
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible quantitative guest sensing via spin crossover of an iron(II) triazole†
Reece G.
Miller
and
Sally
Brooker
*
Department of Chemistry and MacDiarmid Institute for Advanced Materials and Nanotechnology, University of Otago, PO Box 56, Dunedin, 9054 New Zealand. E-mail: sbrooker@chemistry.otago.ac.nz; Fax: +64 3 4797906; Tel: +64 3 4797919
First published on 9th February 2016
Abstract
A new phenyl-triazole-pyrazine ligand, 4-p-tolyl-3-(phenyl)-5-(2-pyrazinyl)-1,2,4-triazole (tolpzph), was prepared in order to enforce pyrazine coordination of the iron(II) centre in the resulting complex, [FeII(tolpzph)2(NCS)2]·THF (1·THF). Structure determinations carried out on this discrete mononuclear complex, 1·THF, at 273 K (mostly high spin) and 100 K (mostly low spin) demonstrate this was successful, and that spin crossover (SCO) occurred on cooling. Subsequent magnetic measurements on 1·THF revealed that it shows highly sensitive and reversible solvent-dependent SCO, with T1/2(1·THF) = 255 K vs. T1/2(1) = 212 K (with SCO of 1 more abrupt and occurring with a 4 K hysteresis loop), a drop of 43 K due to THF loss. This is reversible over at least 10 cycles of re-solvating with THF followed by re-drying, so 1 ↔ 1·THF can be considered an ‘on–off’ THF sensor, monitored by the T1/2 reversibly shifting (by 43 K). Furthermore, quantitative sensing of the fractional amount of THF present in 1·nTHF, 0 ≤ n ≤ 1, is demonstrated. Monitoring the T1/2 and using TGA to quantify n(THF) revealed a linear dependence (25 data points; Pearson r2 = 0.93): T1/2 = 41.1n(THF) + 219. Finally, 1 is also shown to take up CHCl3 [T1/2(1·CHCl3) = 248 K], with a logarithmic T1/2 dependence on the fractional amount of CHCl3 present (10 data points; Pearson r2 = 0.98): T1/2 = 27.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10[n(CHCl3)] + 243. This study is a proof of principle that a (multi-use) quantitative sensor material based on spin crossover is feasible.
log10[n(CHCl3)] + 243. This study is a proof of principle that a (multi-use) quantitative sensor material based on spin crossover is feasible.
Introduction
Spin crossover (SCO) can be observed in d4–d7 transition metal complexes when the ligand field around the metal centre is perfectly tuned, such that application of an external stimulus causes the complex to switch between the low spin (LS) and high spin (HS) states.1–6 SCO-active materials have primarily been of interest due to the potential to store information when very slow relaxation (HS ↔ LS) allows a metastable state (within a hysteresis loop) to be observed for a finite lifetime.4–8Due to the sensitivity of SCO-active materials to their environment, sensor applications are also of considerable interest, as exemplified by a wide range of recent high profile reports.9–22 These examples have focussed on the use of SCO as a qualitative on/off-sensor,9,11,13,16,19–24 in some cases showing remarkable tuning of the spin crossover T1/2 (which can be monitored by many techniques besides magnetic, such as IR and UV-Vis spectroscopy) by varying the type of guest molecule within the crystal lattice.5,9,11,13,19–22,25 Metal–organic frameworks (MOFs) with permanent porosity have been prominent in such studies.9,11,13,22,26–28 Similarly, stepwise tuning of T1/2 by post-synthetic modification of a porous MOF has been reported.29 Whilst on/off sensing is inherently useful, for many applications a quantitative sensor would have significant advantages. A couple of solution iron(II) SCO-systems have shown some promise in this regard, including proof of concept studies showing sensing of anions in non-protic solvents30–32 or of sensing temperature in aqueous solution [for PARACEST (paramagnetic chemical exchange saturation transfer) MRI thermometers].33
Reported herein is the synthesis, structure and magnetic properties of [FeII(tolpzph)2(NCS)2]·THF, 1·THF, which can be reversibly desolvated to give 1. Both 1·THF and 1 are shown to be SCO-active but the T1/2 values differ by 43 K. To the best of our knowledge we report herein the first demonstration of reversible quantitative guest sensing by an SCO-active material.
Our previous work with the ligand 4-p-tolyl-3-(2-pyrazinyl)-5-(2-pyridyl)-1,2,4-triazole (tolpzpy, Fig. S7†) resulted in an unexpected preference of the iron(II) centre for the better σ-donor but poorer π-acceptor34,35 pyridine donor over the pyrazine donor.36 This led us to develop an analogue, the pyrazine-phenyl ligand tolpzph (Scheme 1), in order to facilitate coordination of the iron(II) centre to the pyrazine moiety by removing the competition provided by the pyridine moiety, replacing it with a phenyl ring.
