
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient metal-free photochemical borylation of aryl halides under batch and continuous-flow conditions†
Kai
Chen
,
Shuai
Zhang
,
Pei
He
and
Pengfei
Li
*
Center for Organic Chemistry, Frontier Institute of Science and Technology (FIST), Xi'an Jiaotong University, 99 Yanxiang Road, Xi'an, Shaanxi 710054, China. E-mail: lipengfei@mail.xjtu.edu.cn
First published on 1st February 2016
Abstract
A rapid, chemoselective and metal-free C–B bond-forming reaction of aryl iodides and bromides in aqueous solution at low temperatures was discovered. This reaction is amenable to batch and continuous-flow conditions and shows exceptional functional group tolerance and broad substrate scope regarding both the aryl halide and the borylating reagent. Initial mechanistic experiments indicated a photolytically generated aryl radical as the key intermediate.
Introduction
Arylboronic acids and esters have found broad applications in chemical, medicinal and materials sciences. In synthetic organic chemistry, in particular, they are versatile synthons for the formation of carbon–carbon or carbon–heteroatom bonds.1 Conventional methods for generating arylboron compounds involve reactions of arylmetallic intermediates with trialkyl borates, followed by transesterification or hydrolysis. These reactions suffer some major drawbacks such as limited functional group tolerance as well as the necessity of rigorous anhydrous conditions (Scheme 1a).2 In recent decades, transition metal-catalyzed borylation reactions using palladium, nickel, copper and zinc have emerged as highly useful methods for the conversion of C–X bonds to C–B bonds (Scheme 1b).3 More recently, direct C–H borylation methods based on transition-metal catalysts have also been developed.4 In order to reduce the costs and the amount of heavy metal residue in the final products, several transition-metal-free methods for C–B bond formation have been developed. Ito and coworkers discovered an alkali alkoxide-mediated borylation of aryl halides with a silylborane as the unique borylating reagent (Scheme 1c).5 Zhang and coworkers reported that aryl iodides could be borylated with 4.0 equivalents of bis(pinacolato)diboron in refluxing methanol using 2.0 equivalents of Ce2CO3 as the promoter. The reaction time ranged from several hours to days and the yields were generally moderate (Scheme 1d).6 Fernándes and Muñiz transformed diaryliodonium acetates to arylboronates under mild conditions.7 Using aryl amines as the starting material, Wang developed a mild and efficient Sandmeyer-type borylation process.8a–c Borylation of aryl diazonium salts8d–f and aryl triazenes8g has also been reported. In addition, innovative methods for direct C–H borylation under transition metal-free conditions have been reported,9 although the substrates were limited to either electron rich arenes or heterocycles, and air and moisture sensitive reagents were needed. Consequently, a practical, metal-free method that is rapid and effective, works under mild conditions with various readily available borylating reagents, shows high functional group tolerance and avoids strong acids, bases and hazardous reagents is still highly desirable. Herein, we wish to report our discovery and development of a new borylation reaction of aryl halides using light as a clean reagent (Scheme 1e).10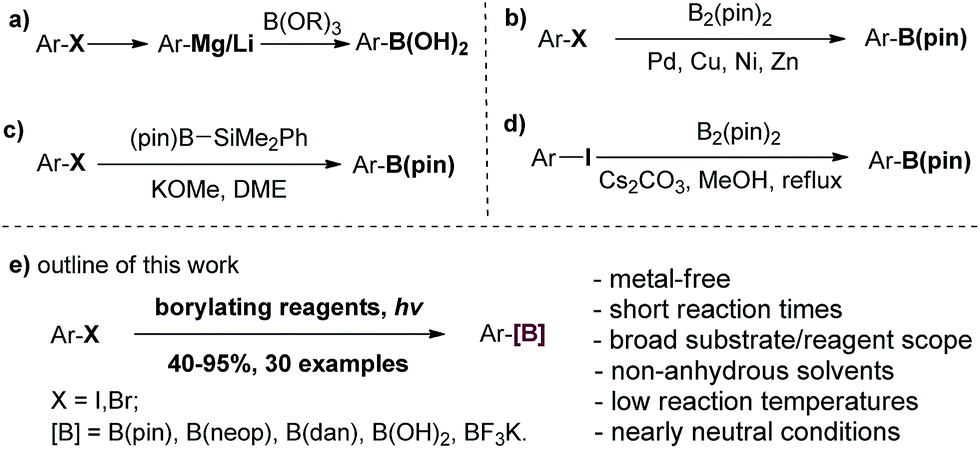 | ||
| Scheme 1 Summary of borylation reactions of aryl halides and outline of this work. | ||
Results and discussion
