
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free electrocatalytic hydrogen oxidation using frustrated Lewis pairs and carbon-based Lewis acids†
Elliot J.
Lawrence
 a,
Ewan R.
Clark‡
b,
Liam D.
Curless
b,
James M.
Courtney
a,
Robin J.
Blagg
a,
Michael J.
Ingleson
*b and
Gregory G.
Wildgoose
*a
a,
Ewan R.
Clark‡
b,
Liam D.
Curless
b,
James M.
Courtney
a,
Robin J.
Blagg
a,
Michael J.
Ingleson
*b and
Gregory G.
Wildgoose
*a
aEnergy & Materials Laboratory, School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: G.Wildgoose@uea.ac.uk
bSchool of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: Michael.Ingleson@manchester.ac.uk
First published on 6th January 2016
Abstract
Whilst hydrogen is a potentially clean fuel for energy storage and utilisation technologies, its conversion to electricity comes at a high energetic cost. This demands the use of rare and expensive precious metal electrocatalysts. Electrochemical-frustrated Lewis pairs offer a metal-free, CO tolerant pathway to the electrocatalysis of hydrogen oxidation. They function by combining the hydrogen-activating ability of frustrated Lewis pairs (FLPs) with electrochemical oxidation of the resultant hydride. Here we present an electrochemical–FLP approach that utilises two different Lewis acids – a carbon-based N-methylacridinium cation that possesses excellent electrochemical attributes, and a borane that exhibits fast hydrogen cleavage kinetics and functions as a “hydride shuttle”. This synergistic interaction provides a system that is electrocatalytic with respect to the carbon-based Lewis acid, decreases the required potential for hydrogen oxidation by 1 V, and can be recycled multiple times.
Introduction
As the demand for sustainable and carbon-neutral sources of electricity increases, there is a need for new technologies that allow the efficient storage and utilization of energy.1 H2 is attractive as an energy vector since energy from renewable sources may be stored in its chemical bond, and then cleanly and safely released as electricity using fuel cell technology.2Unfortunately, in the absence of a suitable electrocatalyst, the conversion of H2 into two protons and two electrons is slow and must be driven by a large overpotential (voltage). Precious metal electrodes (such as Pt) provide an electrocatalytic effect that is indicated by a marked increase in current and a shift in the electrode reaction to a lower potential (voltage).3,4 However, the high cost and low abundance of such materials presents a significant barrier to the wide-spread adoption of current H2 fuel cell technology. There is clearly a need to develop new H2 oxidation electrocatalysts that are free from precious metals. Progress has been made in this area using bioinspired catalysts5–7 that contain either Ni8–10 or Fe11–13 centres. However, a significant weakness of existing electrocatalysts (Pt and the majority of hydrogenase enzyme mimics) is that they are highly sensitive to CO binding and inhibition.5,14 Trace amounts of CO are inevitably present in H2 that is commercially produced from hydrocarbon feedstocks. Worse still, for indirect methanol fuel cells (a combined H2 fuel cell and MeOH reformer) a CO removal process is often necessary to prevent electrocatalyst poisoning.15
An alternative metal-free strategy uses frustrated Lewis pairs (FLPs) to activate H2. Since their discovery by Stephan's group in 2006,16 research involving FLPs has grown apace.17–23 FLPs, formed from the combination of suitably sterically encumbered Lewis acids (LA) and bases, are precluded from forming classical Lewis adducts; such systems can heterolytically cleave H2 to generate hydridic and protic components. The hydrogenation of a wide range of functional groups including imines, enamines, nitriles,24–27 aldehydes,28 and ketones29–33 using FLPs has been reported.
In 2014, Wildgoose and Ashley pioneered a new metal-free route to H2 oxidation using a combined “electrochemical–FLP” approach.34,35 This enables the conversion of H2 into two protons and two electrons at cheap and ubiquitous carbon electrodes. Using the archetypal tBu3P/B(C6F5)3 system (Fig. 1a),34,36 the voltage (driving energy) required to oxidize H2 was decreased by 610 mV (ca. 118 kJ mol−1). Later, we applied this “electrochemical–FLP” approach to Stephan's NHC-stabilized borenium cation (Fig. 1a),35,37 which decreased the voltage required for H2 oxidation by 910 mV (ca. 176 kJ mol−1). However, a detailed mechanistic study of both these electrochemical–FLP systems revealed several limitations that significantly hindered their catalytic turnover, efficiency, and application as replacement electrocatalysts for energy applications. This included the side-reaction of radical intermediates with solvent/electrolyte during electrolysis, and the deactivation of electrocatalyst via its reaction with electrogenerated protons. Whilst the borenium cation offered an improvement over the borane system, the rate of H2 cleavage by this borenium–FLP is far too slow.