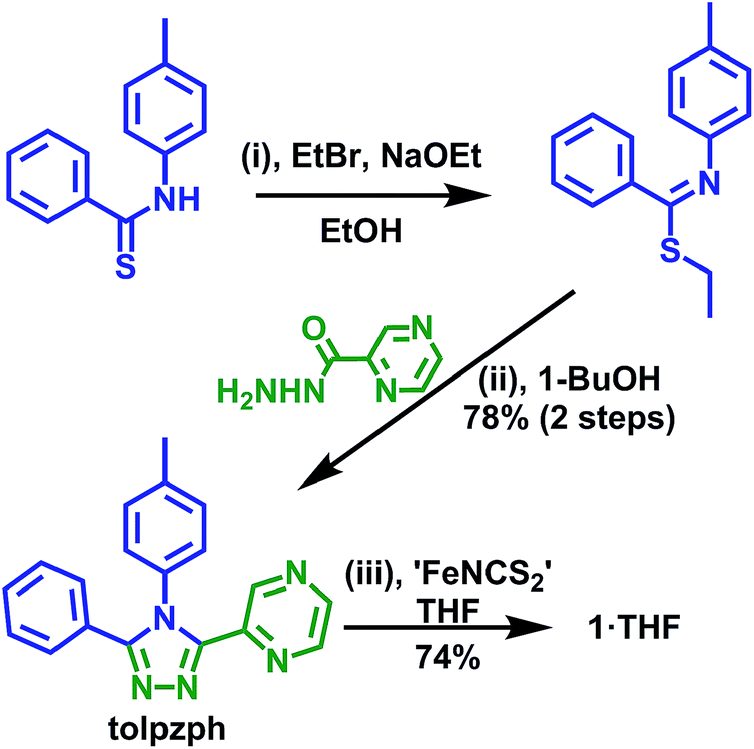 | ||
| Scheme 1 Synthesis of the novel ligand tolpzph and the iron(II) isothiocyanato complex of it [FeII(tolpzph)2(NCS)2]·THF (1·THF). | ||
Results and discussion
Synthesis
The new ligand tolpzph is prepared in excellent yield (78% over two steps) from the known precursor N-(p-tolyl)-benzenethioamide using methods adapted from our previously reported syntheses (Scheme 1).36,37The complex 1·THF is prepared in good yield (74%), under Schlenk conditions, by the in situ preparation of ‘FeII(NCS)2’ in dry THF followed by the addition of a dry THF solution of tolpzph. Interestingly, unlike the previously reported complex of the pyrazine-pyridine ligand, [FeII(tolpzpy)2(NCS)2],36 the synthesis of 1·THF is highly moisture sensitive. The complex also readily decomposes in solution in the presence of coordinating solvents such as MeOH or MeCN. These observations are consistent with the reduced σ-donor character of pyrazine relative to pyridine making the Fe–N bond more labile, at least in solution. In contrast to these solution state findings, 1·THF is stable in air in the solid state.
Structure determinations at 273 and 100 K
Forest green, rod shaped, single crystals of 1·THF were grown by vapour diffusion of dry, degassed diethyl ether into a THF solution of the reaction mixture under an inert atmosphere. The complex crystallises in the orthorhombic space group Pbca, with half of the complex in the asymmetric unit, and the other half generated by a centre of inversion at the iron(II) centre (Fig. 1).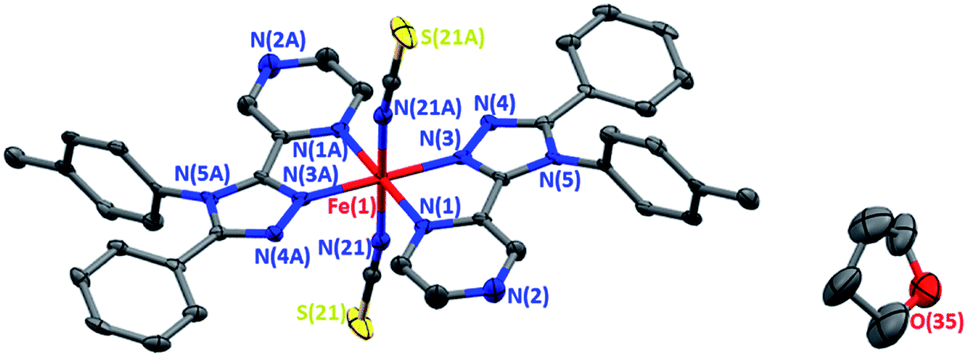 | ||
| Fig. 1 Solid state structure of [FeII(tolpzph)2(NCS)2]·THF (1·THF) at 100 K. Only one of the disordered 1/4 occupancy THF molecules shown; H atoms omitted for clarity; thermal ellipsoids drawn at 70% probability. | ||
The Fe–N bond lengths (average 2.08 Å) and the high value of the octahedral distortion parameter (Σ = 63.6°) observed for 1·THF at 273 K (Table 1) are consistent with HS iron(II). On decreasing the temperature to 100 K the Fe–N bond lengths (average 1.97 Å) and the decreased value of the octahedral distortion parameter (Σ = 54.9°) are consistent with the iron(II) centre being in the LS state (Table 1). The large decrease (8%) in the unit cell volume between 350 K and 100 K (Table 1, Fig. S17†) provides additional confirmation of the change in spin state.