Initially, a solution of 4-iodoanisole (1a) and bis(pinacolato)-diboron (2) in acetonitrile was placed in a quartz test tube and irradiated with a 300 W high pressure mercury lamp (maximum at 365 nm) for 4 hours. Encouragingly, the desired aryl-B(pin) product 3a was formed in 29% yield based on 1H NMR analysis of the crude product (Table 1, entry 1). Other polar solvents such as trifluoroethanol and methanol did not improve the reaction (entries 2 and 3). Adding water and acetone as co-solvents was beneficial in both cases and increased the yield to 46% (entries 4 and 5). Screening of various organic and inorganic additives revealed that an organic base, N,N,N′,N′-tetramethyldiaminomethane (TMDAM), could further improve the yield to 58% (entry 9). By comparison, other bases led to inferior results (entries 6–8). Interestingly, a greater amount of TMDAM led to a significantly lower yield (entry 10). Using two equivalents of B2(pin)2 could improve the yield to 72% (entry 11). Further optimization by changing the reaction concentration of 1a resulted in a higher yield (c = 0.1 M, 81% yield) (entry 12 vs. 11 and 13).|
|
||||
|---|---|---|---|---|
| Entry | 2 (eq.) | Solvent | Additive (mol%) | Yieldc [%] |
| a Batch conditions: 1a (0.1–0.2 mmol, c = 0.05 M/0.1 M), 2 (0.1–0.4 mmol), RT, 4 h. b Flow conditions: 1a (c = 0.1 M), −5 °C, residence time 15 min. c Determined by 1H NMR with 1,3,5-trimethoxybenzene as an internal standard. d c = 0.1 M. e c = 0.2 M; TMEDA: N,N,N,N-tetramethylethylenediamine; TMDAM: N,N,N′,N′-tetramethyldiaminomethane. | ||||
| Batch conditions | ||||
| 1 | 1.0 | MeCN | None | 29 |
| 2 | 1.0 | TFE | None | 26 |
| 3 | 1.0 | MeOH | None | 15 |
| 4 | 1.0 | MeCN/H2O | None | 42 |
| 5 | 1.0 | MeCN/H2O/acetone | None | 46 |
| 6 | 1.0 | MeCN/H2O/acetone | Cs2CO3 (100) | 16 |
| 7 | 1.0 | MeCN/H2O/acetone | KOtBu (100) | 12 |
| 8 | 1.0 | MeCN/H2O/acetone | TMEDA (50) | 52 |
| 9 | 1.0 | MeCN/H2O/acetone | TMDAM (50) | 58 |
| 10 | 1.0 | MeCN/H2O/acetone | TMDAM (100) | 39 |
| 11 | 2.0 | MeCN/H2O/acetone | TMDAM (50) | 72 |
| 12 | 2.0 | MeCN/H 2 O/acetone | TMDAM (50) | 81 |
| 13e | 2.0 | MeCN/H2O/acetone | TMDAM (50) | 55 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Flow conditions | ||||
| 14 | 2.0 | MeCN/H2O/acetone | TMDAM (50) | 87 |
| 15 | 1.5 | MeCN/H 2 O/acetone | TMDAM (50) | 88 |
During the study, we observed gradual decomposition of B2(pin)2. We felt that continuous-flow photolytic conditions might help in reducing the amount of B2(pin)2 by competitively accelerating the desired reaction. In comparison with a typical batch photoreactor, microchannel photochemical reactors have significant benefits for reaction efficiency, yield, reproducibility, material throughput and scale-up.11–13 Based on the method developed by Booker-Milburn11a and our own experience in flow chemistry,14 we designed and assembled a continuous-flow photochemical reactor. Thus, transparent fluorinated ethylene propylene (FEP) tubing (reaction volume 780 μL) was coiled around a jacketed quartz immersion well in which the mercury lamp was situated. The reaction temperature was regulated by a cooling liquid circulating pump (see ESI†). A stock solution containing all reactants and reagents was introduced into the tubing using a syringe pump. To our delight, running the reaction under the same conditions as entry 12 but in continuous-flow mode gave 3a in excellent yield (87%, entry 14) with a residence time of only 15 minutes. Indeed, the amount of B2(pin)2 could be reduced to 1.5 equivalents without affecting the reaction efficiency (88% yield, entry 15).
