
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoselective nitro reduction and hydroamination using a single iron catalyst†
Kailong
Zhu
,
Michael P.
Shaver
* and
Stephen P.
Thomas
*
School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: michael.shaver@ed.ac.uk; stephen.thomas@ed.ac.uk
First published on 26th January 2016
Abstract
The reduction and reductive addition (formal hydroamination) of functionalised nitroarenes is reported using a simple and bench-stable iron(III) catalyst and silane. The reduction is chemoselective for nitro groups over an array of reactive functionalities (ketone, ester, amide, nitrile, sulfonyl and aryl halide). The high activity of this earth-abundant metal catalyst also facilitates a follow-on reaction in the reductive addition of nitroarenes to alkenes, giving efficient formal hydroamination of olefins under mild conditions. Both reactions offer significant improvements in catalytic activity and chemoselectivity and the utility of these catalysts in facilitating two challenging reactions supports an important mechanistic overlap.
Introduction
The chemoselective production of aniline and aniline derivatives is a cornerstone of modern industrial chemistry. The global aniline market was valued at £6.25 billion in 2013 and will reach a gross market value of £10.17 billion by 2020.1 Aniline and aniline derivatives find use in polymers and materials (e.g. polyurethane and rubber), herbicides, bulk chemicals and dyes and pigments (e.g. indigo).2 Current preparation methods rely on the precious metal-catalysed reduction of nitroarenes using high pressures of hydrogen gas, and suffer from low chemoselectivity.3 Silanes have emerged as a hydrogen gas alternative.4Earth abundant metal catalysis is at the fore of academic and industrial research due to the inherent availability, sustainability, low cost and low toxicity of these metals. Iron holds a unique position, offering the lowest environmental and societal impact as it is the most abundant transition metal, non-toxic and environmentally benign. Although great strides have been made in the use of iron catalysts in organic synthesis,5 the reduction of nitro groups has received relatively little attention.6 The current state-of-the-art methods often require high catalyst loadings and long reaction times to transform a limited scope of substrates with low chemoselectivity.
We recently reported the reduction of carbonyl compounds using an iron(III)-amine-bis(phenolate) catalyst 4b and silane as the stoichiometric reductant (Scheme 1a).7 While these complexes were originally used as mediators of controlled radical polymerisations,8 (Scheme 1b) we were surprised that they also showed excellent activity and chemoselectivity for the hydrosilylation of aldehydes and ketones over a wide range of other functionalities.
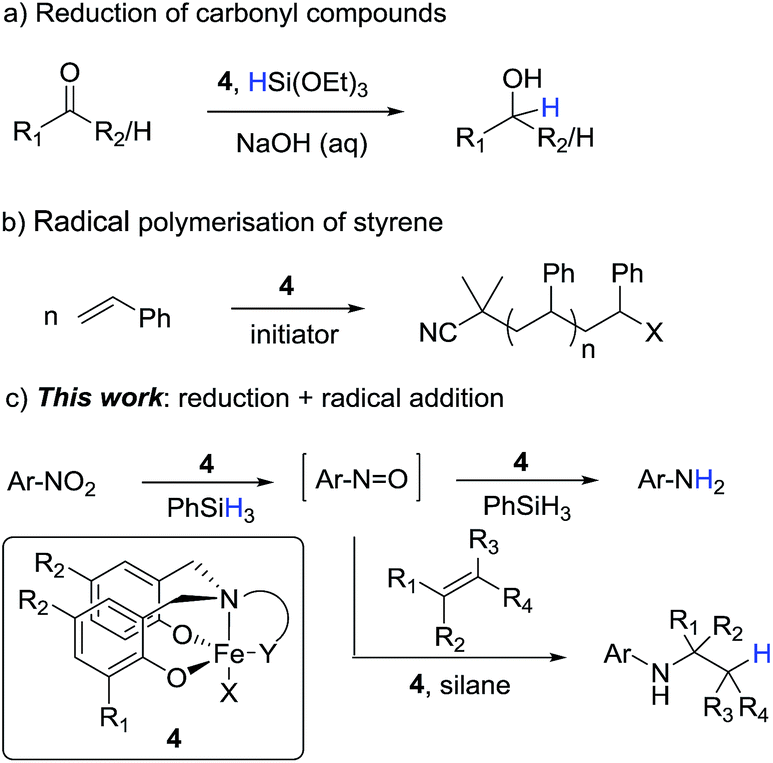 | ||