| 100 K | 273 K | |
|---|---|---|
| a Within a ligand strand. | ||
| Cell volume [Å3] | 4184.24 (16) | 4377.1 (2) |
| Fe–Ntriazole [Å] | 1.971 (3) | 2.070 (2) |
| Fe–Nazine [Å] | 1.999 (3) | 2.125 (2) |
| Fe–NCS [Å] | 1.951 (3) | 2.033 (3) |
| Average Fe–N [Å] | 1.97 | 2.08 |
| Ntriazole–Fe–Nazine [°]a | 80.60 (9) | 77.25 (8) |
| Ntriazole–Fe–NCS [°] | 92.35 (9) | 92.11 (9) |
| Nazine–Fe–NCS [°] | 91.97 (9) | 91.03 (9) |
| Σ [°] | 54.9 | 63.6 |
The THF molecule of solvation is disordered over two unique, overlapping, quarter occupancy sites in the asymmetric unit (Fig. S5†). This gives a total of four, one quarter occupancy, positions per iron(II) centre. By symmetry, these form a continuous line down a ‘pseudo-channel’ along the crystallographic a-axis (Fig. 2, S4 and S5†).
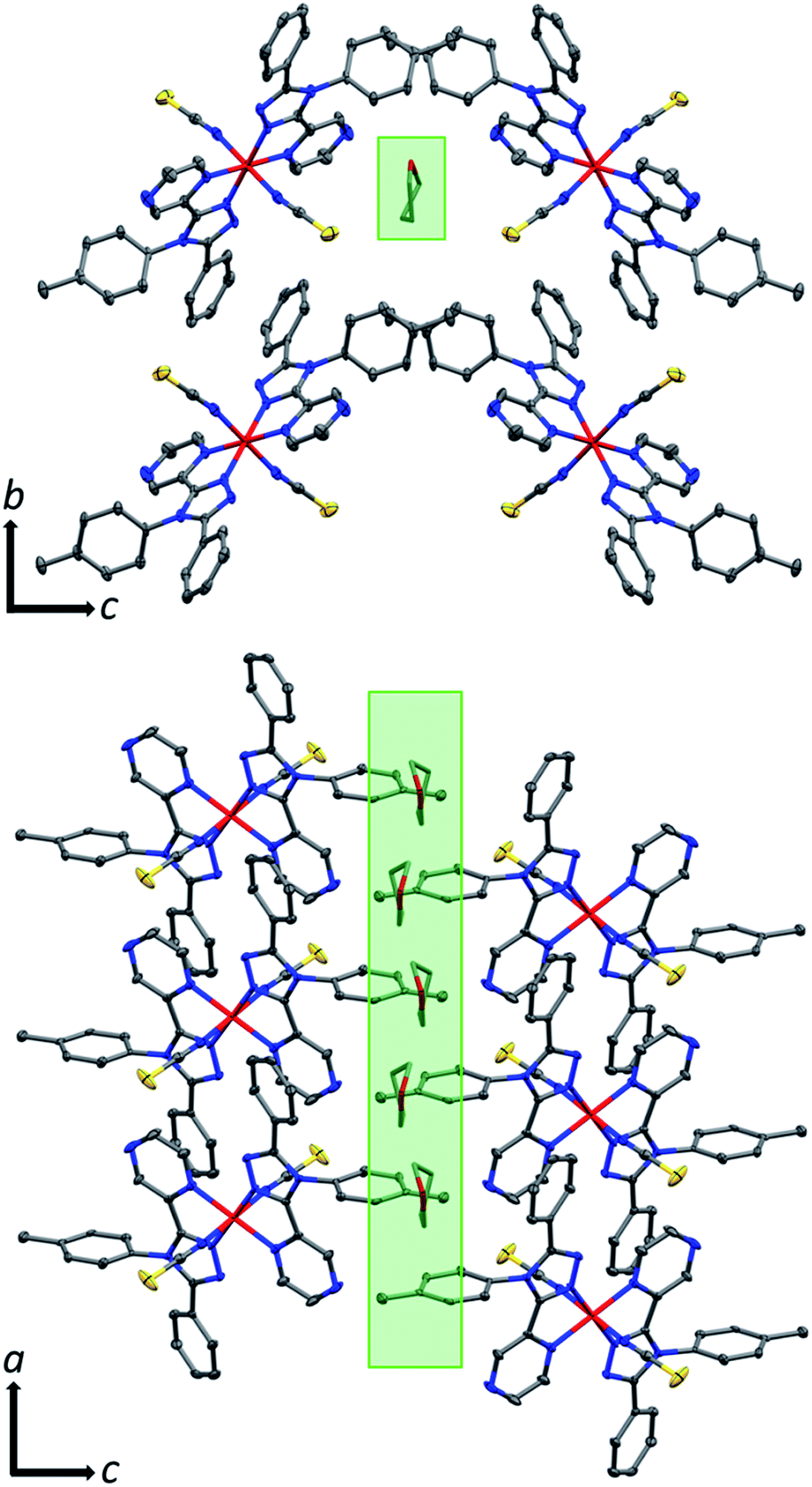 | ||
| Fig. 2 Crystal packing in 1·THF as viewed down the crystallographic (top) a-axis (just 1 of the 4 overlapping (each on average 1/4 occupancy) disordered THF positions is shown for clarity) and (bottom) b-axis (just 2 of the 4 overlapping disordered THF positions are shown for clarity). Both views show the pore-like ‘pseudo-channels’ along the a-axis that contain the THF of solvation (indicated by green highlight) which can be reversibly removed (also see Fig. S4 and S5†). Hydrogen atoms omitted for clarity; thermal ellipsoids drawn at 70% probability. THF molecules drawn as capped sticks for clarity. | ||
Magnetic characterisation
