deficient porphyrins 1c and 1d gave complicated mixtures consisting of various fused isomers. More electron-deficient 5-nitroporphyrin 1e and 5-diphenylphosphinylporphyrin 1f7 afforded the corresponding doubly phenylene-fused porphyrins 2e and 2f regioselectively in 29 and 61% yields, respectively, while singly phenylene-fused porphyrin 3 was isolated as a side product in 18% yield in the reaction of 1e. The structures of 2e, 2f and 3 were all unambiguously determined by X-ray diffraction analyses (Fig. 1).8 In 2e and 2f, the aryl groups at the 10- and 20-positions were fused onto the porphyrin periphery, as is the case of 2a. The preferential formations of 2e and 2f suggest that a strongly electron-withdrawing meso-substituent plays an important role in the regioselective fusion reactions.
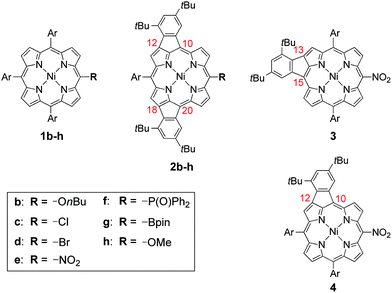 |
| | Scheme 2 Structures of 1b–h, 2b–h, 3 and 4. Ar = 3,5-di-tert-butylphenyl. | |
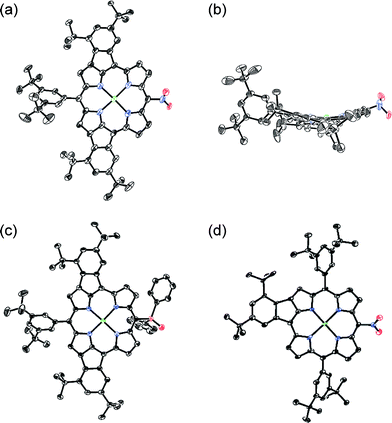 |
| | Fig. 1 X-Ray crystal structures. (a) Top view and (b) side view of 2e, (c) top view of 2f, and (d) top view of 3. Thermal ellipsoids are drawn at the 50% probability level. Solvent molecules and all hydrogen atoms are omitted for clarity. | |
Investigation into the regioselectivity
Recently, Kadish and Gryko et al. proposed, on the basis of detailed electrochemical studies, that the oxidative fusion reaction of meso-naphthylporphyrin might occur via one-electron oxidations of both the porphyrin and naphthalene moieties followed by an intramolecular radical–radical coupling between generated porphyrinyl radical cation and naphthyl radical cation.9,10 In this intramolecular radical coupling mechanism, the spin density of the porphyrinyl radical cation should govern the selectivity.
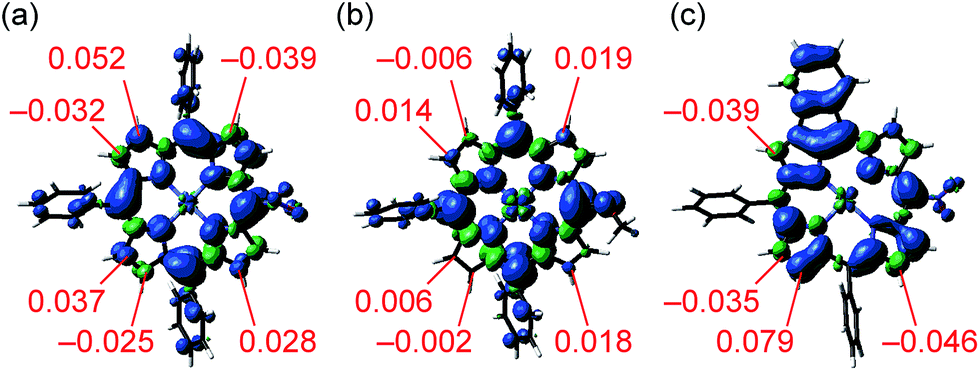
To find a clue about the regioselectivity, we performed density functional theory (DFT) calculations.11 Spin density maps of radical cations of 1e and 1h were calculated at the B3LYP/6-311G* level using the Gaussian 09 package12 (Fig. 2a and b). 5-Substituted porphyrins possess three possible reaction sites to connect with meso-phenyl groups, namely 8-, 12- and 13-positions. In the case of 5-nitroporphyrin 1e, the largest spin density value of 0.052 was calculated at the 12-position. On the other hand, in the case of 5-methoxyporphyrin 1h, spin density values at the possible reaction sites are almost similar. These results accord with our experimental observation that a strong electron-withdrawing group promoted the regioselective fusion of meso-phenyl groups at the 12- and 18-positions. Moreover, we also calculated the spin density of singly 10,12-phenylene-fused 5-nitroporphyrin 4, which is a possible intermediate to provide doubly 10,12- and 18,20-phenylene-fused porphyrin 2e (Fig. 2c). Curiously, compared to the case of radical cation of 1e, the difference of spin density values between 12- and other positions became more distinguishable, which might imply that electronic perturbation by the fused phenylene unit urged the opposite meso-phenyl group to react with the 18-position.
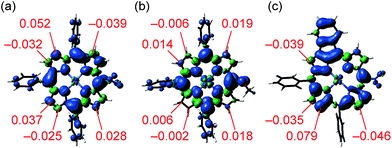 |
| | Fig. 2 Total spin density of radical cations of (a) 1e, (b) 1h and (c) 4 obtained by DFT calculations at the B3LYP/6-311G* level (isovalue: 0.001). tert-Butyl groups were replaced with hydrogen atoms to simplify the calculations. | |
Another possible mechanism, nucleophilic attack of the peripheral aryl group to the cationic porphyrin core, has been proposed.13 According to this mechanism, we also performed DFT calculations of dications of 1e and 1h. However, both in the case of radical cations and dications of 1e and 1h, the obtained molecular orbitals or charge distributions showed no clear difference at the 8-, 12- and 13-positions to explain the regioselectivity (ESI, Fig. S46–S50†). From the point of view of these calculations, an intramolecular radical–radical coupling mechanism seems plausible for our phenylene-fusion reactions.
It is of note to mention that numerous studies have proven that electron-rich aromatic compounds easily undergo the oxidative fusion reactions.13 In the case of small aromatic compounds, the presence of an electron-withdrawing group decreases the HOMO level to prevent the initial oxidation. On the other hand, for large aromatic compounds including porphyrin, such a decrease of the HOMO will be too negligible to stop the oxidative fusion reactions. In our reactions, electron-withdrawing groups might strongly reorganize the overall electron-density to make the 12-position relatively reactive.
Synthesis of a meso-chloro doubly phenylene-fused Ni(II)-porphyrin
In the next step, we chose a pinacolatoboryl group as an interconvertible electron-withdrawing substituent (Scheme 3). The reaction of 5-pinacolatoborylporphyrin 1g14 afforded doubly phenylene-fused porphyrin 2g with partial production of meso-chloroporphyrin 2c. Thus, we subjected this crude reaction mixture of 2c and 2g directly to Cu-mediated chlorination conditions,15 which allowed for isolation of 2c in 37% yield. It is of note that 2c is a useful precursor for further fabrications but is inaccessible through the direct oxidation of 1c.