| Scheme 1 Iron-catalysed reduction and radical reactions supported by Fe(III) amine-bis(phenolate) catalysts and the extension of these reactions to the reduction and reductive addition of nitroarenes. | ||
Inspired by this ability of the amine-bis(phenolate) catalyst to mediate both radical and reduction events we sought to extend this reaction manifold to the reduction and reductive functionalisation of nitroarenes. If the chemoselectivity and high catalyst activity observed in the carbonyl reductions could be transferred to nitroarene reductions, the amine-bis(phenolate) system would represent a clear advance of the current iron-based reduction manifolds. Moreover, if nitro reduction is combined with radical hydrogen-atom transfer (HAT) to an alkene,9 a formal hydroamination would be possible. As recently reported,9 the iron-catalysed formal hydroamination of alkenes is an extremely powerful reaction accessible with high iron catalyst loadings (>30 mol%) and elevated reaction temperatures (60 °C) to achieve moderate yields. We postulated that increasing the rate of nitroarene reduction, and thus increasing the concentration of the intermediate nitrosoarene, the overall efficiency of this process could be increased and the catalyst loading lowered significantly.
In the context of the catalytic flexibility of the Fe(III) amine-bis(phenolate) framework, there is an obvious opportunity to develop a single catalytic system that is able to both reduce nitro compounds under mild conditions with high chemoselectivity and access a formal hydroamination of olefins at low catalyst loadings and temperatures (Scheme 1c).
Results and discussion
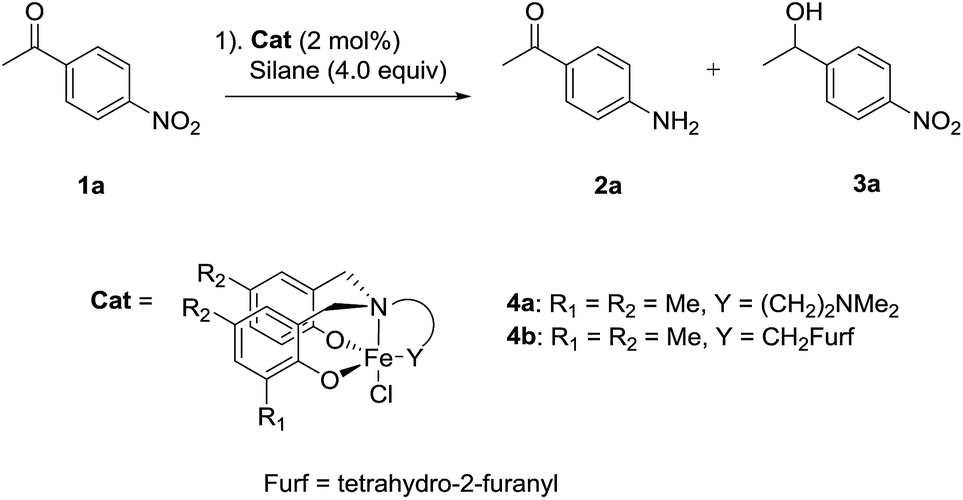
Our investigations began with the development of the chemoselective nitro reduction of 4-nitroacetophenone 1a (Table 1). Triethoxysilane was selected as the stoichiometric reductant due to its low cost, ease of handling and wide availability.10 Amine-bis(phenolate) iron(III) catalysts 4a gave amine 2a in 79% yield with 7% reduction of the ketone to give alcohol 3a (entry 1). Catalyst 4b, bearing the tetrahydro-2-furanyl donating group gave both high reactivity and chemoselectivity for amine 2a (entry 2). Solvent had a significant influence on the reaction in terms of both reactivity and selectivity. Replacing MeCN with toluene gave a significant drop in both conversion and chemoselectivity (entry 3). Using THF gave an even lower conversion and reduction of the carbonyl group was observed (entry 4). Ethyl acetate gave a similar result to that of toluene with regard to conversion and selectivity (entry 5). The use of PhSiH3 or Ph2SiH2 as the reducing agent, significantly reduced conversion and chemoselectivity (entries 6 and 7). The sterically bulky silanes Ph2MeSiH and (Me3SiO)2MeSiH showed no reactivity towards either the nitro or carbonyl groups (entries 8 and 9). Reducing the amount of silane from 4.0 equivalents to 3.0 equivalents led to a longer reaction time of 8 h in order to obtain comparable reduction (entry 10). Using diethoxymethylsilane, a widely used surrogate for triethoxysilane with greater thermal stability, gave product 2a in 90% yield together with 5% of 3a using 5 mol% catalyst (entry 11).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Silane | SMb (%) | 2a (%) | 3a (%) |