The magnetic susceptibility of 1·THF was initially studied from 300 to 50 K. At 300 K the μeff is 4.98 B.M., consistent with the complex being almost completely HS. On cooling, the μeff drops, revealing an abrupt spin transition with a T1/2 of 250 K (Fig. 3, black data points; Fig. S22†). Consistent with the crystal structure determined at 100 K (Table 1), the μeff of 1.63 B.M at 100 K is indicative of the complex being mostly LS.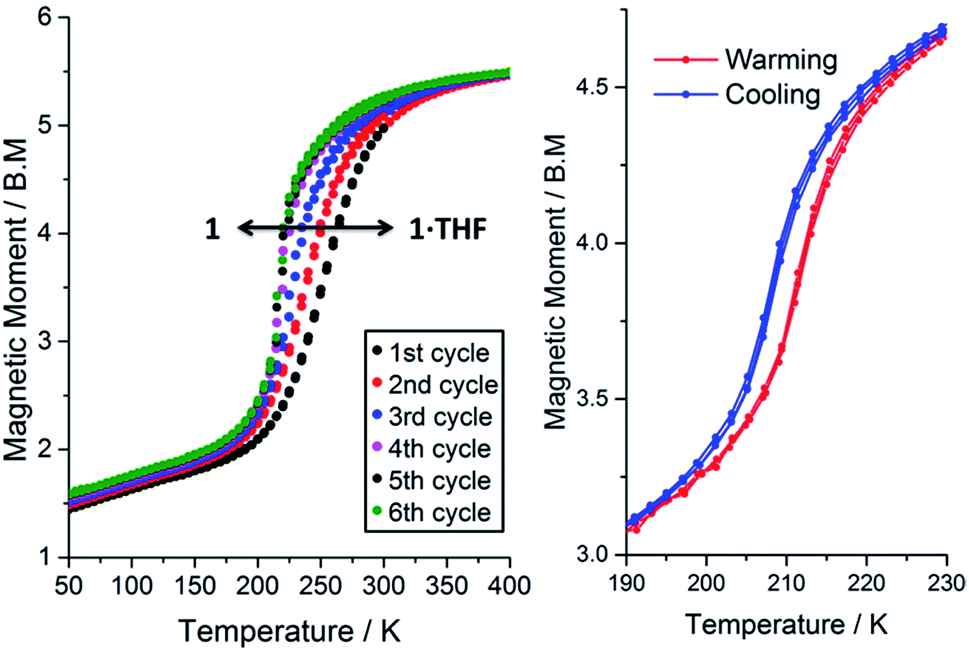 | ||
| Fig. 3 Left: Magnetic moment of 1·n(THF) (where 0 ≤ n ≤ 1) as a function of temperature for 300 → 50 K and then repeated cycles of 50 ↔ 400 K showing the solvent dependence of SCO in this compound. Note: TGA shows that n = 1 for the first cycle and n = 0 for the 5th cycle. Right: A zoom in over the temperature range of the SCO event after thorough drying (403 K, N2(g), 2 hours) so that n = 0, shows a reproducible 4 K hysteresis (three cycles shown). All data collected in settle mode (data point taken after constant T to within the lesser of ±0.5% and ±0.5 K for 1 minute, see page S7†); MW and sample mass were revised for each cycle based on the relationship derived in eqn (1) (see later), but please note that using MW (1·THF) for all calculations makes very little difference (ESI, Fig. S28†). | ||
Notably the T1/2 observed for the pyrazine-coordinated iron(II) centre in 1·THF is over 100 K higher than the T1/2 of 145 K observed for the related solvent-free SCO-active polymorph of [FeII(tolpzpy)2(NCS)2] in which it is unclear whether the iron(II) is coordinated to pyrazine or to pyridine.36 If the latter is pyridine bound then this is consistent with pyrazine providing a stronger ligand field than pyridine (stabilising the LS state, hence increasing the T1/2), as expected, but if both are pyrazine bound then this difference in T1/2 may well be due to the differences in crystal packing and/or solvent content.