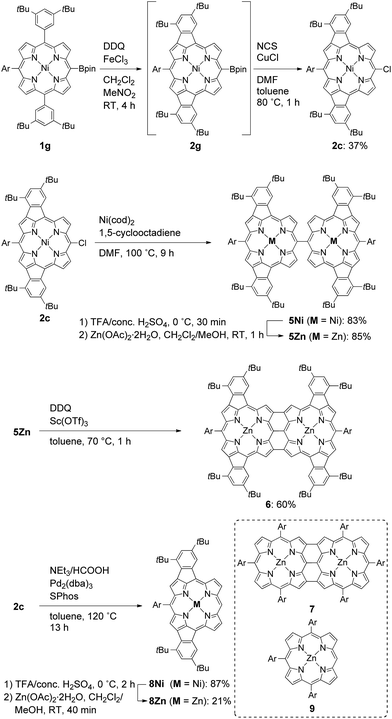 |
| | Scheme 3 Synthesis of quadruply phenylene-fused meso–meso, β–β, β–β triply linked Zn(II)-diporphyrin 6 and structure of meso–meso, β–β, β–β triply linked Zn(II)-diporphyrin 7. Ar = 3,5-di-tert-butylphenyl, NCS = N-chlorosuccinimide, cod = 1,5-cyclooctadiene, dba = dibenzylideneacetone, SPhos = 2-(2′,6′-dimethoxybiphenyl)dicyclohexylphosphine. | |
Synthesis of a quadruply phenylene-fused meso–meso, β–β, β–β triply linked Zn(II)-diporphyrin
With 2c in hand, we envisioned the synthesis of quadruply phenylene-fused meso–meso, β–β, β–β triply linked Zn(II)-diporphyrin 6 as a new π-extended meso–meso, β–β, β–β triply linked porphyrin dyad. The synthetic route to dimer 6 is shown in Scheme 3. Ni(0)-mediated reductive homo-coupling16 of 2c furnished meso–meso linked diporphyrin 5Ni in 83% yield. Removal of the central nickel atoms of 5Ni with concentrated sulfuric acid in trifluoroacetic acid (TFA) proceeded cleanly at 0 °C, and subsequent zincation afforded 5Zn in 85% yield in two steps. Finally, the oxidative fusion reaction of 5Zn with DDQ and Sc(OTf)317 gave 6 in 60% yield. Doubly phenylene-fused Zn(II)-porphyrin monomer 8Zn was synthesized as a reference compound by dechlorination of 2c followed by removal of the nickel and zinc-metalation.
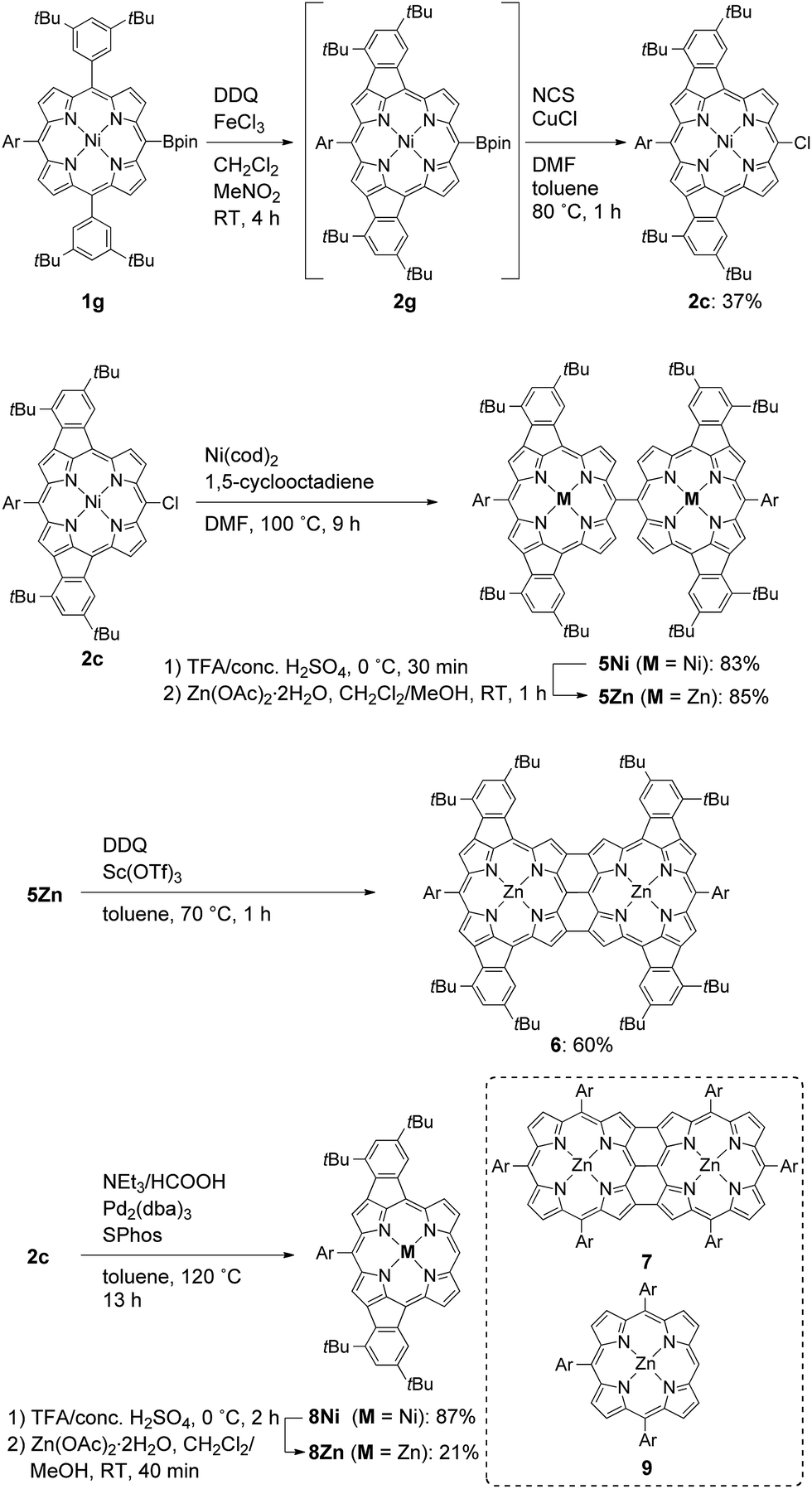
The atmospheric pressure chemical ionization time-of-flight mass spectra of 5Zn and 6 displayed the parent ion peaks at 1862.9249 (calcd for C124H134N864Zn2, m/z 1862.9320 [M]−) and 1858.8967 (calcd for C124H130N864Zn2, m/z 1858.9007 [M]−), respectively, indicating that four hydrogen atoms were removed during the oxidative fusion reaction from 5Zn to 6. The 1H NMR spectrum of 6 in pyridine-d5 showed six signals at 8.19, 8.06, 8.00, 7.86, 7.33 and 7.29 ppm in the aromatic region. The structure of 6 has been revealed by X-ray crystallographic analysis to be fairly coplanar with a small mean plane deviation (MPD) of 0.102 Å (Fig. 3).18,19 The Cmeso–Cmeso bond length is 1.489(3) Å, and the two Cβ–Cβ bond lengths are both 1.442(3) Å. These structural features are similar to those of previously reported porphyrin dyad 7.17f