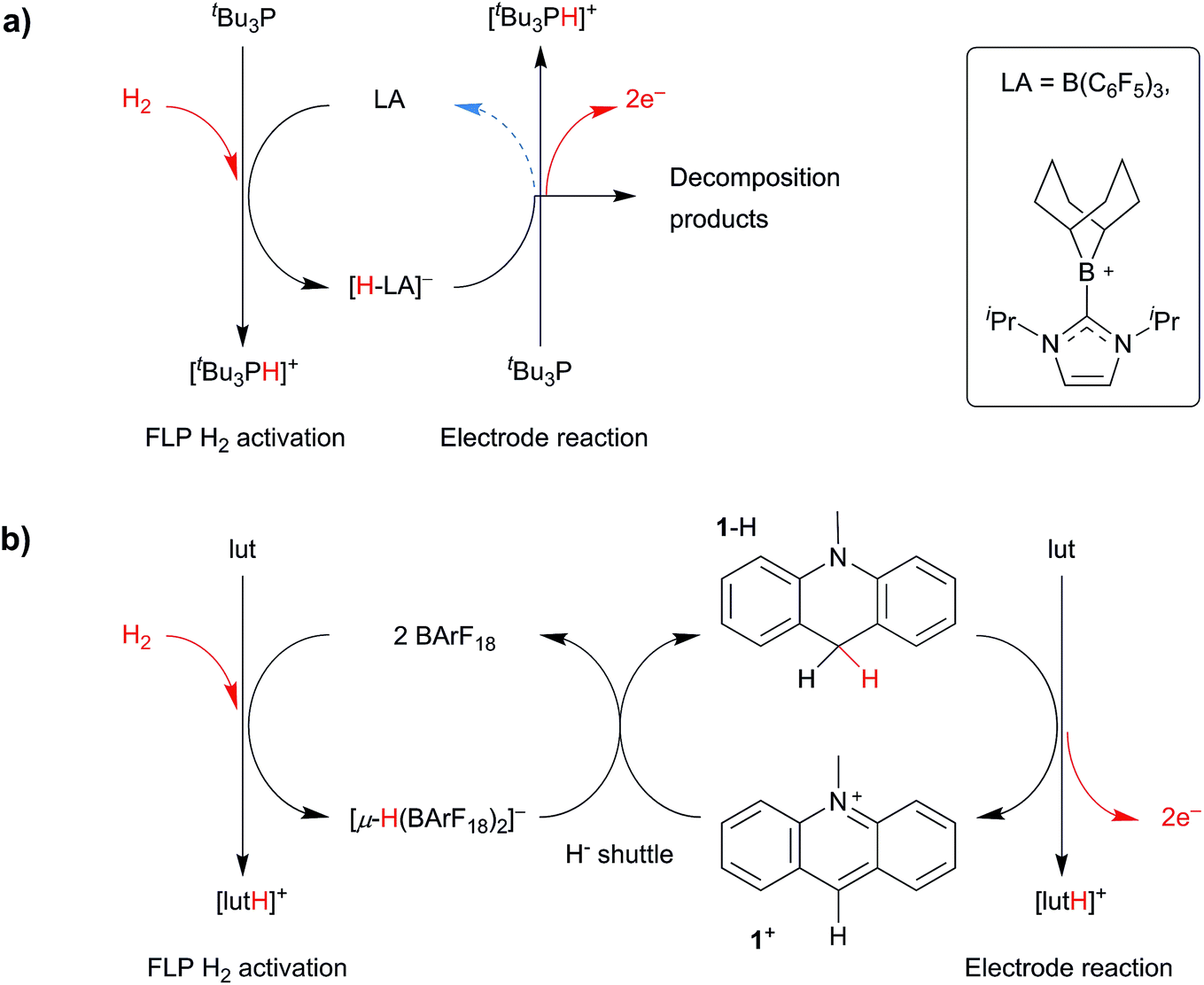 | ||
| Fig. 1 (a) The borane-only electrochemical–FLP system that was limited by the electrochemical stability of the borane, and (b) a carbon-based electrochemical–FLP that uses a redox inactive borane as a hydride shuttle. Note that any residual charges are counterbalanced by the supporting electrolyte [nBu4N][B(C6F5)4], which is present in large excess. | ||
Whilst the majority of research involving FLP H2 activation has been focused on boron-centred Lewis acids, the Ingleson group have recently reported a FLP derived from salts of the N-methylacridinium cation (1+), a carbon-centred Lewis acid, and the Lewis base 2,6-lutidine (lut).38,391+ is inexpensive, easy to synthesise, and is similar in structure to the NADH/NAD+ coenzyme system that is found in biological redox systems.40–44 Furthermore, in 1990, Savéant and co-workers elucidated all the pertinent non-aqueous mechanistic parameters of the 1+/N-methylacridane (1-H) redox couple both in the presence and the absence of a Brønsted base.43,44 The oxidation of 1-H involves an ECE-DISP1 mechanism and results in the net formation of two electrons and an electrogenerated proton (Scheme S1†). Compared to either of the boron-based electrochemical–FLP systems reported previously, the standard potential of the 1-H/[1-H]˙+ couple is relatively low (+0.48 ± 0.01 V vs. Cp2Fe0/+ in MeCN). Also, 1-H is insufficiently hydridic to react with any electrogenerated H+ produced, so no competing H2 evolution reaction (the reverse reaction of H2 cleavage by the FLP) occurs. Together, these attributes (ease of synthesis, high hydride affinity of 1+, favourable oxidation potential of 1-H and the lack of side-reactions during electrolysis) combine to make the carbon-based 1-H/1+ system a highly attractive candidate for electrochemical–FLP studies. The only limitation of the 1+/lut FLP is that the rate of H2 cleavage is very slow – requiring >9 days for almost complete H2 activation at 60 °C and 4 bar.38
Fortunately, a solution to this final problem is available to us. We have recently examined the possibility of using tris[3,5-bis(trifluoromethyl)phenyl]borane (BArF18) as the Lewis acidic component of an electrochemical–FLP system.45 The activation of H2 by BArF18-containing FLPs is rapid and favours the formation of the bridging hydride, [(μ-H) (BArF18)2]−.46,47 However, the oxidation potential of [(μ-H) (BArF18)2]− is too positive to be useful for electrochemical–FLP applications (ca. +1.55 V vs. Cp2Fe0/+) and resembles that of molecular H2 – BArF18 is not electrocatalytic towards H2 oxidation.