Interestingly, on repeated variable temperature cycles in the range 50–400 K, in settle mode, a small decrease in the T1/2 was observed with each cycle (Fig. 3). This was coupled with an increasingly abrupt SCO event, which by the 6th cycle also shows a small (4 K) but reproducible thermal hysteresis. The changes are confirmed to be due to the loss of some THF of solvation with each cycle to high temperature by thermogravimetric analysis (TGA) after each cycle (Fig. S17†). Overall the T1/2 decreases 43 K, from T1/2(1·THF) = 255 K to T1/2(1) = 212 K. A significant solvochromic effect is also observed with a change from forest green (1·THF) to violet (1) on drying (Fig. 4, right).
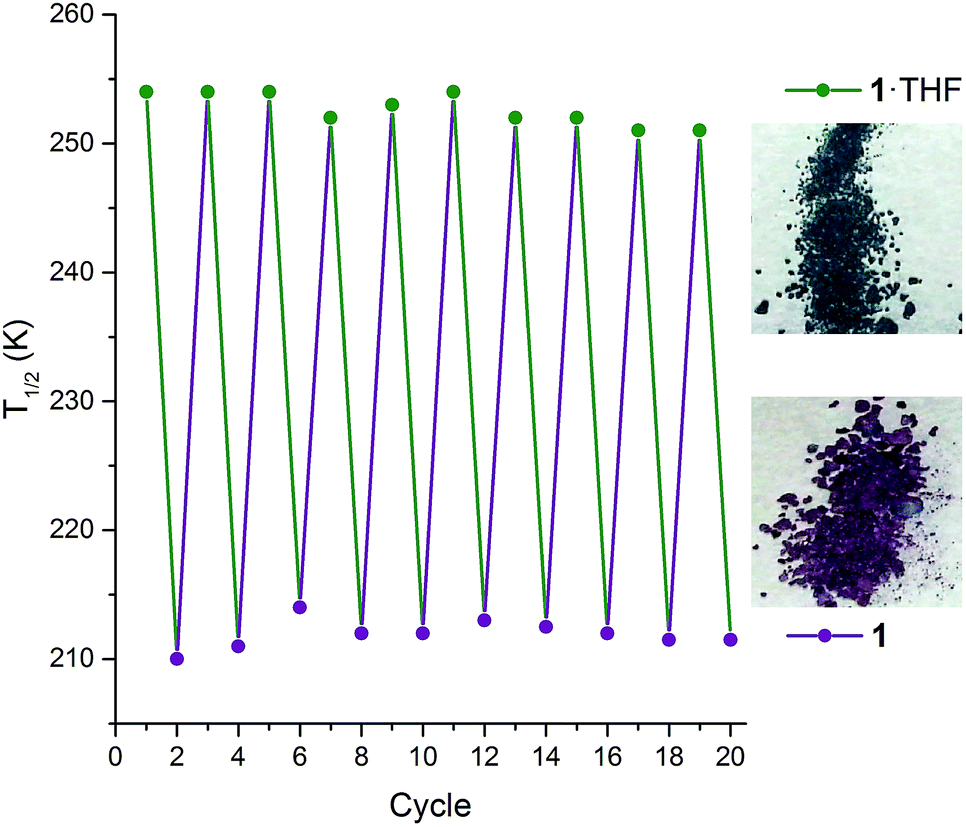 | ||
| Fig. 4 Left: The T1/2 as monitored over 10 consecutive cycles‡ of interconversions 1·THF (green points) ↔ 1 (purple points), whereby 1·THF is heated at 400 K for two hours to give 1 then exposed to THF vapour for 2 hours to regenerate 1·THF (lines are simply guides to the eyes). Note: appropriate molecular weight (either 1·THF or 1), and revised sample mass, used for each cycle. Right: A distinct change in colour is observed between 1·THF (forest green) and 1 (violet). | ||
The observed shift in T1/2 as the guest concentration is decreased is likely to be due to internal lattice pressure effects6,13,38 whereby the larger HS state becomes more favourable when the a-axis channels are less occupied. The observed shifting of T1/2 is reversible on exposure to THF vapour, for as little as 2 hours, restoring the original magnetic response vs. temperature. Heating the sample for 2 hours at 400 K, either under a weak vacuum (4 mbar), or under a N2(g) flow, regenerates 1 and the associated magnetic response. This cycling, between 1·THF and 1, is reproducible over at least 10 cycles with minimal fatigue (Fig. 4, left). In summary, this 1·THF ↔ 1 spin crossover system is a robust ‘on–off’, or qualitative, sensor for THF.