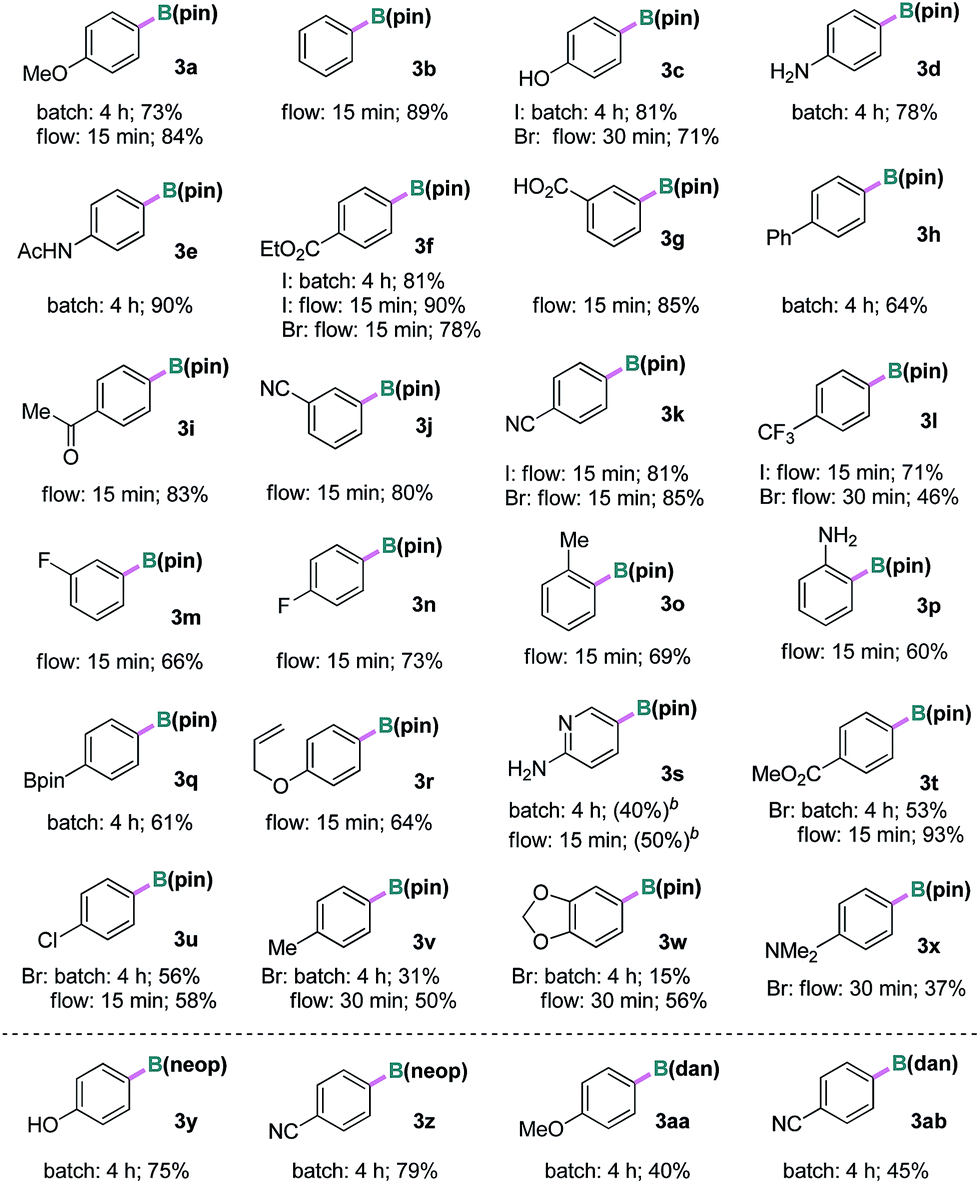
With the optimized conditions in hand, we examined the substrate scope of the current borylation reaction under batch and/or continuous-flow conditions, as summarized in Table 2. Iodoarenes with various electron-donating, -neutral and -withdrawing groups at the para-, meta-, or ortho-positions, including hydroxyl, amino, amide, ester, acid, ketone, cyano, fluorine, boronate and trifluoromethyl groups, were all efficiently converted to the corresponding aryl pinacol boronates in good to excellent yields (3a–3r). Groups potentially reactive under UV light such as aryl ketone (for 3i) and biaryl (for 3h) were compatible. A substrate containing an allyl ether group was also viable (3r), which is interesting considering that the reaction might involve a reactive carbon-based radical and the double bond could be attacked. In addition, the borylation of 2-amino-5-iodopyridine was possible, and a moderate yield of the corresponding boronate 3s was observed by 1H NMR spectroscopic analysis. Attempts to purify 3s were unsuccessful due to its decomposition on silica gel. Furthermore, when aryl bromides were subjected to the same reaction conditions, the desired products were produced in comparable or slightly lower yields than the iodides (3c, 3f, 3k, 3l and 3t–3x). Finally, different borylating reagents were utilized under otherwise identical conditions. Reactions using bis(neopentanediolato)diboron B2(neop)2 successfully afforded the desired products in good yields (3y and 3z). Interestingly, when an unsymmetrical diboron (pin)B–B(dan) was employed, selective introduction of the B(dan) moiety was realized (3aa and 3ab) and no aryl pinacol boronate was observed.15 To demonstrate the stability and usefulness of this reaction in larger