In this paper we combine the rapid H2 cleavage kinetics of BArF18-derived FLPs with the stability and efficiency of the carbon-centred Lewis acid, 1+. Using this approach, the bridging hydride, [(μ-H) (BArF18)2]−, effectively functions as a redox inactive “hydride shuttle” to generate 1-H from 1+ (Fig. 1b). As we demonstrate herein, the “hydride shuttle” combines the rapid cleavage of H2 by the BArF18/lut FLP with the favourable electrochemical properties of 1-H. This provides an improved electrocatalytic system, with numerous advantages over previous electrochemical–FLP systems: a ca. 1 V decrease in the voltage for H2 oxidation at a carbon electrode; a metal-free system that is catalytic in 1+, turns over efficiently and can be recharged multiple times; no undesirable H2 evolution side-reactions and a marked improvement in FLP H2 cleavage kinetics compared to carbon-based Lewis acids alone. We also demonstrate that, in stark contrast to conventional H2 oxidation electrocatalysts, this electrochemical–FLP system is tolerant of CO.
Results and discussion
BArF18 as a hydride shuttle
Bridging borohydrides are generally considered to be less hydridic than their terminal analogues. However, NMR experiments show that [(μ-H) (BArF18)2]− is capable of transferring hydride to B(C6F5)3. When a suspension of [tmpH][(μ-H) (BArF18)2] (tmp = 2,2,6,6-tetramethylpiperidine) in CD2Cl2 is treated with B(C6F5)3, the formation of a clear, colourless solution is observed, indicating that the sparingly soluble starting material has undergone reaction. Indeed, 1H, 19F{1H} and 11B NMR spectra of the reaction mixture indicate the formation of [tmpH][HB(C6F5)3] and two equivalents of BArF18 (Fig. S1–S3†).28,46,47 The sequestration of hydride by B(C6F5)3 likely reflects the greater electrophilicity of B(C6F5)3 (E° = −1.52 V vs. Cp2Fe0/+) compared to BArF18 (E° = −1.61 V vs. Cp2Fe0/+).45Given that 1+ has a higher hydride ion affinity than B(C6F5)3,38 which has a greater hydride ion affinity than BArF18, one would expect salts of 1+ to abstract hydride from [(μ-H) (BArF18)2]−. This was confirmed experimentally by the formation of 1-H when 1[BArCl] {[BArCl]− = tetra(3,5-dichlorophenyl)borate} was treated with an equivalent of [tmpH][(μ-H) (BArF18)2] (Fig. 2c). This suggests that BArF18 is highly suitable as a hydride shuttle for carbon-based electrochemical–FLPs derived from 1-H/1+, and may provide a means of overcoming the high kinetic barrier for H2 activation by FLPs comprised of this carbon-based Lewis acid alone.