| a Unless otherwise noted, all reactions were carried out using 4.0 eq. of hydrosilane (1.20 mmol), 1.0 eq. of 4-nitroacetophenone (0.3 mmol), 0.02 eq. of catalyst 4 (0.006 mmol) in 0.3 mL solvent at 80 °C for 4 h. b Determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. c Isolated yield. d 3.0 eq. of triethoxysilane was used, 8 h. e 0.05 eq. of catalyst was used. | ||||||
| 1 | 4a | MeCN | HSi(OEt)3 | 7 | 79 | 7 |
| 2 | 4b | MeCN | HSi(OEt)3 | Trace | >95(91)c | Trace |
| 3 | 4b | PhMe | HSi(OEt)3 | 46 | 40 | 14 |
| 4 | 4b | THF | HSi(OEt)3 | 84 | 14 | 2 |
| 5 | 4b | EtOAc | HSi(OEt)3 | 51 | 35 | 14 |
| 6 | 4b | MeCN | PhSiH3 | 22 | 60 | 11 |
| 7 | 4b | MeCN | Ph2SiH2 | 55 | 26 | 10 |
| 8 | 4b | MeCN | Ph2MeSiH | >95 | 0 | 0 |
| 9 | 4b | MeCN | (Me3SiO)2MeSiH | >95 | 0 | 0 |
| 10d | 4b | MeCN | HSi(OEt)3 | Trace | >95 | Trace |
| 11e | 4b | MeCN | HSiMe(OEt)2 | Trace | 90 | 5 |
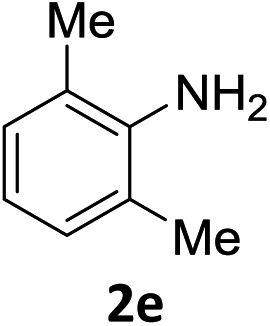
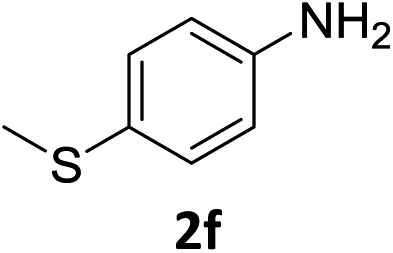
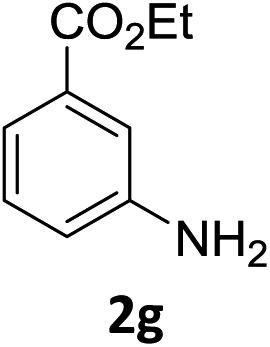
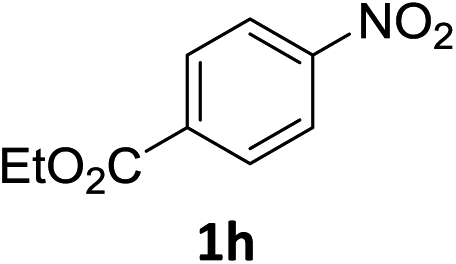
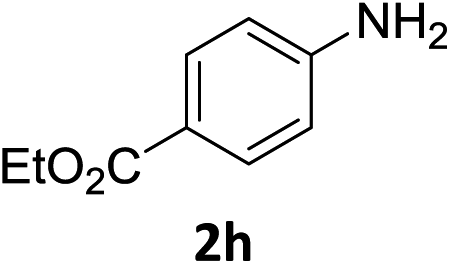
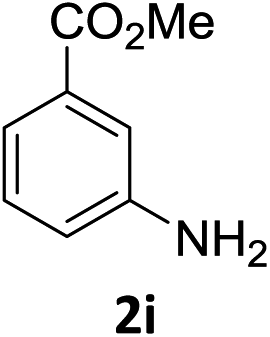
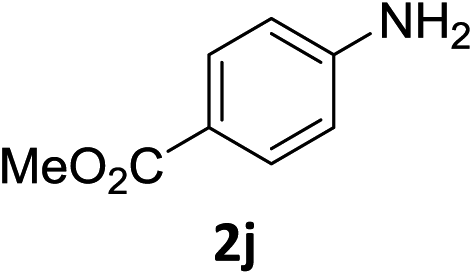
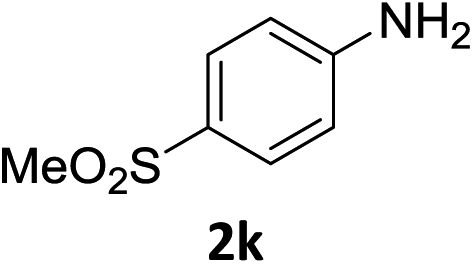
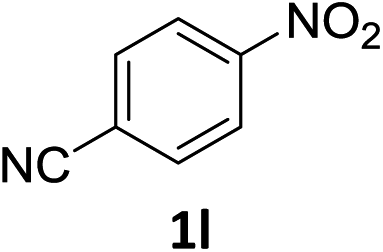
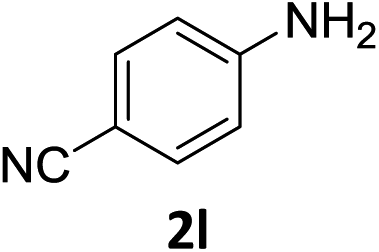
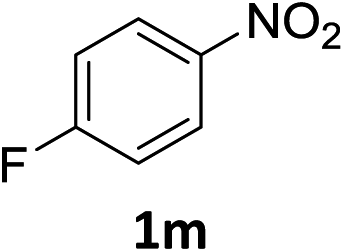
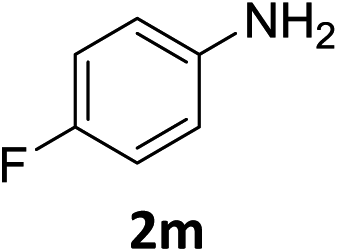
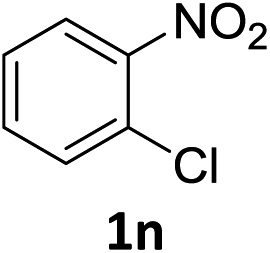
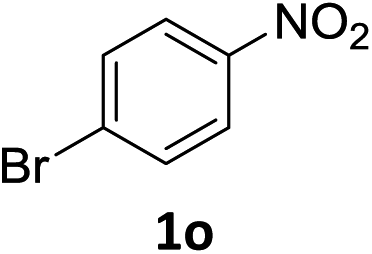
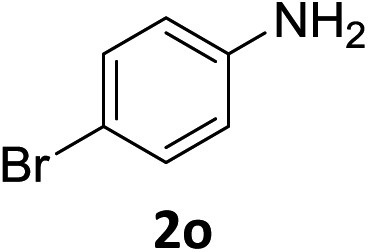
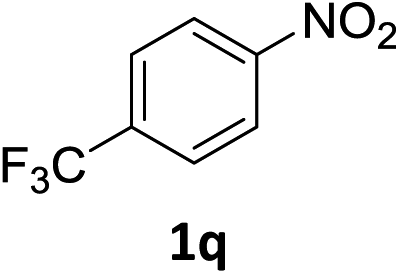
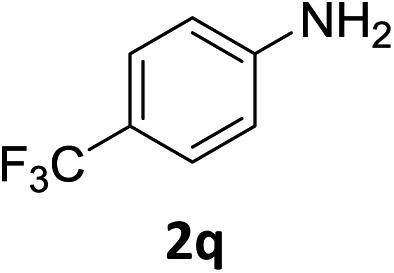
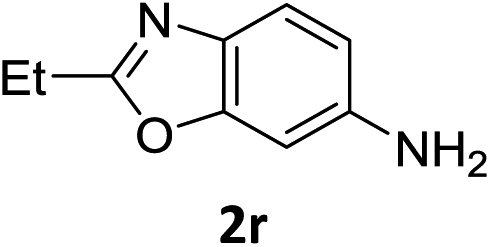
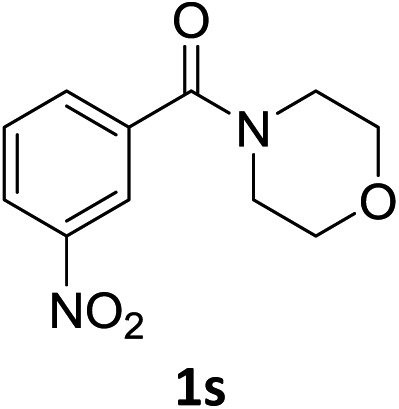
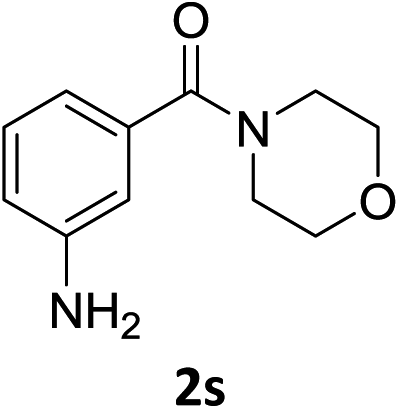
The substrate scope of this newly developed catalytic system was investigated using the optimised reaction conditions of 2 mol% catalyst 4b, 4.0 equivalents of triethoxysilane in MeCN (1.0 M) at 80 °C (Table 2). In all cases, good to excellent yield and chemoselectivity were obtained, demonstrating the utility of this methodology in synthesis. Industrially important aniline could be prepared from nitrobenzene in 84% isolated yield (entry 1). o-Methyl, m-methyl and, in contrast to other methods,6 even the sterically hindered 2,6-dimethyl nitrobenzene were all successfully reduced in good yields (entries 2, 3 and 4). p-(Methylthio)aniline 2f was produced from the corresponding nitroarene in excellent yield, although an 8 hour reaction time was needed (entry 5). The selective reduction of the nitro group in the presence of an ester functionality proceeded smoothly in all cases with excellent yields (entries 6, 7, 8, and 9), indicating the