Next, we probed whether the magnetic response could be used to determine the solvent content in 1·n(THF) where this was fractional rather than simply n = 0 or 1, in other words whether this system can act as a quantitative sensor for THF. A comparison of the magnetic and thermogravimetric data reveals that the T1/2 has an approximately linear dependence on the concentration of THF guest (Fig. 5).
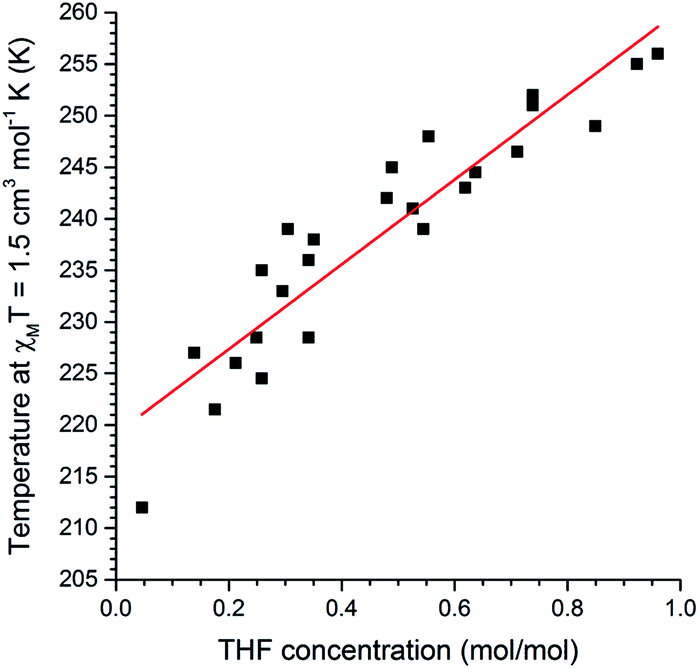 | ||
| Fig. 5 A plot of mole fraction of THF, n(THF) as mol(THF)/mol(Fe), as a function of the T1/2 (determined as the temperature at which χMT = 1.5 cm3 mol−1 K). Pearson r2 = 0.93 for the linear fit of the 25 data points. See page S7 (and Fig. S27†) for a full description of the collection of these data. | ||
The relationship between T1/2 and n(THF) is described by the equation:
| T1/2 = 41.1n(THF) + 219 | (1) |
Taking this a step further, this SCO system was also tested as a quantitative sensor for another solvent. Exposure of either the dry compound 1, or the THF solvated 1·THF, to CHCl3 vapours for 24 hours results in the CHCl3 solvated analogue 1·CHCl3 (confirmed by CHN and TGA analysis, see ESI†), for which the T1/2 is 248 K. Interestingly, for CHCl3 the T1/2 dependence of the SCO event on n(CHCl3) appears tobe more logarithmic in nature (Fig. 6 and S26†). The relationship between T1/2 and n(CHCl3) is described by the equation:
| T1/2 = 27.0log10[n(CHCl3)] + 243 | (2) |
log10[n(CHCl3)], significantly different from 0 (p < 0.001). Presumably this logarithmic response is because the inter-guest interactions are stronger for CHCl3 than for THF, and these guest–guest interactions are becoming more significant at higher loadings. This is consistent with CHCl3 having a higher propensity to hydrogen bond than THF. A logarithmic dependence on guest concentration could be beneficial in applications where a higher sensitivity over a narrow range is desired. Hence this SCO system is also a quantitative sensor for CHCl3.