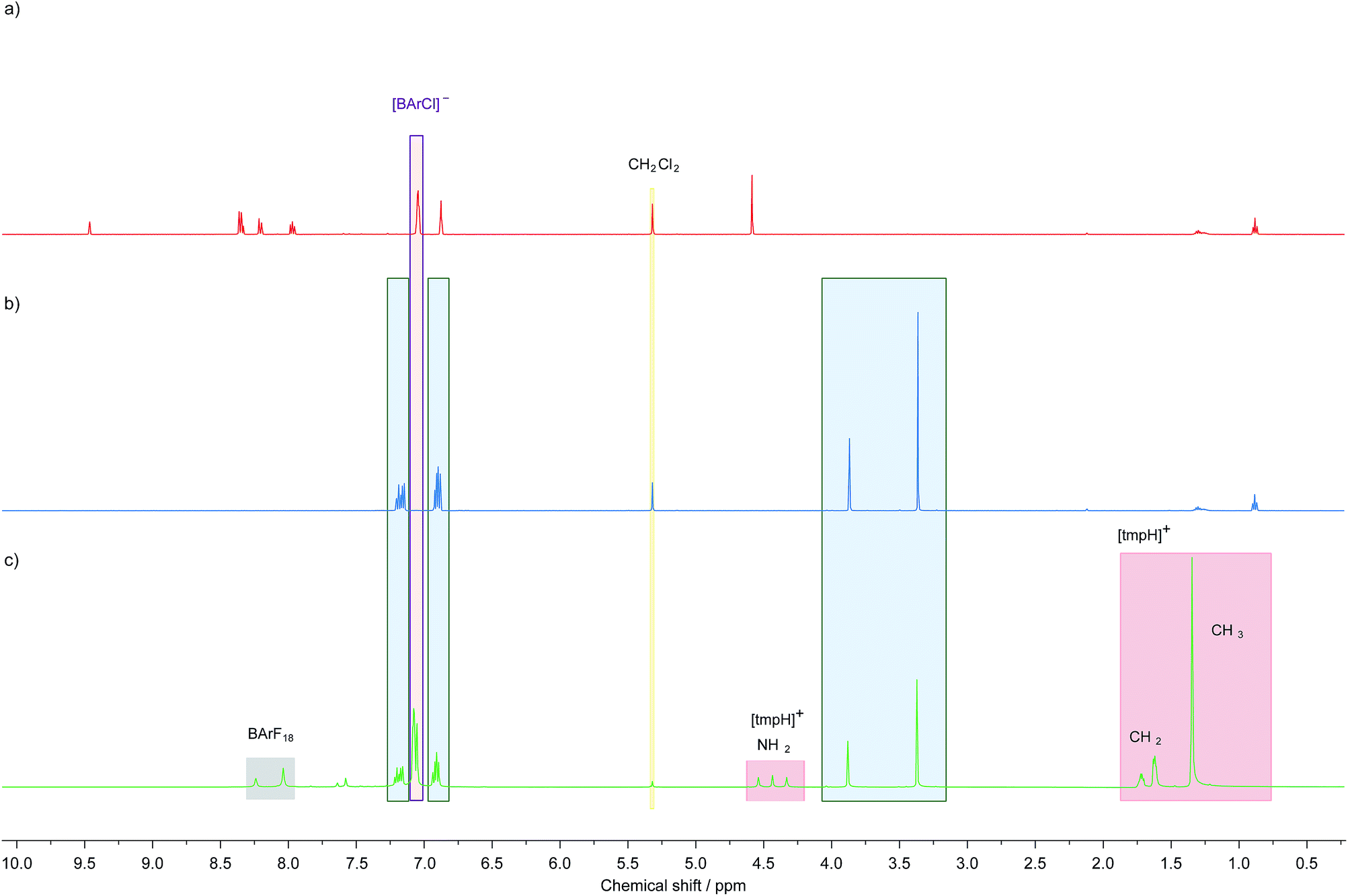 | ||
| Fig. 2 1H NMR spectra demonstrating the ability of [μ-H(BArF18)2]− and [H-BArF18]− to transfer hydride to 1[BArCl] in CD2Cl2, (a) 1[BArCl], (b) authentic 1-H, and (c) an equimolar mixture of 1[BArCl] and [tmpH][μ-H(BArF18)2] after 30 minutes at 20 °C. | ||
For proof of concept, a sample of 1[BArCl] (1.0 equivalent), BArF18 (2.3 equivalents) and 2,6-lutidine (1.7 equivalents) in CD2Cl2 were combined. 2,6-Lutidine was chosen as the Lewis base because it is known to be compatible with 1+ and allows direct comparison to previous work.38 Importantly, in a control experiment 2,6-lutidine was found to be compatible with BArF18 as a FLP, with no evidence for adduct formation observed by NMR spectroscopy when an equimolar mixture of BArF18 and 2,6-lutidine was left for 2 days in CD2Cl2 (Fig. S8–S10†). On exposure of the three component mixture to H2 (4 bar) at room temperature, the progress of 1-H formation was monitored by the disappearance of the CH signal of 1+ (at δ 9.4 ppm) and the appearance of the CH2 signal of 1-H (at δ 3.9 ppm) in the 1H NMR spectrum (Fig. 3 and S4†). After only 25 minutes at 20 °C, 30% of 1[BArCl] had been converted to 1-H; quantitative conversion was achieved after 17 hours. This represents a significant improvement in H2 cleavage rate compared to 1+/lut in the absence of BArF18, which requires over 9 days of heating at 60 °C before it approaches completion.38 Additionally, no evidence for CO binding was observed via NMR spectroscopy when the three component mixture (1+/BArF18/lut) was sparged with pure CO gas for 30 seconds (Fig. S5 and S7b†). On admission of excess H2 to the sample headspace, the usual formation of 1-H occurred with no discernible signals corresponding to a formyl-borate species (Fig. S7†).48 This suggests that, in contrast to Pt or bioinspired organometallic electrocatalysts for H2 oxidation,5,14 our electrochemical–FLPs are CO tolerant and are not poisoned or otherwise inhibited, even in the presence of significant CO.