complete chemoselectivity and reactivity of the developed system for these challenging substrates.11 Methylsulfonyl substituted nitrobenzene 1k was also reduced with excellent chemoselectivity for the NO2 group (entry 10). In the case of 4-nitrobenzonitrile, 65% yield of amine 2l was obtained; however, 30% of the starting material was recovered, once again demonstrating the high chemoselectivity of the catalyst for the nitro group over nitrile (entry 11). The low yield in the reaction of 4-nitrobenzonitrile is attributed to the strong coordination of the nitrile group to the catalyst which inhibits catalyst activity. To demonstrate this, a standard reduction of ethyl 4-nitrobenzoate was doped with 4-nitrobenzonitrile (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and the yield of nitro reduction at ethyl 4-nitrobenzoate 1h dropped from 98% to 30%.12 Halogenated nitrobenzene derivatives were also reduced chemoselectively and in good yield without any protodehalogenation13 or homocoupling14 observed (entries 12, 13 and 14). Nitroarenes 1p and 1q, bearing the strongly electron-withdrawing CF3 group, were reduced to the corresponding anilines, 2p and 2q, in 90% and 85% yield, respectively (entries 15 and 16). Nitro-substituted benzoxazole 1r was selectively reduced to corresponding amine 2r in 88% yield without any ring-opening or arene hydrogenation observed (entry 17).15 Nitrobenzene 1s, bearing an amide functionality, was also well tolerated to give the product in excellent yield (entry 18). When a substrate bearing a free alcohol was used, decreased catalytic activity was observed (entry 19).
:1) and the yield of nitro reduction at ethyl 4-nitrobenzoate 1h dropped from 98% to 30%.12 Halogenated nitrobenzene derivatives were also reduced chemoselectively and in good yield without any protodehalogenation13 or homocoupling14 observed (entries 12, 13 and 14). Nitroarenes 1p and 1q, bearing the strongly electron-withdrawing CF3 group, were reduced to the corresponding anilines, 2p and 2q, in 90% and 85% yield, respectively (entries 15 and 16). Nitro-substituted benzoxazole 1r was selectively reduced to corresponding amine 2r in 88% yield without any ring-opening or arene hydrogenation observed (entry 17).15 Nitrobenzene 1s, bearing an amide functionality, was also well tolerated to give the product in excellent yield (entry 18). When a substrate bearing a free alcohol was used, decreased catalytic activity was observed (entry 19).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time | Yieldb |
| a Unless otherwise noted, all reactions were carried out using 4.0 eq. of triethoxysilane (2.40 mmol), 1.0 eq. of nitro substrate (0.6 mmol), 0.02 eq. of catalyst (0.012 mmol) in 0.6 mL MeCN at 80 °C. b Isolated yield. c Using 4.0 eq. of HSiMe(OEt)2, 0.05 eq. of 4b, 5 h. d Starting material recovered. e Yield determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. f 0.04 eq of catalyst was used. | ||||
| 1 |
|