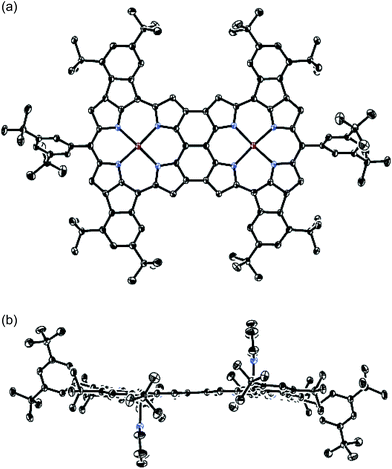 |
| | Fig. 3 X-Ray crystal structure of 6. (a) Top view and (b) side view. Thermal ellipsoids are drawn at the 50% probability level. Solvent molecules and all hydrogen atoms are omitted for clarity. In the top view, pyridine ligands on the central Zn atoms are also omitted for clarity. | |
UV/vis absorption spectra and density functional theory calculations
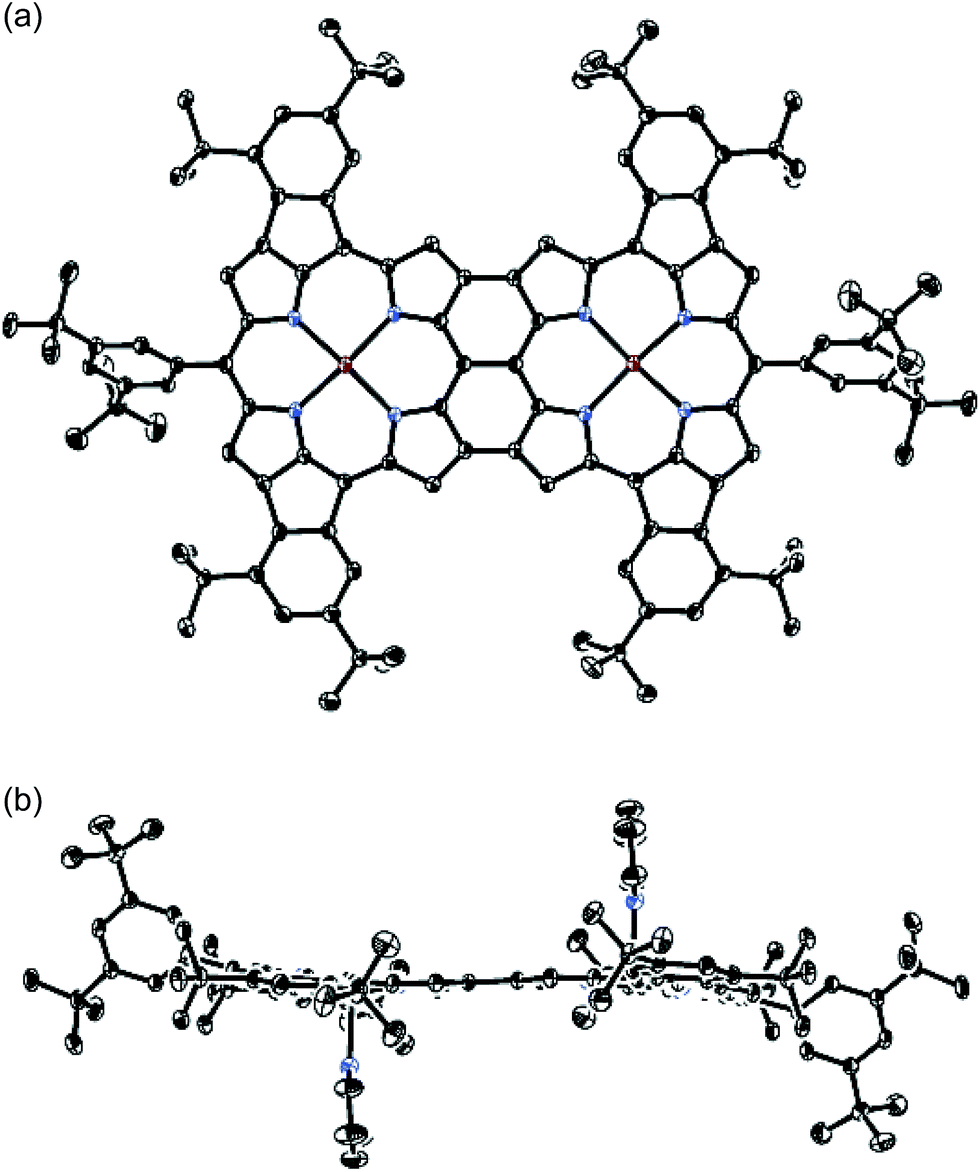
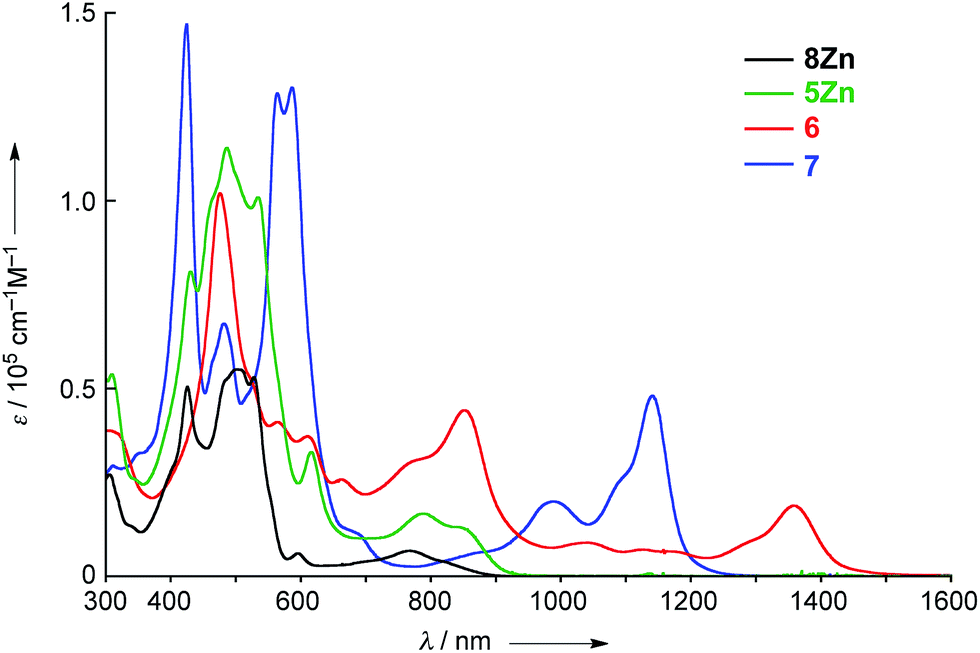
The UV/vis/NIR absorption spectra of 8Zn, 5Zn, 6 and 7 are shown in Fig. 4. CH2Cl2 containing 1% of pyridine was employed as a solvent to increase the solubility of these compounds by the axial coordination of pyridine to the central Zn atom. As compared with normal Zn(II)-porphyrins, doubly phenylene-fused porphyrin 8Zn exhibits a red-shifted and broad absorption spectrum, probably due to the π-extension and pseudo 20π-antiaromatic contribution. In line with this, the anisotropy of the induced current density (ACID) plot20 around the fused five-membered ring of 8Zn was calculated to be counterclockwise (Fig. 5). In addition, the NICS(0) value21 at the center of the fused five-membered ring was calculated to be a large positive value of +17.0 at the B3LYP/6-31(d) level using the Gaussian 09 package.12 The meso–meso linked dimer 5Zn shows an absorption spectrum which is slightly red-shifted, but otherwise similar to that of 8Zn, indicating small electronic interaction between the two doubly phenylene-fused porphyrin units. In contrast, the absorption spectrum of 6 is strongly altered and red-shifted, showing bands at 476, 851 and 1358 nm. The Q-like band at 1358 