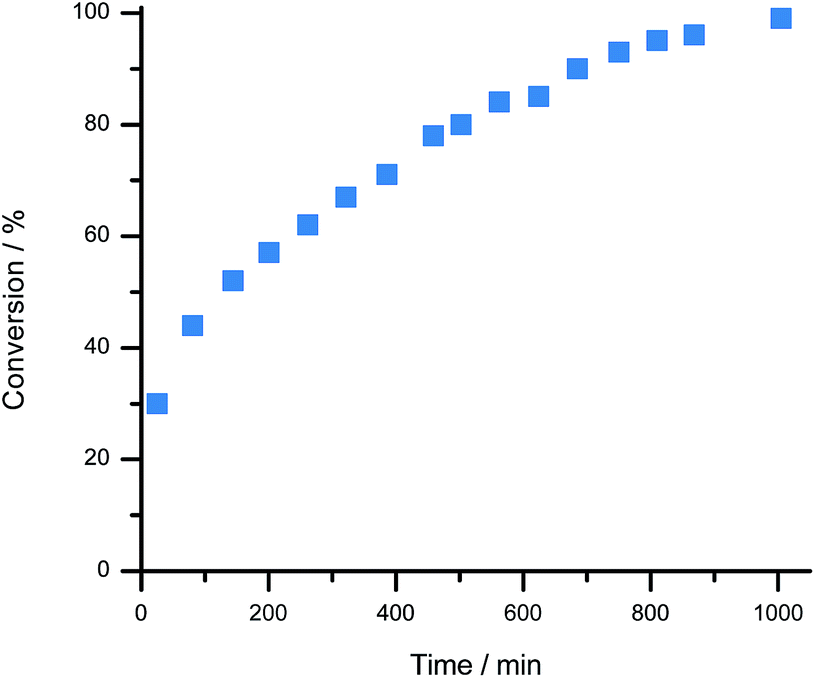 | ||
| Fig. 3 Progress of H2 activation by 1[BArCl] in the presence of the mediator, BArF18. H2 (4 bar) was admitted to a sample of 1[BArCl] (0.028 mmol, 1.0 equivalents), BArF18 (0.064 mmol, 2.3 equivalents), and 2,6-lutidine (0.048 mmol, 1.7 equivalents) in CD2Cl2 and the formation of 1-H product was monitored using 1H NMR spectroscopy. | ||
Electrochemical–FLP experiments
Cyclic voltammetry was performed at a glassy carbon electrode (GCE) on solutions of 1-H in CH2Cl2 containing 0.1 M [nBu4N][B(C6F5)4] as a weakly-coordinating supporting electrolyte. In the absence of Brønsted base, cyclic voltammograms (CVs) of 1-H exhibit a single-electron oxidation wave that is devoid of a back-peak (appears to be irreversible) until scan rates exceed 300 mV s−1 (Fig. S11†). In the presence of excess 2,6-lutidine, electrochemical reversibility is lost at all scan rates (Fig. S12†) and the peak current obtained for 1-H approximately doubles (Fig. 4) – a 2-fold increase in peak current is observed at 50 mV s−1 and a 1.7-fold increase is observed at 2000 mV s−1. This effect is highly indicative of an underlying ECE-DISP1 mechanism, as reported by Savéant and co-workers previously.43 A peak potential of +0.47 V vs. Cp2Fe0/+ was obtained for 1-H at the 100 mV s−1 scan rate. This is represents a 1 V decrease in the potential that is required for H2 oxidation at a GCE, a very significant energy saving that is equivalent to ca. 197 kJ mol−1, and provides a further 110 mV improvement over the previous most suitable borenium-based electrochemical–FLP system.35 | ||
| Fig. 4 CVs comparing the electrochemical behaviour of 1-H (1.8 mM) with (red line) and without (blue line) the addition of excess Brønsted base (2,6-lutidine; 6.7 mM) at a scan rate of 100 mV s−1. | ||
Note that 2,6-lutidine has the added benefit of being electrochemically inactive within the potential window of our electrolyte system. This is unlike the phosphine and aliphatic amine bases used in our previous electrochemical–FLP studies which oxidize at similar potentials to the borohydrides, leading to electrode passivation and failure of the system.