scale preparation, the borylation reactions of iodobenzene and 4-iodophenol were carried out at gram scale (10.0 mmol) employing a commercial automated flow chemistry system (reactor volume 7.8 mL, see ESI†). Without any further optimization, the reactions produced the desired arylboronate products in excellent isolated yields (3b 90% and 3c 93%) and the productivity corresponded to ∼3 mmol h−1.
|
|
|---|
| a Batch conditions: 1a (0.2 mmol, c = 0.1 M), 2 (0.4 mmol, 2.0 eq.), TMDAM (0.5 eq.), RT, 4 h; flow conditions: 1a (c = 0.1 M), 2 (1.5 eq.), TMDAM (0.5 eq.), −5 °C, residence time 15–30 min. b Determined by 1H NMR with 1,3,5-trimethoxybenzene as an internal standard; TMDAM: N,N,N′,N′-tetramethyldiaminomethane. |
|
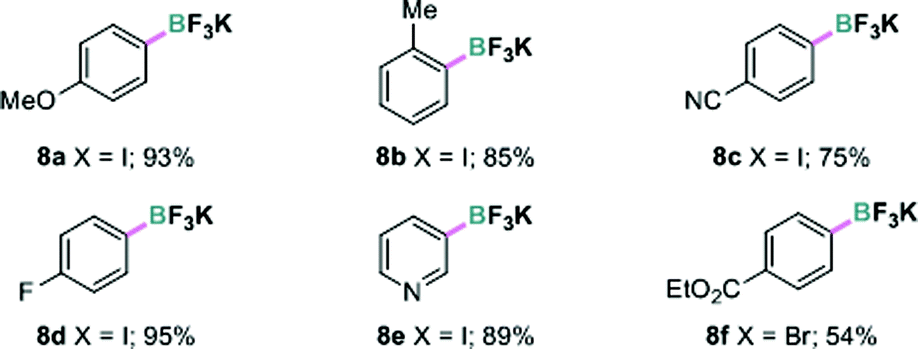
Encouraged by the above results, we further investigated the possibility of using a more atom economical borylating reagent, bis-boronic acid (BBA, 6). Largely because its polar protic properties may not be amenable to most known borylation methods, this reagent has only recently been successfully used in palladium or nickel-catalyzed Miyaura borylation by Molander and coworkers.16 In the present borylation, pleasingly, we were able to convert 4-iodoanisole 1a to the corresponding boronic acid 7a under continuous-flow conditions in quantitative yield based on 1H NMR analysis (residence time 10 minutes). The key variation from the previous conditions was using aqueous methanol (MeOH:H2O = 4:1 v/v) as the solvent. Due to the inconvenience of isolating the pure arylboronic acid, aqueous KHF2 was added and the resulting potassium aryltrifluoroborate 8a was obtained in 93% yield. Other aryl and heteroaryl iodides and a bromide were also transformed to the boronates in good to excellent yields in this manner (Table 3).