nm in 6 is red-shifted by 217 nm (1.4 × 103 cm−1) as compared with that of 7 at 1141 nm, as an indication of a more π-extended network in 6. Interestingly, 6 shows a characteristic absorption band at 851 nm, while such a band was not observed in other π-extended meso–meso, β–β, β–β triply linked diporphyrins.22
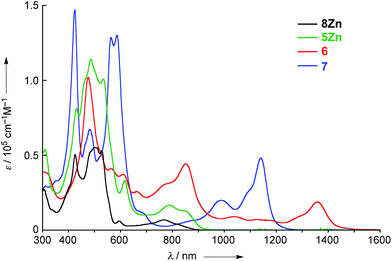 |
| | Fig. 4 UV/vis/NIR absorption spectra of 8Zn, 5Zn, 6 and 7 in CH2Cl2 containing 1% pyridine. ε = molar extinction coefficient. | |
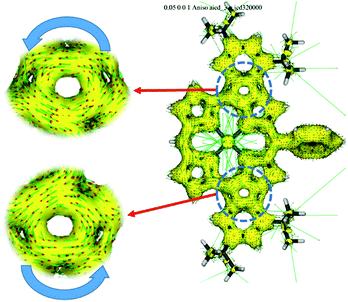 |
| | Fig. 5 ACID plot for 8Zn at an isosurface value of 0.05 at the B3LYP/6-31(d) level. | |
Time-dependent density functional theory (TD-DFT) calculations on 6 and 7 were performed at the B3LYP/6-31G*(C,H,N) + LANL2DZ(Zn) level using Gaussian 09 package12 (See ESI). The TD-DFT calculations roughly reproduced the absorption spectra of 6 and 7. The calculations suggested that the absorption at 851 nm in 6 is mainly ascribed to the transition from the HOMO−1 to LUMO+1. The same transition was calculated around 600 nm for 7. This difference may be ascribed to the largely stabilized LUMO+1 in 6 through the interaction with the fused five-membered rings.
Electrochemical properties
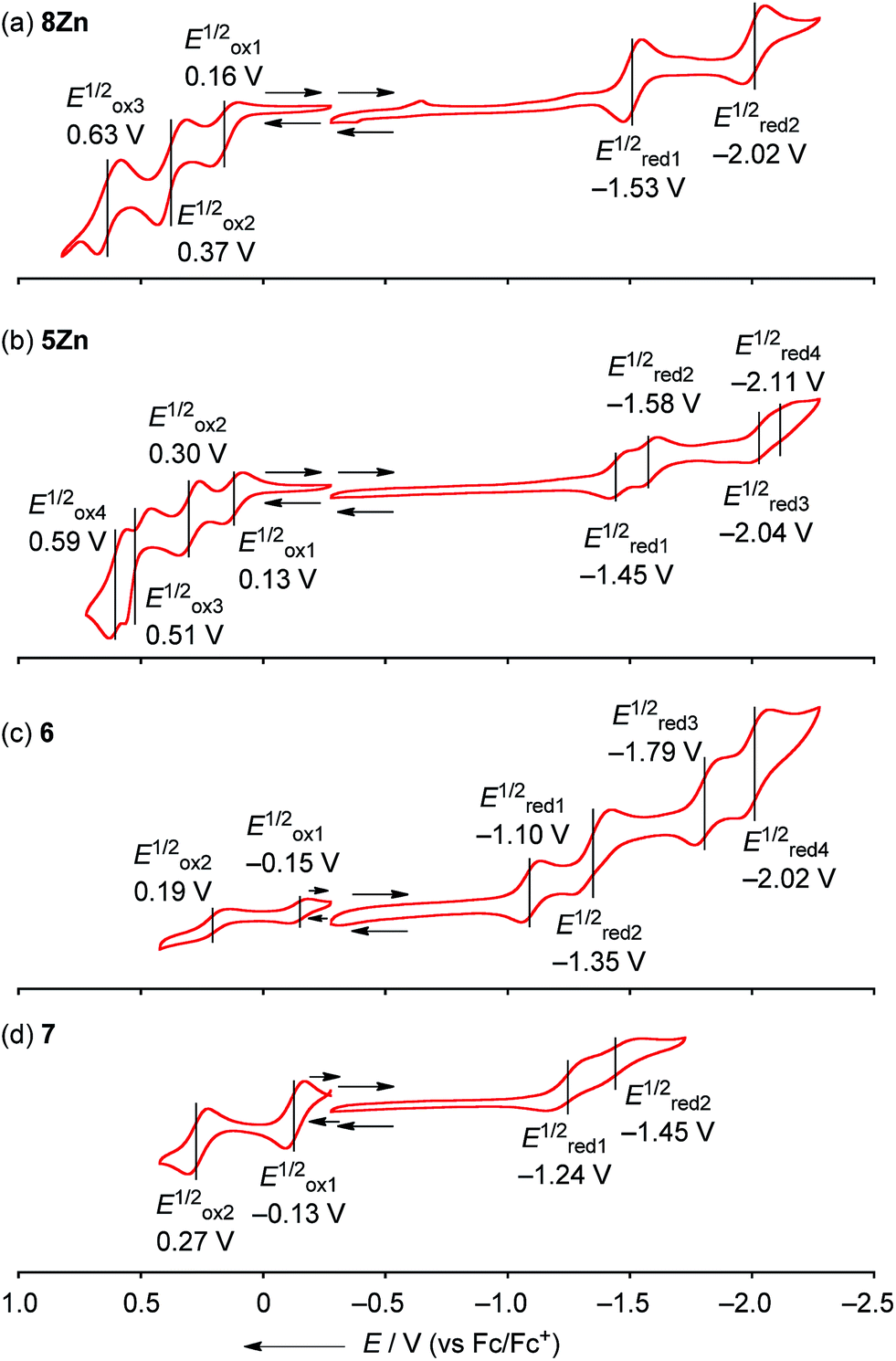
The electrochemical properties of 8Zn, 5Zn, 6 and 7 were studied by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in CH2Cl2 containing 1% pyridine with 0.1 M nBu4NPF6 as a supporting electrolyte (Fig. 6). Doubly phenylene-fused porphyrin monomer 8Zn showed three reversible oxidation waves at 0.16, 0.37 and 0.63 V, and two reversible reduction waves at −1.53 and −2.02 V. The electrochemical HOMO–LUMO gap ΔE (Eox11/2 − Ered11/2) of 8Zn was calculated to be 1.69 eV. This ΔE value is much smaller than that of triaryl Zn(II)-porphyrin 9 (2.22 eV),23 which can be ascribed to the effective π-extension caused by the pseudo 20π-antiaromatic contribution.