|
4 h | 84% |
| 2 |

|

|
6 h | 84% |
| 3 |

|
|
6 h | 70% |
| 4 |

|
|
6 h | 82% |
| 5 |

|
|
8 h | 93% |
| 6 |

|
|
5 h | 91% |
| 7 |
|
|
5 h | 98%(85%)c |
| 8 |

|
|
5 h | 94% |
| 9 |

|
|
5 h | 93% |
| 10 |

|
|
4 h | 80%(15%)d |
| 11 |
|
|
6 h | 65%(30%)d |
| 12 |
|
|
5 h | 82% |
| 13 |
|

|
8 h | 87%e |
| 14 |
|
|
5 h | 90% |
| 15 |

|

|
3 h | 90% |
| 16 |
|
|
3 h | 85% |
| 17 |

|
|
6 h | 88% |
| 18f |
|
|
4 h | 98% |
| 19 |
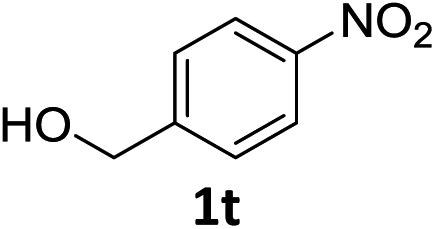
|

|
6 h | 55% |

To showcase the applicability of the developed nitro reduction, we explored the reduction of academically- and industrially-relevant targets. Bis(2-aminophenyl)amine 2u, which is widely used in the synthesis of N,N,N-type pincer16 or triamido17 ligands, was obtained in 80% isolated yield. This was directly comparable to the yield reported using palladium-catalysed reduction with H2.16 Drug precursor 1v was chemoselectively reduced to amine 2v in 90% yield. This late stage reduction with a non-toxic metal greatly simplifies purification and trace metal removal, so making the developed method ideal for targets to be tested in vivo.18 Amidation of 2v would give 4-(pyrazole-1-yl)carboxanilides, a family of drugs that tune the activity of canonical transient receptor potential channels (TRPC) and thus control the influx of intracellular Ca2+ into a plethora of mammalian cell types (Scheme 2).19
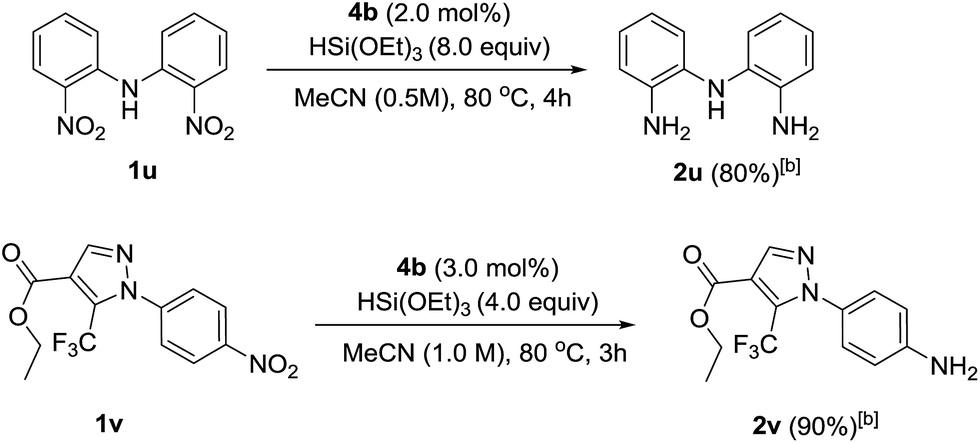 | ||
| Scheme 2 Selective nitro reduction in the synthesis of ‘real-world’ targets. [a] Reactions were carried out using 0.6 mmol of nitro compounds. [b] Isolated yield. | ||
Having successfully developed a highly efficient silane-mediated reduction of nitroarenes, we were keen to explore if this could be combined with radical HAT for a formal hydroamination.9,20 We hypothesised that the high reactivity of 4b for nitroarene reduction would increased rates of reactions with alkenes. Strikingly, application of catalyst 4b in the formal hydroamination of alkenes using nitroarene 1w and alkene 5a gave amine 6a in 75% isolated yield after Zn/HCl work-up21 using just 2.0 mol% catalyst, a 15-fold reduction in catalyst loading compared to the previous system.20 Additionally, the starting nitro substrate was fully consumed at room temperature in just 2 h. 1-Chloro-2-nitrobenzene 1n gave the corresponding amine product 6b in relatively lower yield, potentially due to the halogenophilicity of the parent Fe complexes.8 A methyl substituted nitroarene 1c was well tolerated to give the formal hydroamination product 6c while