|
|
|---|
|
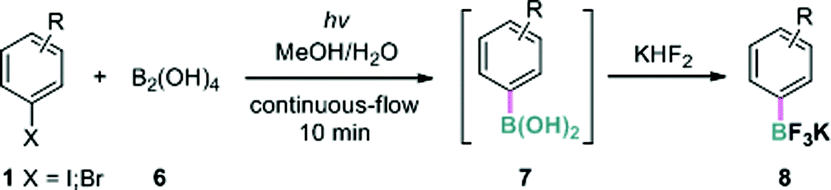
To gain insight into the reaction mechanism, and particularly to probe the role of additives and light, we conducted a series of control experiments (Table 4). When the batch reaction of 1f with B2(pin)2 was run under the standard conditions, deiodination product 9 was formed in 7% yield in addition to the borylation product 3f (entry 1). In the absence of both TMDAM and light (entry 2), no conversion was observed. However, the reaction with 0.5 equivalents of TMDAM in the dark led to a small amount of 3f (entry 3); higher reaction temperatures and prolonged reaction time had little influence on the outcome. A hydrogen atom donor, Bu3SnH, increased the conversion but led to 9 as the major product (entry 4). Furthermore, the reaction with Bu3SnH under UV irradiation afforded 9 in high yield (entry 5). Similarly, using 9,10-dihydroanthracene instead of Bu3SnH, 9 (26%) and concomitant anthracene (11%) were observed (entry 6). Finally, when TEMPO was added as a radical scavenger, the conversion was low and four products including 3f (15%), 9 (11%), the aryl-TEMPO adduct 10 (14%) and ethyl 4-hydroxybenzoate 11 (26%) were formed (entry 7).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Light | TMDAM | Additive | Conversion [%] | Yield of 3f [%] | Yield of 9 [%] |
| a Reactions were run in batch and yields were determined by 1H NMR spectroscopic analysis with 1,3,5-trimethoxybenzene as an internal standard. b 11% of anthracene was formed. TMDAM: N,N,N′,N′-tetramethyldiaminomethane; DHA: 9,10-dihydroanthracene; TEMPO: (2,2,6,6-tetramethylpiperidin-1-yl)oxyl. | ||||||
| 1 | + | + | − | 100 | 81 | 7 |
| 2 | − | − | − | 0 | 0 | 0 |
| 3 | − | + | − | 13 | 13 | 0 |
| 4 | − | + | Bu3SnH | 46 | 17 | 26 |
| 5 | + | + | Bu3SnH | 100 | 18 | 80 |
| 6 | + | + | DHA | 68 | 42 | 26b |
| 7 | + | + | TEMPO | 69 | 15 | 11 |
Based on the experimental results and related reports on photolytic reactions of aryl iodides,17 we propose two pathways both involving an aryl radical intermediate as the possible reaction mechanism (Scheme 2). The excited state 12 is generated by UV irradiation of aryl iodide 1. In path A, 12 undergoes homolytic C–I bond cleavage to form aryl radical 13 and an iodine atom. Under aqueous conditions, TMDAM activates a water molecule, combining with B2(pin)2 (2) to form a sp3–sp2 diboron species 14.7,8f,18 Aryl radical 13 then reacts with 14 to produce arylboronate 3 and a boryl radical anion 15.1915 can also be viewed as an anionic base-stabilized boryl radical.20 Alternatively, in path B, the excited state 12 or the starting aryl iodide 1 (when in darkness, although with low efficiency) is reduced by TMDAM via a single electron transfer (SET) process to form radical anion 16 and TMDAM-derived radical cation 17. 16 then undergoes C–I bond cleavage to generate aryl radical 13 and iodide anion. Finally, 15 is oxidized by the iodine atom from path A or TMDAM-derived radical cation 17 from path B to form borate 18 as a byproduct.