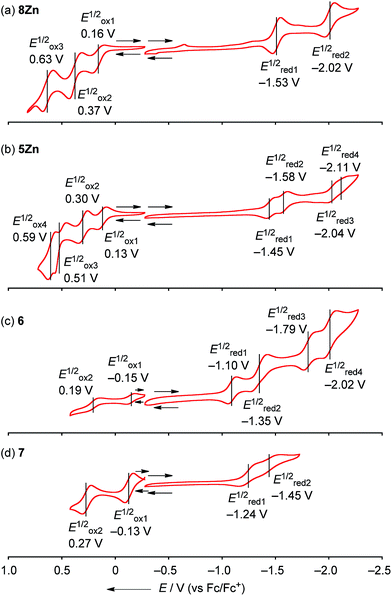 |
| | Fig. 6 Cyclic voltammograms of (a) 8Zn, (b) 5Zn, (c) 6 and (d) 7. The redox potentials were measured by cyclic voltammetry in anhydrous CH2Cl2 containing 1% pyridine with 0.1 M nBu4NPF6 as a supporting electrolyte, and Ag/AgNO3 as a reference electrode. Ferrocene/ferrocenium ion couple was used as an external reference. | |
The corresponding meso–meso linked dimer 5Zn displayed four reversible oxidation waves at 0.13, 0.30, 0.51 and 0.59 V, and four reversible reduction waves at −1.45, −1.58, −2.04 and −2.11 V. The first and second reduction waves can be assigned as split first reduction waves (one electron per porphyrin unit) as judged from the results of other electronically coupled diporphyrins.24 The ΔE value of 5Zn was calculated to be 1.58 eV, which is very similar to that of 8Zn, suggesting little electronic communication between the two phenylene-fused porphyrin units. On the other hand, quadruply phenylene-fused meso–meso, β–β, β–β triply linked Zn(II)-diporphyrin 6 showed remarkably perturbed potentials. 6 exhibited two reversible oxidation waves at −0.15 and 0.19 V, and four reversible reduction waves at −1.10, −1.35, −1.79 and −2.02 V. Then, 7 showed two reversible oxidation waves at −0.13 and 0.27 V, and two reversible reduction waves at −1.24 and −1.45 V. The first reduction potential of 6 showed a large positive shift of 0.14 V compared to that of 7, while the first oxidation potential displayed a small negative shift of 0.02 V. The ΔE value of 6 is calculated to be 0.95 V. It is apparently smaller than that of meso–meso, β–β, β–β triply linked Zn(II)-diporphyrin 7 (1.11 eV), indicating the effective π-delocalization over the fused phenylene units. These results indicate that introduction of fused phenylene units is effective to enhance the electron-accepting property, which is in accord with the calculated molecular orbital diagrams of 6 and 7 (ESI, Fig. S41†). The obtained ΔE values for 8Zn, 5Zn, 6 and 7 were almost close in value to the observed optical gaps.
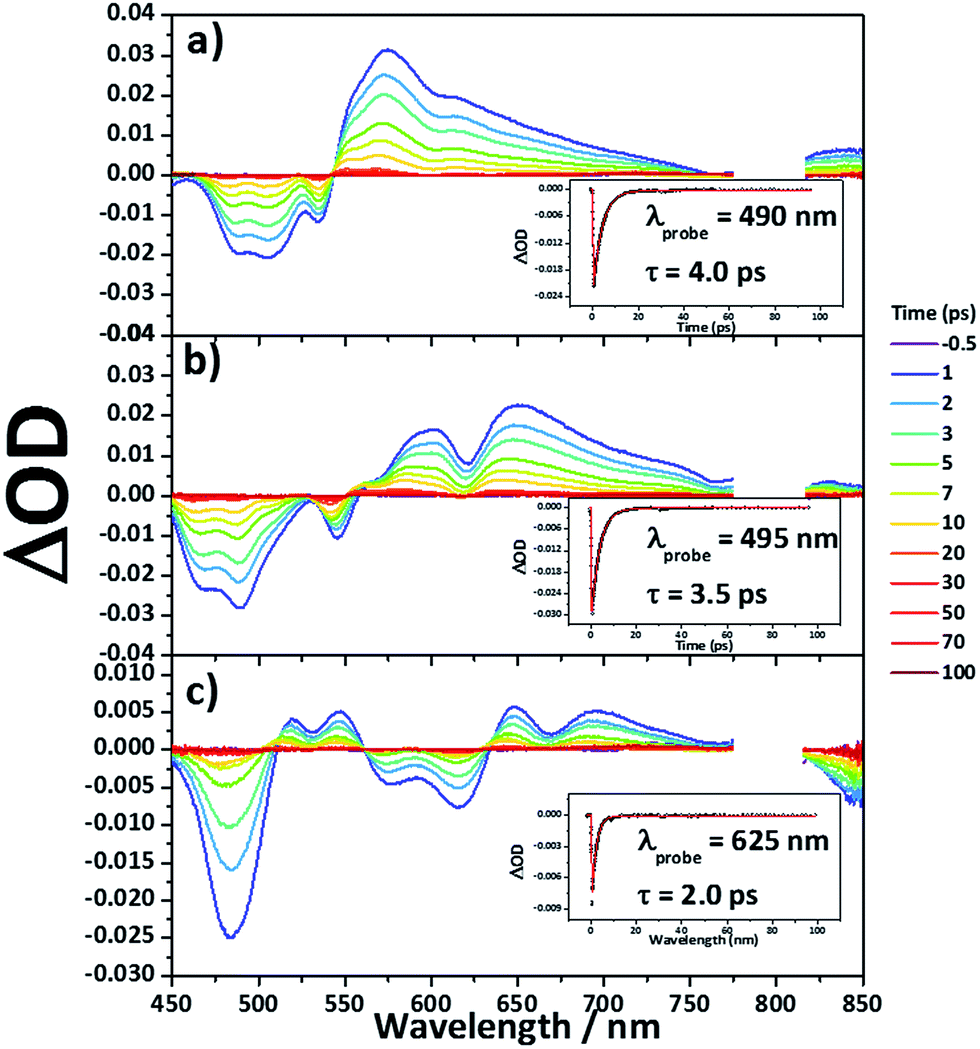
Femtosecond transient absorption measurements
Femtosecond transient absorption measurements were carried out for 8Zn, 5Zn and 6 in toluene containing 1% of pyridine (Fig. 7). Excitation wavelengths are 800 nm for 8Zn and 5Zn, and 1350 nm for 6, which correspond to the lowest Q-like bands. All the compounds exhibit single exponential decay profiles of the ground-state-bleaching signals (4.0, 3.5 and 2.0 ps, respectively). Compound 7 shows a similar decay time of 4.5 ps.25 The observed short S1-state lifetime of 8Zn may be ascribed to the pseudo 20π-antiaromatic contribution of 7,8-dehydropurpurin-like structure2e and accelerated non-radiative relaxation associated the narrow energy gap. The decay constant of 5Zn is very similar to that of 8Zn due to small interaction between the two doubly phenylene-fused porphyrin cores. An observation that the S1-state lifetime of 6 is shorter than that of 7 can be interpreted in terms of the reduced HOMO–LUMO gap arising from the extended π-conjugation and contribution of 20π-antiaromaticity.26