Applied H2 oxidation and electrocatalyst recyclability
The 1+/BArF18/lut system was next applied towards the in situ oxidation of H2 with the intention of investigating whether the electrocatalyst (1+) could participate in successive charging and discharging cycles. The advantage of using BArF18 as a hydride shuttle is that the oxidation potential of [(μ-H) (BArF18)2]− is on the limit of the oxidative potential window, and does not interfere with the measurement of 1-H concentration at the electrode surface.A sample of 1[B(C6F5)4] was electrosynthesised via the controlled-potential bulk electrolysis of 1-H (0.1 equivalent) in the presence of excess 2,6-lutidine (11 equivalents) at a Toray carbon paper electrode. An initial CV scan of 1-H (recorded at a GCE) produced a peak current of 153 μA, and 8.03C of charge was passed during the initial bulk electrolysis step – this data is represented by the dotted line in Fig. 5. The formation of 1+ was further indicated by the solution turning bright yellow.
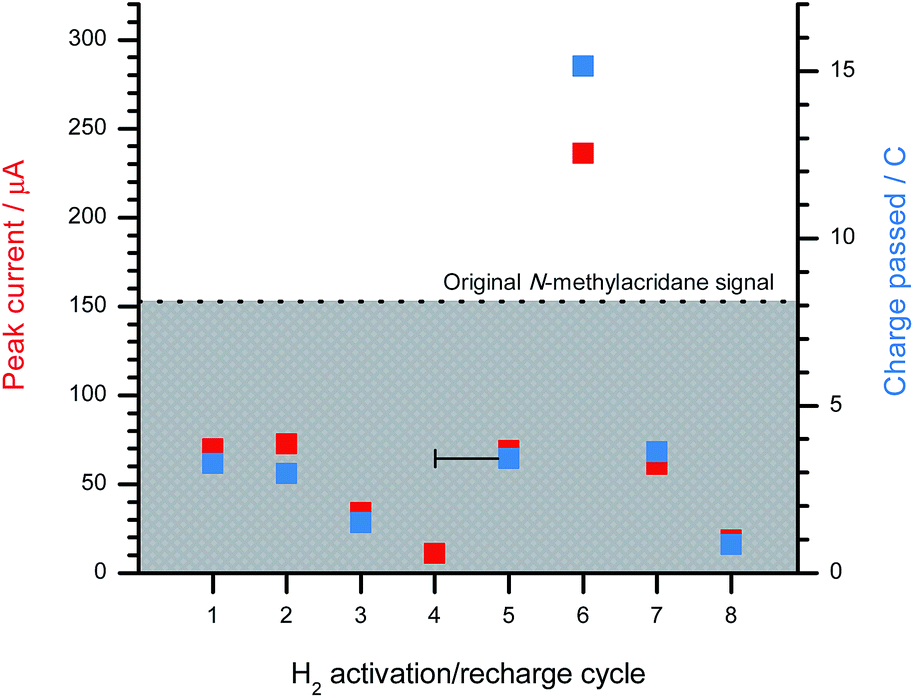 | ||
| Fig. 5 The peak current obtained at a GCE (left y-axis, red) and the charge passed at a Toray carbon paper electrode (right y-axis, blue) after sparging a freshly generated 1[B(C6F5)4] (3.7 mM, 10 mol%) solution in CH2Cl2 with H2 for 20 minutes in the presence of 2,6-lutidine (41 mM, 11 equivalents) and BArF18 (37 mM, 1 equivalent). The dotted line represents the peak current/charge passed for the original 1-H sample, which was converted to 1[B(C6F5)4] via bulk electrolysis, and provides a reference point for the following H2 activation cycles. Additional 2,6-lutidine was added from cycle 4 onwards. | ||
An equivalent of BArF18 was added (relative to the catalyst, 1+, which is present at 10 mol%) and the sample was sparged with H2 gas for 20 minutes before a CV was recorded at the GCE. The CV clearly demonstrated the regeneration of considerable amounts of 1-H, even at this short sparging time, with the peak current for this first H2 activation cycle at 46% (in agreement with the NMR studies above) of that passed for the original 1-H sample prior to bulk electrolysis. The sample was electrolyzed back to 1[B(C6F5)4], passing 3.28C of charge (41% of that passed for the original sample). H2 activation was then repeated for the second time (again, with only a 20 minute sparge) at which point the observed peak current was comparable to that obtained for the first H2 activation. On repeating the H2 activation a third time, the peak current and charge passed for 1-H during bulk electrolysis was somewhat diminished compared to the initial two attempts (to ca. 20% of the original sample values). A fourth H2 activation attempt was unsuccessful, with no regeneration of 1-H.