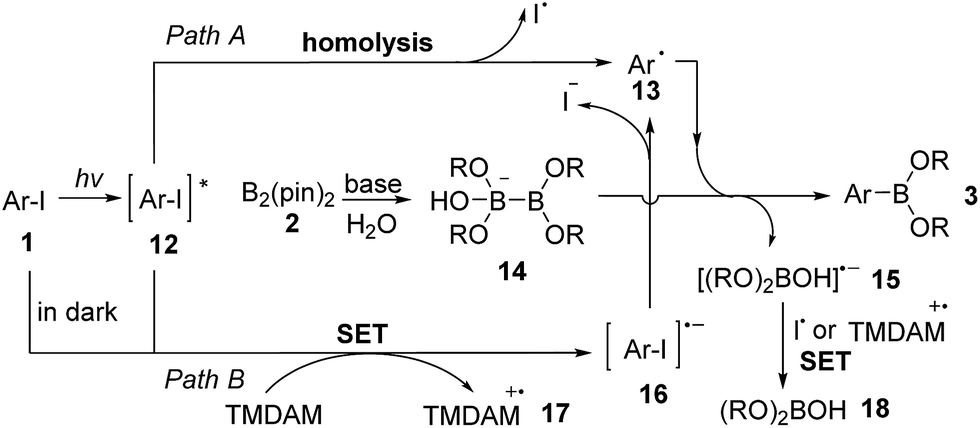 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
In summary, we have discovered a novel and efficient photolytic borylation reaction of aryl halides using diboron reagents. This metal-free reaction features very mild conditions, short reaction times, generally high yields and broad functional group tolerance. Considering the reaction conditions, borylating reagent types and possible reaction mechanism, this work represents an important complementary approach to the existing C–B bond formation methods. Further studies on the mechanism and synthetic applications of this reaction are ongoing.Acknowledgements
This work was financially supported by the NSFC (No. 21472146), the Department of Science and Technology of Shaanxi Province (No. 2015KJXX-02) and the Ministry of Science and Technology of the People's Republic of China (No. 2014CB548200). We thank Prof. Que (Xi'an Jiaotong University) and Dr Duncan Guthrie (Vapourtec) for their generous sharing of the batch and flow photochemistry equipment.Notes and references
- For reviews on arylboronic acid derivatives, see: (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) D. G. Hall, in Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2011, pp. 1–134 CrossRef; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (d) L. Xu, S. Zhang and P. Li, Chem. Soc. Rev., 2015, 44, 8848 RSC.
- (a) H. C. Brown and T. E. Cole, Organometallics, 1983, 2, 1316 CrossRef CAS; (b) H. C. Brown, M. Srebnik and T. E. Cole, Organometallics, 1986, 5, 2300 CrossRef CAS; (c) O. Baron and P. Knochel, Angew. Chem., Int. Ed., 2005, 44, 3133 CrossRef CAS PubMed; (d) C. Pintaric, S. Olivero, Y. Gimbert, P. Y. Chavant and E. Duñach, J. Am. Chem. Soc., 2010, 132, 11825 CrossRef CAS PubMed.
- (a) T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508 CrossRef CAS; (b) W. Zhu and D. Ma, Org. Lett., 2006, 8, 261 CrossRef CAS PubMed; (c) K. L. Billingsley, T. E. Barder and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 5359 CrossRef CAS PubMed; (d) C. M. So, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 8059 CrossRef CAS PubMed; (e) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350 CrossRef CAS PubMed; (f) D. A. Wilson, C. J. Wilson, C. Moldoveanu, A. M. Resmerita, P. Corcoran, L. M. Hoang, B. M. Rosen and V. Percec, J. Am. Chem. Soc., 2010, 132, 1800 CrossRef CAS PubMed; (g) Y. Nagashima, R. Takita, K. Yoshida, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2013, 135, 18730 CrossRef CAS PubMed; (h) C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754 CrossRef CAS PubMed; (i) S. K. Bose and T. B. Marder, Org. Lett., 2014, 16, 4562 CrossRef CAS PubMed; (j) W. K. Chow, O. Y. Yuen, P. Y. Choy, C. M. So, C. P. Lau, W. T. Wong and F. Y. Kwong, RSC Adv., 2013, 3, 12518 RSC; (k) T. Niwa, H. Ochiai, Y. Watanabe and T. Hosoya, J. Am. Chem. Soc., 2015, 137, 14313 CrossRef CAS PubMed; (l) S. K. Bose, A. Deiβenberger, A. Eichhorn, P. G. Steel, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2015, 54, 11843 CrossRef CAS PubMed.
- For selected references on catalytic C–H borylation, see: (a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (b) C. N. Iverson and M. R. Smith III, J. Am. Chem. Soc., 1999, 121, 7696 CrossRef CAS; (c) H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS PubMed; (d) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS PubMed; (e) S. Kawamorita, H. Ohmiya, K. Hara, A. Fukuoka and M. Sawamura, J. Am. Chem. Soc., 2009, 131, 5058 CrossRef CAS PubMed; (f) H. X. Dai and J. Q. Yu, J. Am. Chem. Soc., 2012, 134, 134 CrossRef CAS PubMed; (g) T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258 CrossRef CAS PubMed; (h) L. Xu, S. Ding and P. Li, Angew. Chem., Int. Ed., 2014, 53, 1822 CrossRef CAS PubMed; (i) J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133 CrossRef CAS PubMed; (j) L.-S. Zhang, G. Chen, X. Wang, Q.-Y. Guo, X.-S. Zhang, F. Pan, K. Chen and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 3899 CrossRef CAS PubMed; (k) G. Wang, L. Xu and P. Li, J. Am. Chem. Soc., 2015, 137, 8058 CrossRef CAS PubMed; (l) S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168 CrossRef CAS; (m) H. Tajuddin, P. Harrisson, B. Bitterlich, J. C. Collings, N. Sim, A. S. Batsanov, M. S. Cheung, S. Kawamorita, A. C. Maxwell, L. Shukla, J. Morris, Z. Lin, T. B. Marder and P. G. Steel, Chem. Sci., 2012, 3, 3505 RSC.