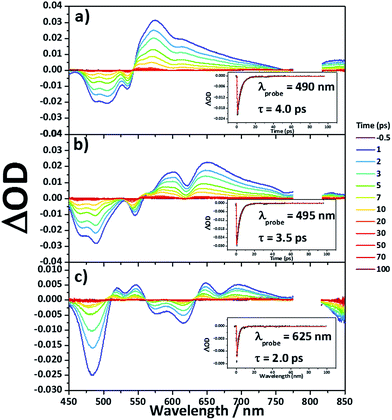 |
| | Fig. 7 Femtosecond transient absorption spectra and decay profiles (inset) of (a) 8Zn, (b) 5Zn and (c) 6 in toluene containing 1% of pyridine with photoexcitation at 800 nm (for 8Zn and 5Zn), and 1350 nm (for 6). | |
Two-photon absorption properties
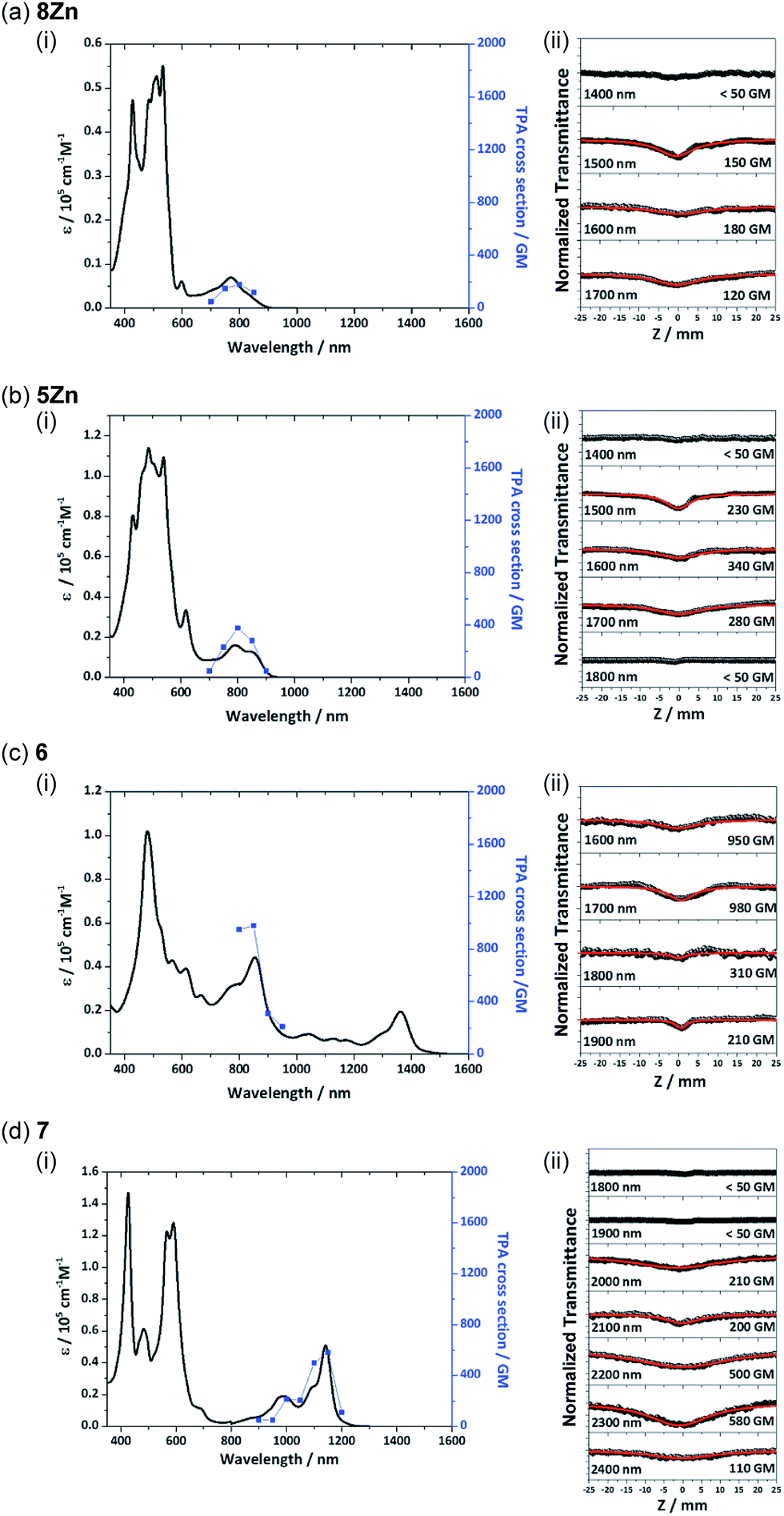
Recently, π-conjugated organic molecules have attracted much attention as soft processable nonlinear optical materials in light of their possible applications such as for photodynamic therapy.27 It has been demonstrated that π-extended porphyrins exhibit large nonlinear optical responses.1e,g,22d Therefore, two-photon absorption (TPA) properties of 8Zn, 5Zn, 6 and 7 were comparatively examined by using an open-aperture Z-scan method in the wavelength range of 1400–2400 nm, where one-photon absorption contributions are negligible (Fig. 8). The TPA cross-section maxima (δ(2)max) were determined to be 180 GM for 8Zn (λex = 1600 nm), 340 GM for 5Zn (λex = 1600 nm), 980 GM for 6 (λex = 1700 nm) and 580 GM for 7 (λex = 2300 nm), respectively. The TPA value of 8Zn is larger than that of usual porphyrin monomer (<100 GM),28 which can be ascribed to the π-extended structure. The observed larger TPA value of 6 as compared with 7 again indicates its extended π-conjugation.
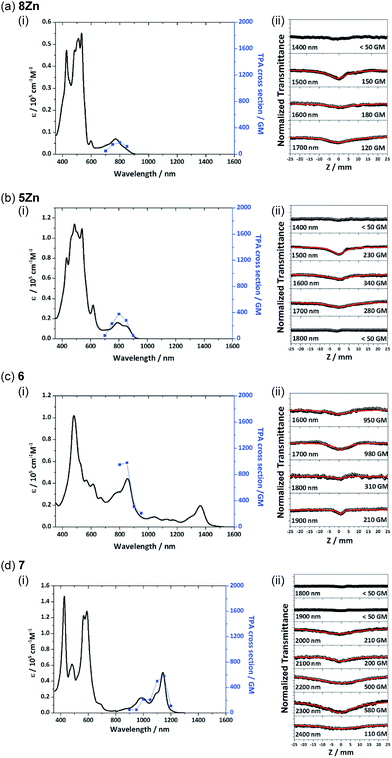 |
| | Fig. 8 (i) One-photon absorption (black) and TPA spectra (blue) and (ii) Z-scan curves of (a) 8Zn, (b) 5Zn, (c) 6 and (d) 7. The TPA spectra are displayed at λex/2 for comparison with the OPA spectra. | |
Conclusions
In summary, the oxidations of porphyrins bearing strongly electron-withdrawing substituents at the 5-position with DDQ and FeCl3 resulted in the selective fusion of the 10- and 20-aryl groups at the 12- and 18-positions, indicating that the electron-withdrawing group dictated the unique selectivity. Doubly phenylene-fused meso-chloroporphyrin 2c was converted to quadruply phenylene-fused meso–meso, β–β, β–β triply linked Zn(II)-diporphyrin 6, which displays red-shifted absorption bands in the near-infrared region, smaller optical and electrochemical HOMO–LUMO gap, and a larger TPA value as compared to the usual meso–meso, β–β, β–β triply linked Zn(II)-diporphyrin 7. These features may be ascribed to its extended π-conjugation and pseudo 20π-antiaromatic contribution. More elaborate systems based on multiply phenylene-fused porphyrins are now actively being pursued in our laboratory.