It was suspected that the system was no longer turning over due to the depletion of 2,6-lutidine via its sequestration by protons generated during the bulk electrolysis of 1-H and also in the H2 activation cycles by the FLP. In a fuel cell, H2 oxidation constitutes only one half-reaction of the redox couple; the other half-reaction, O2 reduction, would consume any protons that are generated during H2 oxidation and regenerate the Brønsted base. Thus, at this point in the experiment, the number of regeneration cycles was limited by the quantity of available 2,6-lutidine. To overcome this issue, an additional 10 equivalents of 2,6-lutidine were added to the sample, which was then subjected to a further 20 minute sparge with H2. Reassuringly, this fifth H2 activation run successfully regenerated 1-H in similar concentrations (ca. 45% of the original sample concentration after a 20 minute sparge) to those obtained during the first two H2 activation attempts. The sample was then subjected to bulk electrolysis.
To investigate the effect of exposing the sample to H2 for longer periods of time, the sample was left sealed under H2 for 2.5 days. To great surprise, the resulting CV (cycle 6) exhibited a 1.5-fold increase in peak current compared to the original 1-H sample. It is likely that excess [lutH][(μ-H) (BArF18)2] builds up in solution once all 1+ (present at 10 mol% cf. the borane) has been converted back to 1-H. As 1-H undergoes oxidation at the electrode surface, the electrogenerated 1+ is rapidly converted back to 1-H via reaction with the excess [(μ-H) (BArF18)2]−. This leads to an enhancement in the peak current of the 1-H oxidation wave i.e. a perceived electrocatalytic effect. This effect was confirmed experimentally by treating a sample of 1-H with increasing quantities (0, 0.5, 1, and 2 equivalents) of the hydride donor [nBu4N][HB(C6F5)3] (Fig. S13a†). The addition of [nBu4N][HB(C6F5)3] resulted in a proportional increase in the peak current of the 1-H wave (Fig. S13b†). Note that whilst [HB(C6F5)3]− is redox active, its peak potential is observed at +0.88 V vs. Cp2Fe0/+ and therefore does not interfere with the 1-H oxidation wave. The fact that the peak current of 1-H increases, with no observable wave corresponding to [HB(C6F5)3]−, suggests that hydride shuttling occurs within the timescale of the electrode process – i.e. the system is not only rechargeable, but it is catalytic and turning over many times per H2-charge cycle. Digital simulation of this electrochemical data determined the turnover frequency of the hydride shuttling process to be 2.7 ± 0.2 × 104 s−1.
Henceforth, excess 2,6-lutidine (10 equivalents) was added after each bulk electrolysis step to ensure that that system recyclability was not limited by the concentration of Brønsted base. Following bulk electrolysis, the sample containing 1+ was subjected to further H2 activation (recharging, cycle 7) and bulk electrolysis cycles (discharging) until 1-H could no longer be regenerated. Only one successful regeneration cycle was performed before no further 1-H formation was observed. Since the 2,6-lutidine concentration was not the limiting factor, it is likely that the deactivation of the electrocatalytic system resulted from the decomposition of the boron-based Lewis acid, BArF18 (whose concentration was not altered from the initial experiment in the series), over the course of several charging and discharging cycles. Indeed, BArF18 is relatively sensitive to trace amounts of adventitious air and moisture. Despite this, the 1+/1-H carbon-based Lewis acid system was confirmed to still be fully active when the addition of [nBu4N][HB(C6F5)3] resulted in successful recovery of the oxidation wave corresponding to 1-H.
Conclusions
The 1+/BArF18/lut system provides a new and improved electrochemical–FLP approach to H2 oxidation by combining the best attributes of two different Lewis acids: one carbon-based with excellent electrochemical attributes, and one boron-based with excellent H2 activating attributes as part of a FLP. Unlike conventional, precious metal or biomimetic electrocatalysts, this system is highly tolerant to CO. The pre-activation of H2 in the form of 1-H results in an astonishing 1 V decrease in the potential that is required for H2 oxidation at ubiquitous carbon electrodes. This represents a significant decrease in the required energetic driving force for H2 oxidation (equivalent to ca. 197 kJ mol−1) and a further 110 mV improvement over previous electrochemical–FLP systems. In addition to this (and in contrast to our previous electrochemical–FLP systems) there are no H2 evolution side-reactions due to the reaction of incoming hydride with electrogenerated H+; this leads to a marked improvement in efficiency and recyclability.The completely metal-free system is electrocatalytic with respect to the carbon-based Lewis acid 1+ and can be turned over multiple times without any loss of activity. The “hydride shuttle” effect provided by the synergistic interaction of BArF18 and 1+ gives rise to a significant improvement in the overall rates of H2 cleavage and the generation of 1-H by the carbon-based FLP.
We see two routes to further improve this electrochemical–FLP system. One, to develop a boron-based FLP that exhibits a greater stability to air and moisture whilst retaining the ability to rapidly cleave H2 and to function as an efficient “hydride shuttle”. Indeed, we have already demonstrated that solutions of B(C6F5)3 in 1,4-dioxane can be rendered water tolerant simply by operating at increased pressures of H2.33 Alternatively, an analogous carbon-based Lewis acid is required that is capable of rapid H2 activation when combined with a suitable Lewis base, without requiring the presence of any additional boron-based Lewis acid as a hydride shuttle. Both approaches form part of our ongoing research efforts.