nitroarene 1f, bearing a methyl thioester group, gave the corresponding amine 6d, both in good yields. Nitroarene 1x with a free carbonyl group was also well tolerated to give the product 6e in lower yield but with the carbonyl functionality unchanged. Importantly, the formal hydroamination of olefin 5d could be further extended under the same reaction conditions. Reaction with 4-nitroanisole 1y followed by an intramolecular reductive amination gave the N-arylpiperidine 7a in 45% yield, indicating the potential application of this method in the preparation of N-heterocycles (Scheme 3). Deprotection of the para-methoxyphenyl (PMP) group of 7a would give 2,2,6-trimethyl piperidine which is a useful reagent for the α-alkylation of aldehydes.22
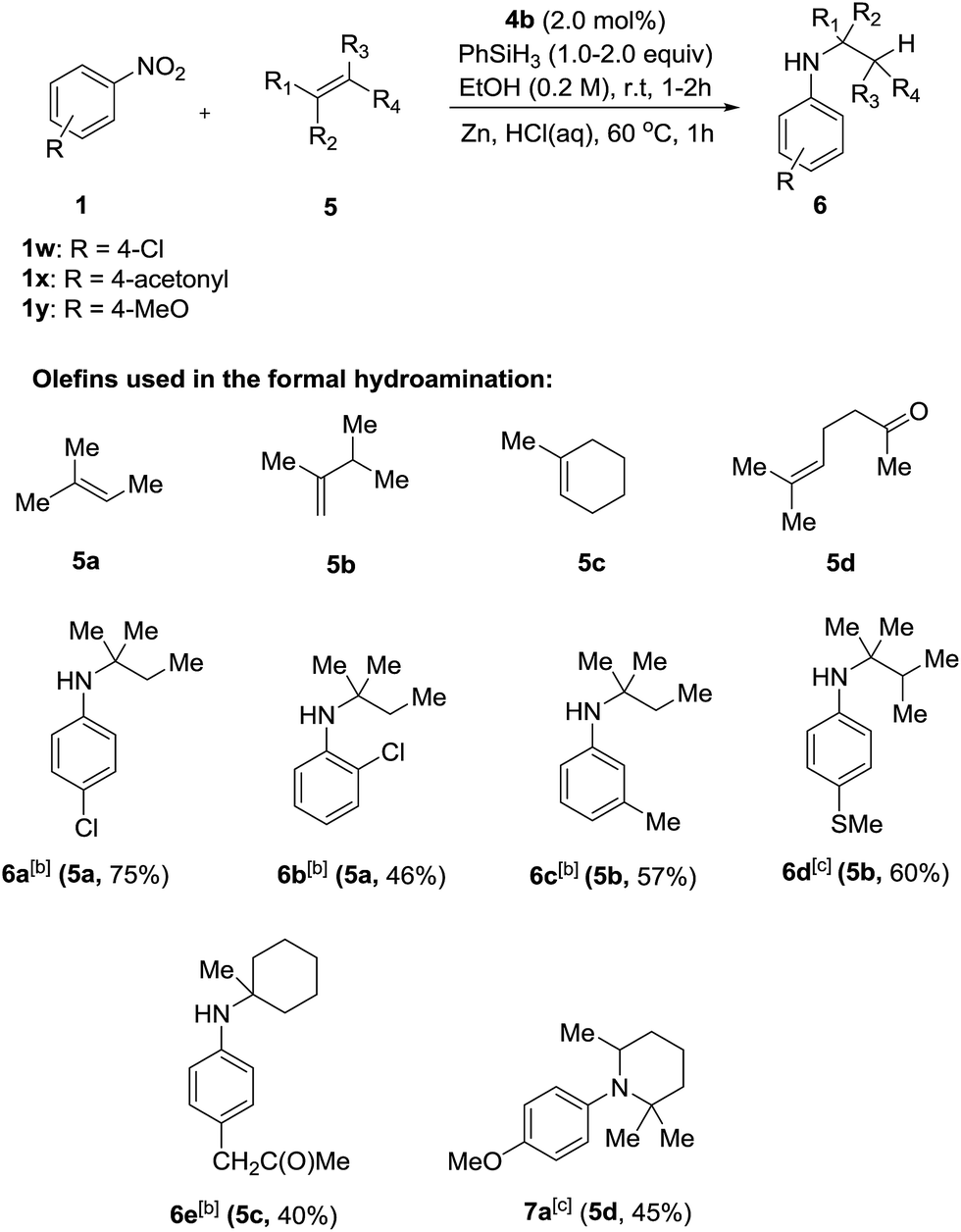 | ||
| Scheme 3 Formal hydroamination of olefins with catalyst 4b. [a] Reactions were carried out using 0.3 mmol of nitro compounds and 0.9 mmol of alkene. [b] 2.0 equiv. of silane, 2 h. [c] 1.0 equiv. of silane, 1 h. Isolated yield are shown in parentheses together with the donor olefin used. | ||
Conclusions
We have developed a highly chemoselective, efficient and operationally simple amine-bis(phenolate) iron(III)-catalysed reduction of nitro compounds using triethoxysilane as the reducing agent. The system chemoselectively reduces aryl nitro groups over carbonyl, ester, imine, sulfonyl and cyano functionalities. The highly efficient formal hydroamination of alkenes has also been developed with excellent activity observed at room temperature with low iron catalyst loadings. Mechanistic studies and the investigation into the scope of catalyst 4b for reductive functionalisation continue.Acknowledgements
MPS and SPT thank the University of Edinburgh for Chancellor's Fellowships and the School of Chemistry for continued support. MPS thanks the Framework Program 7 for a Marie Curie Career Integration Grant. SPT thanks the Royal Society for a University Research Fellowship and a Research Grant. KZ thanks the China Scholarship Council and the University of Edinburgh for an academic scholarship. The authors thank Prof. Jason Love and AJ MacNair for useful discussions.Notes and references
- Transparency Market Research, Aniline Market-Global Industry analysis, Size, Share, Growth, Trends and Forecast 2014-2020.
- R. S. Downing, P. J. Kunkeler and H. van Bekkum, Catal. Today, 1997, 37, 121–136 CrossRef CAS.
- B. Cornils and W. A. Herrmann, Applied Homogeneous Catalysis with Organometallic Compounds, Wiley-VCH, Weinheim, Germany, 2nd edn, 2002 Search PubMed.