Acknowledgements
The work at Kyoto University was supported by Grants-in-Aid from MEXT (No.: 25107002 “Science of Atomic Layers”), from JSPS (No.: 25220802 (Scientific Research (S)), 24685007 (Young Scientists (A)), 26620081 (Exploratory Research)), and from ACT-C, JST. N. F. acknowledges a JSPS Fellowship for Young Scientists. H. Y. acknowledges Kansai Research Foundation for Technology Promotion and Asahi Glass Foundation for financial support. The work at Yonsei University was supported by Global Research Laboratory (2013K1A1A2A02050183) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT (Information and Communication Technologies) and Future Planning. The quantum calculations of ACID and NICS values were performed using the supercomputing resources of the Korea Institute of Science and Technology Information (KISTI).
Notes and references
-
(a) H. L. Anderson, Chem. Commun., 1999, 2323 RSC
 ;
(b) M. Craça, H. Vicente, L. Jaquinod and K. M. Smith, Chem. Commun., 1999, 1771 Search PubMed ;
(c) J. R. Reimers, N. S. Hush and M. J. Crossley, J. Porphyrins Phthalocyanines, 2002, 6, 795 CrossRef CAS ;
(d) S. Fox and R. W. Boyle, Tetrahedron, 2006, 62, 10039 CrossRef CAS ;
(e) N. Aratani, D. Kim and A. Osuka, Chem.–Asian J., 2009, 4, 1172 CrossRef CAS PubMed ;
(f) C. Jiao and J. Wu, Synlett, 2012, 23, 171 CAS ;
(g) J. P. Lewtak and D. T. Gryko, Chem. Commun., 2012, 48, 10069 RSC ;
(h) A. M. V. M. Pereira, S. Richeter, C. Jeandon, J.-P. Gisselbrecht, J. Wytko and R. Ruppert, J. Porphyrins Phthalocyanines, 2012, 16, 464 CrossRef CAS ;
(i) H. Mori, T. Tanaka and A. Osuka, Chem.–Asian J., 2013, 1, 2500 CAS ;
(j) T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943 RSC .
;
(b) M. Craça, H. Vicente, L. Jaquinod and K. M. Smith, Chem. Commun., 1999, 1771 Search PubMed ;
(c) J. R. Reimers, N. S. Hush and M. J. Crossley, J. Porphyrins Phthalocyanines, 2002, 6, 795 CrossRef CAS ;
(d) S. Fox and R. W. Boyle, Tetrahedron, 2006, 62, 10039 CrossRef CAS ;
(e) N. Aratani, D. Kim and A. Osuka, Chem.–Asian J., 2009, 4, 1172 CrossRef CAS PubMed ;
(f) C. Jiao and J. Wu, Synlett, 2012, 23, 171 CAS ;
(g) J. P. Lewtak and D. T. Gryko, Chem. Commun., 2012, 48, 10069 RSC ;
(h) A. M. V. M. Pereira, S. Richeter, C. Jeandon, J.-P. Gisselbrecht, J. Wytko and R. Ruppert, J. Porphyrins Phthalocyanines, 2012, 16, 464 CrossRef CAS ;
(i) H. Mori, T. Tanaka and A. Osuka, Chem.–Asian J., 2013, 1, 2500 CAS ;
(j) T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943 RSC .
-
(a) S. Fox and R. W. Boyle, Chem. Commun., 2004, 1322 RSC ;
(b) D.-M. Shen, C. Liu and Q.-Y. Chen, Chem. Commun., 2005, 4982 RSC ;
(c) S. Hayashi, Y. Matsubara, S. Eu, H. Hayashi, T. Umeyama, Y. Matano and H. Imahori, Chem. Lett., 2008, 37, 846 CrossRef CAS ;
(d) T. D. Lash, B. E. Smith, M. J. Melquist and B. A. Godfrey, J. Org. Chem., 2011, 76, 5335 CrossRef CAS PubMed ;
(e) Y. Mitsushige, S. Yamaguchi, B. S. Lee, Y. M. Sung, S. Kuhri, C. A. Schierl, D. M. Guldi, D. Kim and Y. Matsuo, J. Am. Chem. Soc., 2012, 134, 16540 CrossRef CAS PubMed ;
(f) T. Ishizuka, Y. Saegusa, Y. Shiota, K. Ohtake, K. Yoshizawa and T. Kojima, Chem. Commun., 2013, 49, 5939 RSC .
-
(a) A. Nakano, N. Aratani, H. Furuta and A. Osuka, Chem. Commun., 2001, 1920 RSC ;
(b) A. K. Sahoo, S. Mori, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 7972 CrossRef CAS PubMed ;
(c) N. Fukui, H. Yorimitsu, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 4395 CrossRef CAS PubMed ;
(d) N. Fukui, S. Arai, H. Shinokubo, H. Yorimitsu and A. Osuka, Heterocycles, 2015, 90, 252 CrossRef .
-
(a) H. S. Gill, M. Harmjanz, J. Satamaria, I. Finger and M. J. Scott, Angew. Chem., Int. Ed., 2004, 43, 485 CrossRef CAS PubMed ;
(b) J. H. Heo, T. Ikeda, J. M. Lim, N. Aratani, A. Osuka and D. Kim, J. Phys. Chem. B, 2010, 114, 14528 CrossRef CAS PubMed ;
(c) K. Ota, T. Tanaka and A. Osuka, Org. Lett., 2014, 16, 2974 CrossRef CAS PubMed .
- N. Fukui, W.-Y. Cha, S. Lee, S. Tokuji, D. Kim, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2013, 52, 9728 CrossRef CAS PubMed .
- Q. Chen, Y.-Z. Zhu, Q.-J. Fan, S.-C. Zhang and J.-Y. Zheng, Org. Lett., 2014, 16, 1590 CrossRef CAS PubMed .
- Y. Matano, K. Matsumoto, Y. Terasaka, H. Hotta, Y. Araki, O. Ito, M. Shiro, T. Sasamori, N. Tokitoh and H. Imahori, Chem.–Eur. J., 2007, 13, 891 CrossRef CAS PubMed .