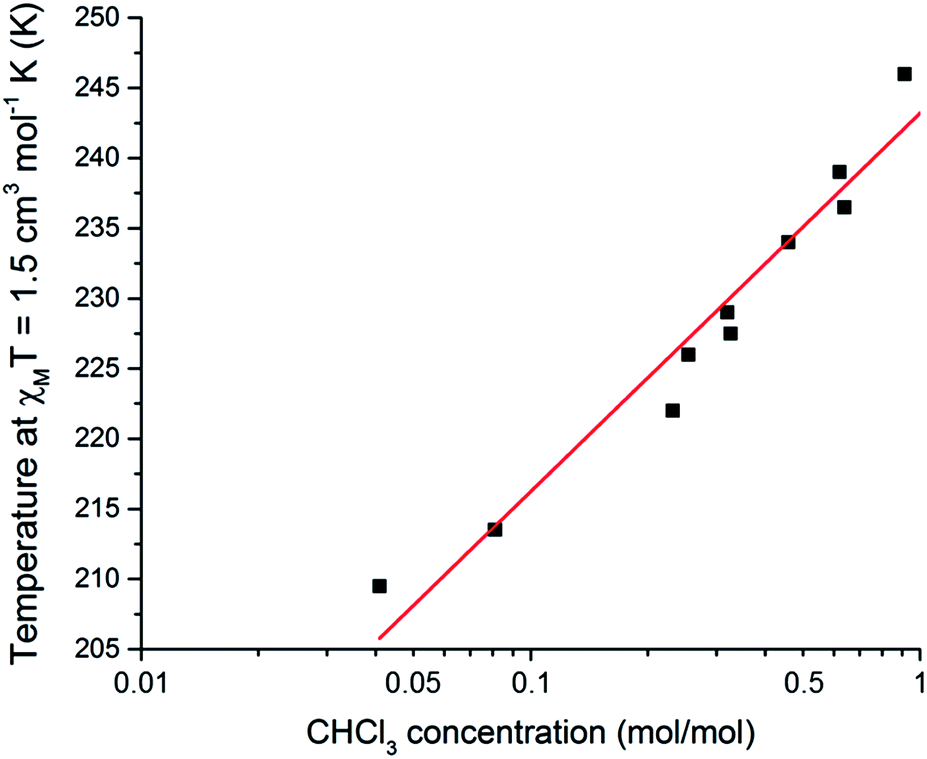 | ||
| Fig. 6 A plot of mole fraction of CHCl3, n(CHCl3) as mol(CHCl3)/mol(Fe), on a log10 scale as a function of the T1/2 (determined as the temperature at which χMT = 1.5 cm3 mol−1 K). Pearson r2 = 0.98 for the linear fit of the 10 data points. See page S7 (and Fig. S27†) for a full description of the collection of these data. | ||
In summary, 1·THF shows an abrupt SCO at 255 K. Stepwise desolvation of 1·THF, via1·nTHF where 0 < n < 1, to 1 (n = 0) is accompanied by a linear shifting of the SCO event by 43 K down to 212 K for 1. The gain and loss of THF guest molecules is also shown to be reversible. These findings demonstrate that this SCO active complex can act as a quantitative sensor. Furthermore, the THF of solvation could be exchanged by CHCl3, by exposure to CHCl3 vapours for 24 hours. The mole fraction of CHCl3 guest molecules present also predictably tunes the T1/2, with it decreasing by 36 K from 1·CHCl3 to 1, but with a logarithmic relationship. Finally, it is important to note that the ability of spin crossover systems to carry out robust, reversible and quantitative solvent sensing is unlikely to be limited to 1. Hence a wide range of both new and previously reported compounds, both discrete and polymeric, both porous and non-porous should be investigated for their sensor potential.
Acknowledgements
We thank the Marsden Fund (RSNZ) and the University of Otago (including the award of a postgraduate scholarship and a publishing bursary to RGM; and the 2015 purchase of a Versalab magnetometer which enabled in house 50–400 K magnetic measurements, which in turn enabled this study) for supporting this research. We are grateful to Dr John C. McAdam (Otago) for assistance with the collection of the solid state reflectance data.Notes and references
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC.
- P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, 233, 1–47 CrossRef.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC.
- M. A. Halcrow, Spin-Crossover Materials: Properties and Applications, John Wiley & Sons, Ltd, 1st edn, 2013 Search PubMed.
- P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef PubMed.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC and front cover feature.
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- K. S. Murray, H. Oshio and J. Real, Eur. J. Inorg. Chem., 2013, 577–580 CrossRef CAS.
- G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and C. S. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS PubMed.
- M. Hostettler, K. W. Törnroos, D. Chernyshov, B. Vangdal and H.-B. Bürgi, Angew. Chem., Int. Ed., 2004, 40, 4589 CrossRef PubMed.
- M. Quesada, V. A. de la Pena-O'Shea, G. Aromi, S. Geremia, C. Massera, O. Roubeau, P. Gamez and J. Reedijk, Adv. Mater., 2007, 19, 1397–1402 CrossRef CAS.
- S. M. Neville, G. J. Halder, K. W. Chapman, M. B. Duriska, P. D. Southon, J. D. Cashion, J.-F. Letard, B. Moubarak, K. S. Murray and C. J. Kepert, J. Am. Chem. Soc., 2008, 130, 2869–2876 CrossRef CAS PubMed.
- P. D. Southon, L. Llu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 CrossRef CAS PubMed.
- S. M. Neville, G. J. Halder, K. W. Chapman, M. B. Duriska, B. Moubaraki, K. S. Murray and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 12106–12108 CrossRef CAS PubMed.