- (a) I. Ojima, in The Chemistry of Organosilicon Compounds, ed. S. Patai and Z. Rapport, Wiley, New York, 1989 Search PubMed; (b) M. A. Brook, in Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000 Search PubMed; (c) Comprehensive Handbook on Hydrosilylation, ed. B. Marciniec, Pergamon, Oxford, 1992 Search PubMed; (d) B. Marciniec, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 1996, vol. 1, ch. 2 Search PubMed; (e) J. L. Speier, Adv. Organomet. Chem., 1979, 17, 407–447 CrossRef CAS; (f) B. Marciniec and J. Gulinski, J. Organomet. Chem., 1993, 446, 15–23 CrossRef CAS.
- For selected reviews, see: (a) K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC; (b) B. A. F. Le Bailly and S. P. Thomas, RSC Adv., 2011, 1, 1435–1445 RSC; (c) C. Bolm, J. Legros, J. L. Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed; (d) M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190–222 CrossRef CAS; (e) A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC; (f) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- (a) Y. Sunada, H. Kawakami, T. Imaoka, Y. Motoyama and H. Nagashima, Angew. Chem., Int. Ed., 2009, 48, 9511–9514 CrossRef CAS PubMed; (b) K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769–1771 RSC; (c) L. Pehlivan, E. Metay, S. Laval, W. Dayoub, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron Lett., 2010, 51, 1939–1941 CrossRef CAS; (d) L. Pehlivan, E. Metay, S. Laval, W. Dayoub, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron, 2011, 67, 1971–1976 CrossRef CAS.
- K. Zhu, M. P. Shaver and S. P. Thomas, Eur. J. Org. Chem., 2015, 2119–2123 CrossRef CAS.
- (a) H. Schroeder, B. R. M. Lake, S. Demeshko, M. P. Shaver and M. Buback, Macromolecules, 2015, 48, 4329–4338 CrossRef CAS; (b) L. E. N. Allan, J. P. MacDonald, A. M. Reckling, C. M. Kozak and M. P. Shaver, Macromol. Rapid Commun., 2012, 33, 414–418 CrossRef CAS PubMed; (c) L. E. N. Allan, J. P. MacDonald, G. S. Nichol and M. P. Shaver, Macromolecules, 2014, 47, 1249–1257 CrossRef CAS.
- (a) J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. L. Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886–891 CrossRef CAS PubMed; (b) M. Villa and A. J. v. Wangelin, Angew. Chem., Int. Ed., 2015, 54, 11906–11908 CrossRef CAS PubMed.
- Triethoxysilane has been reported to form flammable gasses upon exposure to transition metal catalysts: (a) S. C. Berk and S. L. Buchwald, J. Org. Chem., 1992, 57, 3751–3753 CrossRef CAS; (b) S. L. Buchwald, Chem. Eng. News, 1993, 71, 2 CrossRef CAS; (c) S. L. Buchwald, U. S. Pat., 5220020, 1993.