- (a) E. Yamamoto, K. Izumi, Y. Horita and H. Ito, J. Am. Chem. Soc., 2012, 134, 19997 CrossRef CAS PubMed; (b) R. Uematsu, E. Yamamoto, S. Maeda, H. Ito and T. Taketsugu, J. Am. Chem. Soc., 2015, 137, 4090 CrossRef CAS PubMed; (c) E. Yamamoto, S. Ukigai and H. Ito, Chem. Sci., 2015, 6, 2943 RSC.
- J. Zhang, H.-H. Wu and J. Zhang, Eur. J. Org. Chem., 2013, 6263 CrossRef CAS.
- N. Miralles, R. M. Romero, E. Fernándes and K. Muñiz, Chem. Commun., 2015, 51, 14068 RSC.
- (a) F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1846 CrossRef CAS PubMed; (b) W. Erb, A. Hellal, M. Albini, J. Rouden and J. Blanchet, Chem.–Eur. J., 2014, 20, 6608 CrossRef CAS PubMed; (c) D. Qiu, L. Jin, Z. Zheng, H. Meng, F. Mo, X. Wang, Y. Zhang and J. Wang, J. Org. Chem., 2013, 78, 1923 CrossRef CAS PubMed; (d) J. Yu, L. Zhang and G. Yan, Adv. Synth. Catal., 2012, 354, 2625 CrossRef CAS; (e) R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594 RSC; (f) S. Pietch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem.–Eur. J., 2015, 21, 7082 CrossRef PubMed; (g) C. Zhu and M. Yamane, Org. Lett., 2012, 14, 4560 CrossRef CAS PubMed.
- (a) A. Prokofjevs, J. W. Kamf and E. Vedejs, Angew. Chem., Int. Ed., 2011, 50, 2098 CrossRef CAS PubMed; (b) L. Niu, H. Yang, R. Wang and H. Fu, Org. Lett., 2012, 14, 2618 CrossRef CAS PubMed; (c) V. Bagutski, A. D. Grosso, J. A. Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474 CrossRef CAS PubMed; (d) M. A. Légaré, M. A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513 CrossRef PubMed; (e) S. K. Bose and T. B. Marder, Science, 2015, 349, 473 CrossRef CAS PubMed.
- (a) Synthetic Organic Chemistry, ed. A. G. Griesbeck and J. Mattay, Marcel Dekker, New York, 2005 Search PubMed. For selected recent examples, see: (b) S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647 CrossRef CAS PubMed; (c) W. Liu, L. Li and C. J. Li, Nat. Commun., 2015, 6, 6526 CrossRef CAS PubMed; (d) K. G. Maskill, J. P. Knowles, L. D. Elliott, R. W. Alder and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2013, 52, 1499 CrossRef CAS PubMed.
- For selected references on continuous-flow photochemical reactions, see: (a) B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558 CrossRef CAS PubMed; (b) Y. S. M. Vaske, M. E. Mahoney, J. P. Konopelski, D. L. Rogow and W. J. McDonald, J. Am. Chem. Soc., 2010, 132, 11379 CrossRef CAS PubMed; (c) F. Lévesque and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 1706 CrossRef PubMed; (d) D. C. Harrowven, M. Mohamed, T. P. Gonçalves, R. J. Whitby, D. Bolien and H. F. Sneddon, Angew. Chem., Int. Ed., 2012, 51, 4405 CrossRef CAS PubMed; (e) R. S. Andrews, J. J. Becker and M. R. Gagne, Angew. Chem., Int. Ed., 2012, 51, 4140 CrossRef CAS PubMed; (f) J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2012, 51, 4144 CrossRef CAS PubMed; (g) K. G. Maskill, J. P. Knowles, L. D. Elliott, R. W. Alder and K. I. B. Milburn, Angew. Chem., Int. Ed., 2013, 52, 1499 CrossRef CAS PubMed; (h) Y. Zhang, M. L. Blackman, A. B. Leduc and T. F. Jamison, Angew. Chem., Int. Ed., 2013, 52, 4251 CrossRef CAS PubMed; (i) X. Wang, G. D. Cuny and T. Noël, Angew. Chem., Int. Ed., 2013, 52, 7860 CrossRef CAS PubMed.