- X. Bao, H. J. Shepherd, L. Salmon, G. Molnár, M.-L. Tong and A. Bousseksou, Angew. Chem., Int. Ed., 2013, 125, 1236–1240 CrossRef.
- Y.-T. Wang, S.-T. Li, S.-Q. Wu, A.-L. Cui, D.-Z. Shen and H.-Z. Kou, J. Am. Chem. Soc., 2013, 135, 5938–5941 CrossRef PubMed.
- A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathonière, R. Clérac and P. E. Kruger, Chem. Commun., 2013, 49, 1597–1599 RSC.
- R. A. Bilbeisi, S. Zarra, H. L. C. Feltham, G. N. L. Jameson, J. K. Clegg, S. Brooker and J. R. Nitschke, Chem.–Eur. J., 2013, 19, 8058–8062 CrossRef CAS PubMed.
- R. Kulmaczewski, J. Olguín, J. A. Kitchen, H. L. C. Feltham, G. N. L. Jameson, J. L. Tallon and S. Brooker, J. Am. Chem. Soc., 2014, 136, 878–881 CrossRef CAS PubMed.
- J. S. Costa, S. Rodriguez-Jimenez, G. A. Craig, B. Barth, C. M. Beavers, S. J. Teat and G. Aromi, J. Am. Chem. Soc., 2014, 136, 3869–3874 CrossRef CAS PubMed.
- M. Steinert, B. Schneider, S. Dechert, S. Demeshko and F. Meyer, Angew. Chem., Int. Ed., 2014, 53, 6135–6139 CrossRef CAS PubMed.
- A. Lennartson, P. Southon, N. F. Sciortino, C. J. Kepert, C. Frandsen, S. Mørup, S. Piligkos and C. J. McKenzie, Chem.–Eur. J., 2015, 21, 16066–16072 CrossRef CAS PubMed.
- W. Zhang, F. Zhao, T. Liu, M. Yuan, Z.-M. Wang and S. Gao, Inorg. Chem., 2007, 46, 2541–2555 CrossRef CAS PubMed.
- D. Gentili, N. Demitri, B. Schafer, F. Liscio, I. Bergenti, G. Ruani, M. Ruben and M. Cavallini, J. Mater. Chem. C, 2015, 3, 7836–7844 RSC.
- J.-B. Lin, W. Xue, B.-Y. Wang, J. Tao, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Inorg. Chem., 2012, 51, 9423–9430 CrossRef CAS PubMed.
- J. Cirera, Rev. Inorg. Chem., 2014, 34, 199–216 CrossRef CAS.
- E. Coronado and G. Minguez Espallargas, Chem. Soc. Rev., 2013, 42, 1525–1539 RSC.
- E. Coronado, M. Giménez-Marqués, G. Mínguez Espallargas, F. Rey and I. J. Vitórica-Yrezábal, J. Am. Chem. Soc., 2013, 135, 15986–15989 CrossRef CAS PubMed.
- R. Ohtani, K. Yoneda, S. Furukawa, N. Horike, S. Kitagawa, A. B. Gaspar, M. C. Muñoz, J. A. Real and M. Ohba, J. Am. Chem. Soc., 2011, 133, 8600–8605 CrossRef CAS PubMed.
- Z. Ni and M. P. Shores, J. Am. Chem. Soc., 2009, 131, 32–33 CrossRef CAS PubMed.
- Z. Ni, A. M. McDaniel and M. P. Shores, Chem. Sci., 2010, 1, 615–621 RSC.
- M. C. Young, E. Liew and R. J. Hooley, Chem. Commun., 2014, 50, 5043–5045 RSC.
- I.-R. Jeon, J. G. Park, C. R. Haney and T. D. Harris, Chem. Sci., 2014, 5, 2461–2465 RSC.
- P. J. Steel, Coord. Chem. Rev., 1990, 106, 227–265 CrossRef CAS.
- K. B. Wiberg, D. Nakaji and C. M. Breneman, J. Am. Chem. Soc., 1989, 111, 4178–4190 CrossRef CAS.
- R. W. Hogue, R. G. Miller, N. G. White, H. L. C. Feltham, G. N. L. Jameson and S. Brooker, Chem. Commun., 2014, 50, 1435–1437 RSC.
- M. H. Klingele and S. Brooker, Eur. J. Org. Chem., 2004, 3422–3434 CrossRef CAS.
- A. Hauser, Chem. Phys. Lett., 1992, 192, 65–70 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, NMR spectra, instrumentation, plus additional structural, magnetic and TGA information and diagrams. CCDC 1434103 and 1434104. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04583e |
| ‡ Transfer losses between measurements precluded measurement of further cycles. |
| This journal is © The Royal Society of Chemistry 2016 |