Acknowledgements
The research leading to these results has received funding from the European Research Council under ERC Starting Grant Agreement no. 307061 (PiHOMER), ERC PoC Grant Agreement no. 640988 (FLPower) and the Leverhulme Trust. MJI & GGW thank the Royal Society for financial support via University Research Fellowships.Notes and references
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- F. Barbir, PEM Fuel Cells: Theory and Practice, Academic Press, Waltham, MA, 2013 Search PubMed.
- A. J. Bard and L. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2nd edn, 2001 Search PubMed.
- B. E. Conway and B. V. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- M. Rakowski DuBois and D. L. Dubois, in Catalysis Without Precious Metals, ed. R. M. Bullock, Wiley, Weinheim, 2010, pp. 165–180 Search PubMed.
- D. L. DuBois and R. M. Bullock, Eur. J. Inorg. Chem., 2011, 1017–1027 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- J. Y. Yang, S. Chen, W. G. Dougherty, W. S. Kassel, R. M. Bullock, D. L. DuBois, S. Raugei, R. Rousseau, M. Dupuis and M. Rakowski-DuBois, Chem. Commun., 2010, 46, 8618–8620 RSC.
- A. L. Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef PubMed.
- J. Y. Yang, R. M. Bullock, W. J. Shaw, B. Twamley, K. Fraze, M. Rakowski-DuBois and D. L. DuBois, J. Am. Chem. Soc., 2009, 131, 5935–5945 CrossRef CAS PubMed.
- T. Liu, D. L. DuBois and R. M. Bullock, Nat. Chem., 2013, 5, 228–233 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2012, 4, 26–30 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, J. Am. Chem. Soc., 2011, 133, 8098–8101 CrossRef CAS PubMed.
- J. J. Baschuk and X. Li, Int. J. Energy Res., 2001, 25, 695–713 CrossRef CAS.
- Methanol: The Basic Chemical and Energy Feedstock of the Future, ed. M. Bertau, H. Offermanns, L. Plass, F. Schmidt and H.-J. Wernicke, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014 Search PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, in Comprehensive Inorganic Chemistry II, ed. J. R. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 1069–1103 Search PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- D. W. Stephan, Nat. Chem., 2014, 6, 952–953 CrossRef CAS PubMed.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701 RSC.
- D. W. Stephan, Org. Biomol. Chem., 2012, 10, 5740–5746 CAS.
- D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338–12348 CrossRef CAS PubMed.
- P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543–7546 CrossRef CAS PubMed.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2014, 53, 10218–10222 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS PubMed.
- D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter and A. E. Ashley, ACS Catal., 2015, 5, 5540–5544 CrossRef CAS PubMed.
- E. J. Lawrence, V. S. Oganesyan, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, J. Am. Chem. Soc., 2014, 136, 6031–6036 CrossRef CAS PubMed.
- E. J. Lawrence, T. J. Herrington, A. E. Ashley and G. G. Wildgoose, Angew. Chem., Int. Ed., 2014, 53, 9922–9925 CrossRef CAS PubMed.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS PubMed.
- E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2014, 53, 11306–11309 CrossRef CAS PubMed.
- E. R. Clark and M. J. Ingleson, Organometallics, 2013, 32, 6712–6717 CrossRef CAS.
- Y. Lu, D. Endicott and W. Kuester, Tetrahedron Lett., 2007, 48, 6356–6359 CrossRef CAS.
- C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498 RSC.
- X.-Q. Zhu, Y.-C. Liu and J.-P. Cheng, J. Org. Chem., 1999, 64, 8980–8981 CrossRef CAS.
- P. Hapiot, J. Moiroux and J.-M. Savéant, J. Am. Chem. Soc., 1990, 112, 1337–1343 CrossRef CAS.
- A. Anne, S. Fraoua, V. Grass, J. Moiroux and J.-M. Savéant, J. Am. Chem. Soc., 1998, 120, 2951–2958 CrossRef CAS.
- R. J. Blagg, E. J. Lawrence, K. Resner, V. S. Oganesyan, T. J. Herrington, A. E. Ashley and G. G. Wildgoose, Dalton Trans., 2016 10.1039/c5dt01918d.
- T. J. Herrington, A. J. W. Thom, A. J. P. White and A. E. Ashley, Dalton Trans., 2012, 41, 9019 RSC.
- E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Eur. J., 2012, 18, 16938–16946 CrossRef CAS PubMed.
- R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 4974–4977 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04564a |
| ‡ Present address: School of Physical Sciences, Ingram Building, University of Kent, Canterbury, Kent, CT2 7NH, UK. |
| This journal is © The Royal Society of Chemistry 2016 |