- For early pioneering work on flow photochemistry, see: (a) H. Lu, M. A. Schmidt and K. F. Jensen, Lab Chip, 2001, 1, 22 RSC; (b) K. Ueno, F. Kitagawa and N. Kitamura, Lab Chip, 2002, 2, 231 RSC.
- For selected recent reviews on flow photochemistry, see: (a) M. Oelgemöller and O. Shvydkiv, Molecules, 2011, 16, 7522 CrossRef PubMed; (b) J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025 CrossRef CAS PubMed; (c) L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chem.–Eur. J., 2014, 20, 15226 CrossRef CAS PubMed.
- (a) P. Li and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 6396 CrossRef CAS PubMed; (b) P. Li, J. S. Moore and K. F. Jensen, ChemCatChem, 2013, 5, 1729 CrossRef CAS.
- L. Xu and P. Li, Chem. Commun., 2015, 51, 5656 RSC.
- (a) G. A. Molander, S. L. J. Trice and S. D. Dreher, J. Am. Chem. Soc., 2010, 132, 17701 CrossRef CAS PubMed; (b) G. A. Molander, S. L. J. Trice, S. M. Kennedy, S. D. Dreher and M. T. Tudge, J. Am. Chem. Soc., 2012, 134, 11667 CrossRef CAS PubMed; (c) G. A. Molander, L. N. Cavalcanti and C. Garcia-Garcia, J. Org. Chem., 2013, 78, 6427 CrossRef CAS PubMed.
- (a) A. G. Sage, T. A. A. Oliver, D. Murdock, M. B. Crow, G. A. D. Ritchie, J. N. Harvey and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2011, 13, 8075 RSC; (b) M. Budén, J. F. Guastavino and R. A. Rossi, Org. Lett., 2013, 15, 1174 CrossRef PubMed; (c) J. Kan, S. Huang, H. Zhao, J. Lin and W. Su, Sci. China: Chem., 2015, 58, 1329 CrossRef CAS; (d) E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. R. Alaniz, Chem. Commun., 2015, 51, 11705 RSC.
- (a) M. Gao, S. B. Thorpe and W. L. Santos, Org. Lett., 2009, 11, 3478 CrossRef CAS PubMed; (b) A. Bonet, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2010, 49, 5130 CrossRef CAS PubMed; (c) A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158 CrossRef CAS PubMed; (d) H. Wu, J. M. Garcia, F. Haeffner, S. Radomkit, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2015, 137, 10585 CrossRef CAS PubMed; (e) S. B. Thorpe, J. A. Calderone and W. L. Santos, Org. Lett., 2012, 14, 1918 CrossRef CAS PubMed and references therein.
- (a) P. P. Power, Chem. Rev., 2003, 103, 789 CrossRef CAS PubMed; (b) H. Braunschweig, V. Dyakonov, J. O. C. Jimenez, K. Kraft, I. Krummenacher, K. Radacki, A. Sperlich and J. Wahler, Angew. Chem., Int. Ed., 2012, 51, 2977 CrossRef CAS PubMed and references therein.
- (a) S.-H. Ueng, M. M. Brahmi, E. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, J. Am. Chem. Soc., 2008, 130, 10082 CrossRef CAS PubMed; (b) D. Lu, C. Wu and P. Li, Chem.–Eur. J., 2014, 20, 1630 CrossRef CAS PubMed; (c) P. R. Rablen and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 4648 CrossRef CAS; (d) J. A. Baban and B. P. Roberts, J. Chem. Soc., Chem. Commun., 1983, 1224 RSC; (e) D. Lu, C. Wu and P. Li, Org. Lett., 2014, 16, 1486 CrossRef CAS PubMed; (f) J. Lalevée, N. Blanchard, M.-A. Tehfe, A.-C. Chany and J.-P. Fouassier, Chem.–Eur. J., 2010, 16, 12920 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of new compounds are provided. See DOI: 10.1039/c5sc04521e |
| This journal is © The Royal Society of Chemistry 2016 |