- (a) D. Bézier, G. T. Venkanna, L. C. M. Castro, J. Zheng, T. Roisnel, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2012, 354, 1879–1884 CrossRef; (b) K. Junge, B. Wendt, S. Zhou and M. Beller, Eur. J. Org. Chem., 2013, 2061–2065 CrossRef CAS; (c) H. Li, L. C. M. Castro, J. Zheng, T. Roisnel, V. Dorcet, J.-B. Sortais and C. Darcel, Angew. Chem., Int. Ed., 2013, 52, 8045–8049 CrossRef CAS PubMed.
- See ESI† for details.
- W. M. Czaplik, S. Grupe, M. Mayer and A. Jacobi von Wangelin, Chem. Commun., 2010, 46, 6350–6352 RSC.
- R. R. Chowdhury, A. K. Crane, C. Fowler, P. Kwong and C. M. Kozak, Chem. Commun., 2008, 94–96 RSC.
- It has been reported that benzoxazoles were reduced exclusively into ring-opened products using NaBH4, see: A. J. MacNair, M.-M. Tran, J. E. Nelson, G. U. Sloan, A. Ironmonger and S. P. Thomas, Org. Biomol. Chem., 2014, 12, 5082–5088 CAS.
- P. Ren, O. Vechorkin, K. von Allmen, R. Scopelliti and X. Hu, J. Am. Chem. Soc., 2011, 133, 7084–7095 CrossRef CAS PubMed.
- R. R. Schrock, J. Lee, L. C. Liang and W. M. Davis, Inorg. Chim. Acta, 1998, 270, 353–362 CrossRef CAS.
- European Medicines Agency, DOC. Ref. EMEA/CHMP/SWP/4446/2000.
- (a) J. Abramowitz and L. Birnbaumer, FASEB J., 2008, 23, 297–328 CrossRef PubMed; (b) Y. Yonetoku, H. Kubota, Y. Miyazaki, Y. Okamoto, M. Funatsu, N. Yoshimura-Ishikawa, J. Ishikawa, T. Yoshino, M. Takeuchi and M. Ohta, Bioorg. Med. Chem., 2008, 16, 9457–9466 CrossRef CAS PubMed; (c) S. Kyonaka, K. Kato, M. Nishida, K. Mio, T. Numaga, Y. Sawaguchi, T. Yoshida, M. Wakamori, E. Mori, T. Numata, M. Ishii, H. Takemoto, A. Ojida, K. Watanabe, A. Uemura, H. Kurose, T. Mori, T. Kobayashi, Y. Sato, C. Sato, I. Hamachi and Y. Mori, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5400–5405 CrossRef PubMed; (d) D. Obermayer, T. N. Glasnov and C. O. Kappe, J. Org. Chem., 2011, 76, 6657–6669 CrossRef CAS PubMed.
- (a) T. J. Barker and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13588–13591 CrossRef CAS PubMed; (b) E. K. Leggans, T. J. Barker, K. K. Duncan and D. L. Boger, Org. Lett., 2012, 14, 1428–1431 CrossRef CAS PubMed.
- Zn/HCl was added to transfer the N,O-alkylated product into desired hydroamination product as it could not be reduced by silane under the reaction condition even at higher temperature. See ref. 9a for the mechanism of the formation of the N,O-alkylated product.
- D. M. Hodgson and N. S. Kaka, Angew. Chem., Int. Ed., 2008, 47, 9958–9960 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04471e |
| This journal is © The Royal Society of Chemistry 2016 |