- Crystallographic data for 2e: C64H67N5O2Ni·2CHCl3, orthorhombic, space group Pbcn (no. 60), a = 11.0983(14), b = 34.992(5), c = 32.965(4) Å, V = 12802(3) Å3, T = 123 K, Z = 8, R1 = 0.0921 (I > 2σ(I)), Rw = 0.2889 (all data), GOF = 1.047. CCDC number: 1433487. Crystallographic data for 2f: C74H77N4OPNi·C6H5CH3·1.5CH3CH2OH, monoclinic, space group P21/n (no. 14), a = 12.3714(16), b = 28.233(3), c = 41.360(6) Å, β = 96.168(4)°, V = 14363(3) Å3, T = 123 K, Z = 8, R1 = 0.0739 (I > 2σ(I)), Rw = 0.2350 (all data), GOF = 1.036. CCDC number: 1433489. Crystallographic data for 3: C62H69N5O2Ni·C6H5CH3, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 14.734(6), b = 20.554(8), c = 20.788(9) Å, α = 90.073(8), β = 103.209(15), γ = 109.518(5)°, V = 5756(4) Å3, T = 123 K, Z = 2, R1 = 0.1419 (I > 2σ(I)), Rw = 0.3713 (all data), GOF = 1.097. CCDC number: 1433488.
(no. 2), a = 14.734(6), b = 20.554(8), c = 20.788(9) Å, α = 90.073(8), β = 103.209(15), γ = 109.518(5)°, V = 5756(4) Å3, T = 123 K, Z = 2, R1 = 0.1419 (I > 2σ(I)), Rw = 0.3713 (all data), GOF = 1.097. CCDC number: 1433488.
- Y. Fang, D. Koszelewski, K. M. Kadish and D. T. Gryko, Chem. Commun., 2014, 50, 8864 RSC .
- O. Yamane, K.-i. Sugiura, H. Miyasaka, K. Nakamura, T. Fujimoto, K. Nakamura, T. Kaneda, Y. Sakata and M. Yamashita, Chem. Lett., 2004, 33, 40 CrossRef CAS .
-
(a) M. Gałęzowski and D. T. Gryko, J. Org. Chem., 2006, 71, 5942 CrossRef PubMed ;
(b) K. Oda, S. Hiroto and H. Shinokubo, Chem. Lett., 2014, 43, 1444 CrossRef CAS .
-
M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed . The full list of the authors is given in ESI.†.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900 CrossRef CAS PubMed .
- A. G. Hyslop, M. A. Kellett, P. M. Iovine and M. J. Therien, J. Am. Chem. Soc., 1998, 120, 12676 CrossRef CAS .
-
(a) J. M. Murphy, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434 CrossRef CAS PubMed ;
(b) H. Wu and J. Jr. Hynes, Org. Lett., 2010, 12, 1192 CrossRef CAS PubMed ;
(c) K. Fujimoto, H. Yorimitsu and A. Osuka, Org. Lett., 2014, 16, 972 CrossRef CAS PubMed .
-
(a) M. F. Semmelhack, P. M. Helquist and L. D. Jones, J. Am. Chem. Soc., 1971, 93, 5908 CrossRef CAS ;
(b) M. Kitano, J. Sung, K. H. Park, H. Yorimitsu, D. Kim and A. Osuka, Chem.–Eur. J., 2013, 19, 16523 CrossRef CAS PubMed .
-
(a) A. Tsuda, H. Furuta and A. Osuka, Angew. Chem., Int. Ed., 2000, 39, 2549 CrossRef CAS ;
(b) A. Tsuda and A. Osuka, Science, 2001, 293, 79 CrossRef CAS PubMed ;
(c) A. Tsuda, H. Furuta and A. Osuka, J. Am. Chem. Soc., 2001, 123, 10304 CrossRef CAS PubMed ;
(d) T. Ikeue, N. Aratani and A. Osuka, Isr. J. Chem., 2005, 45, 293 CrossRef CAS ;
(e) M. Kamo, A. Tsuda, Y. Nakamura, N. Aratani, K. Furukawa, T. Kato and A. Osuka, Org. Lett., 2003, 5, 2079 CrossRef CAS PubMed ;
(f) T. Tanaka, Y. Nakamura and A. Osuka, Chem.–Eur. J., 2008, 14, 204 CrossRef CAS PubMed .
- Crystallographic data for 6: C124H130N8Zn2·2C5H5N·3C6H5CH3·CH3CN, triclinic, space group P (no. 2), a = 10.0013(12), b = 14.5831(10), c = 24.366(4) Å, α = 75.428(16), β = 87.017(19), γ = 71.26(2)°, V = 3255.6(8) Å3, T = 123 K, Z = 1, R1 = 0.0531 (I > 2σ(I)), Rw = 0.1531 (all data), GOF = 1.018. CCDC number: 1433486.
- The MPD of 6 is defined by the 48 core atoms consisting of eight pyrrole units and eight methine carbons.
-
(a) D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758 CrossRef CAS PubMed ;
(b) R. Herges and D. Geuenich, J. Phys. Chem. A, 2001, 105, 3214 CrossRef CAS .
-
(a) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed ;
(b) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS .
-
(a) Y. Inokuma, N. Ono, H. Uno, D. Y. Kim, S. B. Noh, D. Kim and A. Osuka, Chem. Commun., 2005, 3782 RSC ;
(b) V. V. Diev, K. Hanson, J. D. Zimmerman, S. R. Forrest and M. E. Thompson, Angew. Chem., Int. Ed., 2010, 49, 5523 CrossRef CAS PubMed ;
(c) N. K. S. Davis, A. L. Thompson and H. L. Anderson, Org. Lett., 2010, 12, 2124 CrossRef CAS PubMed ;
(d) J. Luo, S. Lee, M. Son, B. Zheng, K.-W. Huang, Q. Qi, W. Zeng, G. Li, D. Kim and J. Wu, Chem.–Eur. J., 2015, 21, 3708 CrossRef CAS PubMed .
- K. Fujimoto, H. Yorimitsu and A. Osuka, Chem.–Eur. J., 2015, 21, 11311 CrossRef CAS PubMed .
- A. Tsuda, H. Furuta and A. Osuka, J. Am. Chem. Soc., 2001, 123, 10304 CrossRef CAS PubMed .
- H. S. Cho, D. H. Jeong, S. Cho, D. Kim, Y. Matsuzaki, K. Tanaka, A. Tsuda and A. Osuka, J. Am. Chem. Soc., 2002, 124, 14642 CrossRef CAS PubMed .
-
N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, 1991 Search PubMed .
-
(a) A. Nowak-Król, M. Grzybowski, J. Romiszawski, M. Drobizhev, G. Wicks, M. Chotkowski, A. Rebane, E. Górecka and D. T. Gryko, Chem. Commun., 2013, 49, 8368 RSC ;
(b) J. Schmitt, V. Heitz, A. Sour, F. Bolze, H. Ftouni, J.-F. Nicoud, L. Flamigni and B. Ventura, Angew. Chem., Int. Ed., 2015, 54, 169 CrossRef CAS PubMed .
-
(a) T. Ikeda, N. Aratani and A. Osuka, Chem.–Asian J., 2009, 4, 1248 CrossRef CAS PubMed ;
(b) M. Drobizhev, A. Karotki, M. Kruk and A. Rebane, Chem. Phys. Lett., 2002, 355, 175 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1433486–1433489. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04748j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence