DOI:
10.1039/C5SC04319K
(Edge Article)
Chem. Sci., 2016,
7, 1988-1995
Origins of unique gold-catalysed chemo- and site-selective C–H functionalization of phenols with diazo compounds†
Received
12th November 2015
, Accepted 27th November 2015
First published on 27th November 2015
Abstract
In past decade, gold revealed more and more unique properties in carbene chemistry. It was disclosed in our recent communication (J. Am. Chem. Soc. 2014, 136, 6904) that gold carbenes have unprecedented chemo- and site-selectivity and ligand effect toward the functionalization of C–H bonds in phenols. In this full article, we report a comprehensively combined theoretical and experimental study on the mechanism of the insertion of gold carbenes into C–H and O–H bonds in phenol. It significantly revealed that the ligands have an important effect on C–H insertion and the reaction proceeds through a pathway involving the formation of an enolate-like intermediate. Moreover, two water molecules serving as a proton shuttle are believed to be the key issue for achieving chemoselective C–H functionalization, which is strongly supported by the DFT calculations and control experiments. It is the first time that a clear explanation is given about the prominent catalysis of gold carbenes toward C–H functionalization based on a theoretical and experimental study.
Introduction
Phenols are not only frequently found in natural products and pharmaceutics, but are also powerful versatile platforms in organic synthesis.1 Thus, the catalytic functionalization of phenols in a chemo- and site-selective manner is the most encompassing issue in organic chemistry.2 In this context, the transition-metal-promoted decomposition of diazo compounds to generate metal carbenes has broad applications3 in the direct site-selective functionalization of phenols. In most cases, O–H insertion takes place catalysed by a series of transition-metal catalysts such as copper, palladium, iron, and rhodium complexes.4 In contrast, very recently, we and Shi independently found the unprecedented C(sp2)–H bond functionalization of phenols with α-diazoesters,5 furnishing para-substituted phenols Pro-c8via para C–H functionalization rather than the O–H insertion product Pro-o8. Meanwhile, the chemoselectivity is heavily dependent on the nature of the ligand of the gold catalyst6 (Table 1). Although Nakamura and Pérez etc.7 have theoretically studied the mechanism of C(sp3)–H insertion with carbenes, and Shi and co-workers5b analysed the electronic structures of gold carbenes by using Density Functional Theory (DFT) calculations, no comprehensive mechanistic insight on the overall pathways of aromatic C(sp2)–H insertion with carbenes has been provided so far. To further understand the origin of this unique behaviour of gold carbenes8 in site selective C–H functionalization of phenol, we decided to carry out DFT calculations and an experimental study to gain insights on the mechanism. Herein, we present a combined computational and experimental study to elucidate the origin of the ligand effect on the chemoselectivity. Furthermore, the trace amount of water in the reaction mixture serving as a proton shuttle is proposed to be the key to give rise to the product Pro-c8 of C–H insertion, which is supported by our current DFT calculations and control experiments.
Table 1 C(sp2)–H bond functionalization of phenols with α-diazoesters under various conditions
|

|
| Entry |
Catalyst (5 mol%) |
Time |
Pro-c8
|
Pro-o8
|
|
NMR yield.
The conversion of 1 is 100% and the major product is dimer of 1.
Trace amount of dimer from 1 was detected.
|
| 1b |
Cu(OTf)2 |
5 min |
0 |
11 |
| 2b |
Cu(OTf)·toluene |
5 min |
0 |
9 |
| 3b |
[Pd(CH3CN)2Cl2] |
12 h |
0 |
33 |
| 4 |
Fe(OTf)2 |
12 h |
NR |
|
| 5c |
FeCl2 |
12 h |
0 |
0 |
| 6b |
[Rh2(OAc)4] |
5 min |
0 |
36 |
| 7 |
Ph3PAuSbF6 |
5 min |
33 |
45 |
| 8 |
(PhO)3PAuSbF6 |
5 min |
82 |
0 |
In a previous work on rhodium catalysed intramolecular aromatic C(sp2)–H insertion reactions, Padwa and Doyle assumed that the reactions were triggered by the electrophilic addition of the metallocarbene intermediate to the aromatic ring, then followed by a [1,2]-H migration step.9 Nevertheless, the reaction mechanisms for the prominent activity of gold carbenes toward C–H bonds still remain elusive. On the other hand, despite Yu and coworkers10 having performed the mechanistic study of the copper- and rhodium-catalysed O–H insertion of water with carbene using DFT calculations, the mechanism of gold-catalyzed O–H insertion has not yet been explored. In principle, there are three pivotal questions unsolved in this transformation: (1) is the mechanism of C(sp2)–H insertion with gold carbenes the same as that with other metals such as rhodium? (2) What is the origin of the ligand effect on the chemoselectivity? (3) Besides the ligand, any other factors affect the chemoselectivity?
Computational methods
All of the DFT calculations were carried out using the Gaussian 09 software package.11 The geometric structure of the intermediates and transition states were optimized and located by using the M06 functional,12 since the M06 functional has been demonstrated to generate accurate results for organometallics especially in the description of noncovalent interactions. The Lanl2dz basis set13 combined with the relativistic effective core potential for inner electrons and double zeta basis for covalent electrons was used to describe the heavy elements Au and P, and the 6-31G* basis set14 was used to describe the nonmetallic elements C, N, O and H. Frequency analyses were also performed based on the structures obtained in the gas phase to confirm that the intermediates are local minima and the transition states have only one imaginary frequency. Intrinsic reaction coordinate (IRC) calculations15 were performed to make sure that the crucial transition states connect the correct reactants and products. The solvent effect of CH2Cl2 was evaluated with single point calculations using the integral equation formalism model (IEFPCM)16 with the dielectric constant ε = 8.93 based on the structures in the gas phase. All discussed energy values of intermediates and transition states throughout this paper are the Gibbs free energies in units of kcal mol−1 that were calculated at the temperature of 298 K, including the corrections of solvation free energies from the IEFPCM model. The 3D images of the calculated structures were plotted using GaussView.17 More structural details of intermediates and transition states can be found in the ESI.†
Results and discussion
On the pathway of C–H functionalization catalysed by (PhO)3PAuSbF6
It has been widely accepted that metal catalysts could catalyse diazo compounds to release N2 and form reactive Au-carbenes, which were regarded as the precursors responsible for C–H bond insertion.5,8a–e To account for the generation of crucial precursors preceding C–H insertion, we calculated the reaction pathways of α-diazoesters and metal catalysts AuL (L = (PhO)3P and Ph3P) in solution (Fig. S1 of ESI†). The calculated free energy profiles indicate that the generation of Au-carbene species is quite facile and drastically exothermic, with the low barriers of 6.5 and 11.4 kcal mol−1, respectively. Therefore, the gold carbenes and phenol are regarded as the reactants for the subsequent C–H bond insertion reactions and the sum of their free energies is set as the benchmark, with the value of 0.0 kcal mol−1.
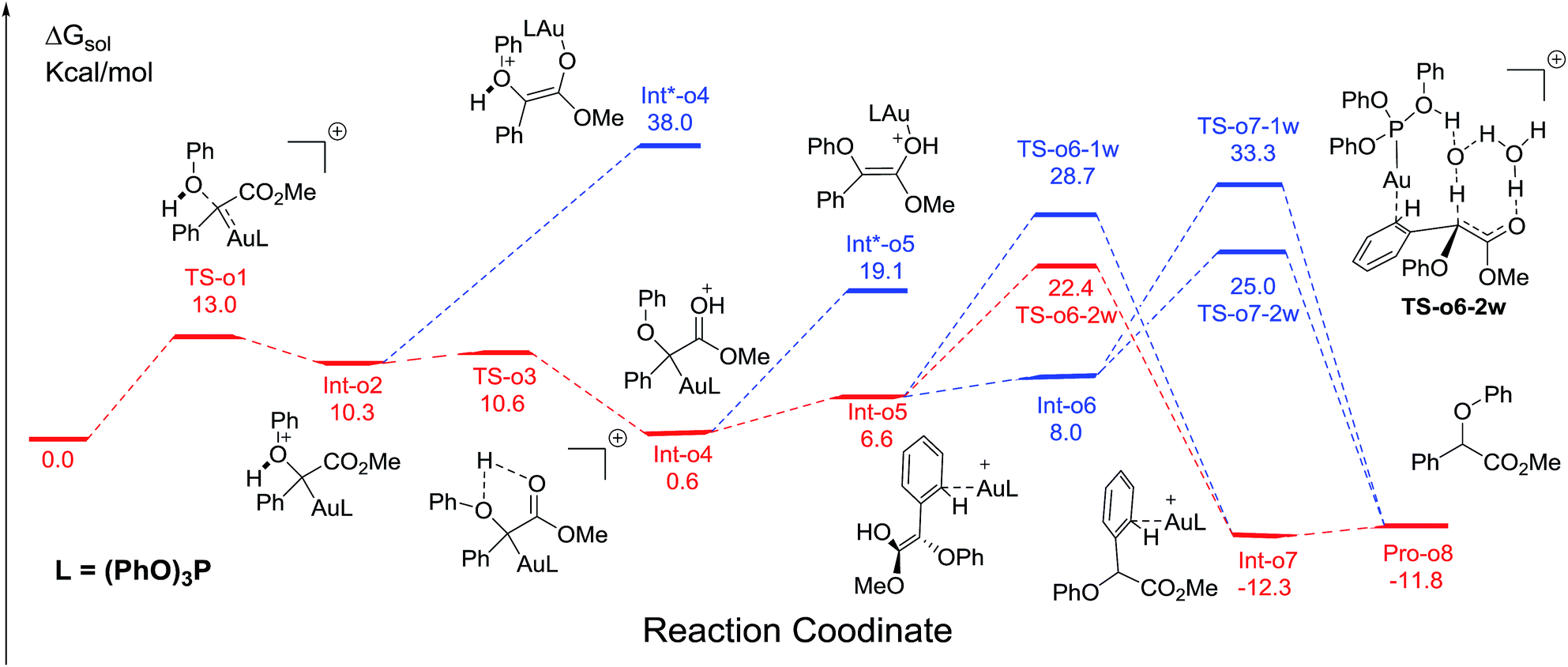
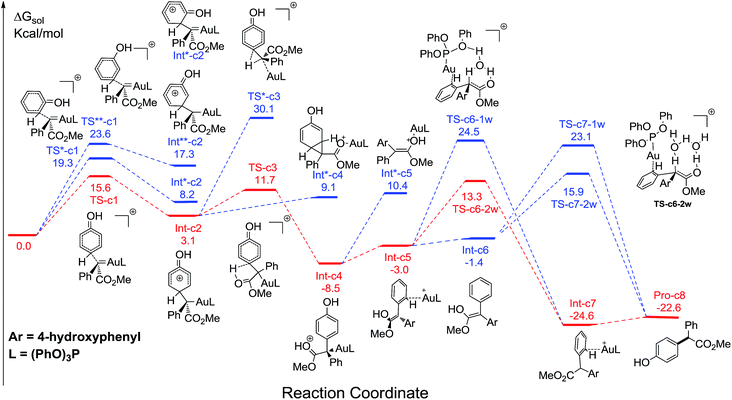
Fig. 1 displays the calculated free energy profiles for the C(sp2)–H functionalization of phenol with a gold carbene with (PhO)3P as the ligand, where the structural details of intermediates and transition states are provided in the ESI.† The previous theoretical investigations on the C(sp3)–H insertion of rhodium, copper or silver carbenes, performed by Nakamura and Pérez, indicate that the first step of the reactions involves the formation of σ-bond complexes of carbenes and the C(sp3)–H bond.7 Recently, Pérez and coworkers suggested that the mechanism of aromatic C(sp2)–H bond insertion might be entirely different from that of aliphatic C(sp3)–H bond insertion.8c In our DFT calculations, a transition state TS-c1 with a barrier of 15.6 kcal mol−1 is located for the addition of the gold carbene at the para-position of phenol. TS-c1 represents the transition state of the direct C(sp2)–C(sp2) coupling of phenol and the gold carbene, leading to the final para-C–H insertion product Int-c2. Accordingly, the addition of the carbene at the ortho site of phenol gives rise to an intermediate Int*-c2 with a higher energy (8.2 kcal mol−1vs.Int-c2's 3.1 kcal mol−1) via the transition state TS*-c1 with 19.3 kcal mol−1. It is not surprising to find that the meta-position of phenol is the most unfavourable site for addition with the gold carbene, which needs an activation energy barrier up to 23.6 kcal mol−1. Such high activation barriers of TS*-c1 and TS**-c1 prevent the carbenes from adding at the ortho- and meta-positions of phenol. The current DFT results are consistent with our experimental observations that the addition at the para-sites of substituted phenols was the predominant even with bulky steric hindrance (i.e. 3,5-dimethyl phenol).5a According to the previous calculations,10 the intermediate Int-c2 may be isomerized to the enolate via the mechanism of [1,3]-metal migration and yield a stable intermediate. However, it is found that such a stable intermediate hardly exists after the [1,3]-Au migration in Int-c2, since a kind of cyclopropanation product of the Büchner reaction is obtained, as shown by Int*-c4.18 Meanwhile, this process of [1,3]-Au migration seems to be energetically unfavorable, with the endothermicity of 9.1 kcal mol−1 to give Int*-c4. Additionally, in order to examine whether the commonly accepted mechanism of direct [1,2]-H shift is feasible, we located the transition state TS*-c3 with the corresponding structure displayed in Fig. 2. Our calculated results imply that TS*-c3 has a considerable barrier of 30.1 kcal mol−1, which could definitely rule out the mechanism of direct [1,2]-H shift in Int-c2. During the course of optimization, we happened to hypothesize that the carbonyl oxygen atom may intend to directly abstract the proton at the original para-site of phenol via the five-membered transition state TS-c3 in Fig. 2, to generate Int-c4. This process is quite facile according to the current calculations, merely with a small barrier of 8.6 kcal mol−1. Also, Int-c4 is more stable than Int*-c4 by 17.6 kcal mol−1. Subsequently, the intermediate Int-c4 undergoes an unexpected isomerization to the enol Int-c5 through [1,3]-migration of the metal complex (PhO)3PAu+ to the phenyl ring rather than to Int*-c5.
 |
| | Fig. 1 The calculated free energy profiles ΔG and the corresponding structures of intermediates and transition states along different possible C–H insertion pathways catalyzed by (PhO)3PAuSbF6. The reasonable pathways are shown in red and other possible pathways are in blue. All values of free energies are relative to the gold carbene and phenol reactants and in the units of kcal mol−1. | |
 |
| | Fig. 2 Optimized structures of transition states TS-c3 and TS*-c3 corresponding to Fig. 1. The distances are in the units of angstroms and the values of free energies ΔG are in kcal mol−1. | |
Two possible pathways might exist between the enol Int-c5 and the final product Pro-c8. One is the gold-associated pathway, where the proton transfers through TS-c6-1w or TS-c6-2w to yield the intermediate Int-c7. Then, the metal complex (PhO)3PAu+ dissociates from Int-c7 to yield Pro-c8. The other is the gold-free pathway, where the catalyst (PhO)3PAu+ first dissociates from Int-c5 to yield the gold-free Int-c6 and then proton transfer occurs viaTS-c7-1w or TS-c7-2w to yield Pro-c8. As far as the two pathways are concerned, the key question is: how does the proton transfer from the hydroxyl to the final carbon in the enol? In this work, we propose that the water in the solvent plays a crucial role in undertaking the remote proton shuttle. Based on this proposal, we located the one-water assisted transition states TS-c6-1w and TS-c7-1w, as well as the two-water assisted transition states TS-c6-2w and TS-c7-2w along the reaction pathways, with the corresponding activation energy barriers of the elementary steps displayed in Fig. 3. The DFT results indicate that TS-c6-1w and TS-c7-1w involving one water molecule has higher energy barriers than two-water assisted TS-c6-2w and TS-c7-2w. Fig. 3 shows that the carboxyl group in TS-c6-1w and TS-c7-1w has to distort from the planar structures to form hydrogen bonds with H2O, but they still remain planar in TS-c6-2w and TS-c7-2w. The structural distortion in TS-c6-1w and TS-c7-1w might be the reason why they are in high energy states. Interestingly, it is found that the oxygen atoms in the ligand (PhO)3P form hydrogen bonding interactions with the shuttle water molecules in TS-c6-2w, while this effect does not exist in the other located transition state TS*-c6-2w (Fig. S2 in ESI†). The calculated distance between the oxygen and hydrogen atoms in TS-c6-2w of Fig. 3 is 1.87 Å and the energy of the transition state is 2.2 kcal mol−1 lower than that of TS*-c6-2w, indicating a possible stabilization effect of the metal complex (PhO)3PAu+ for the water chain in TS-c6-2w.
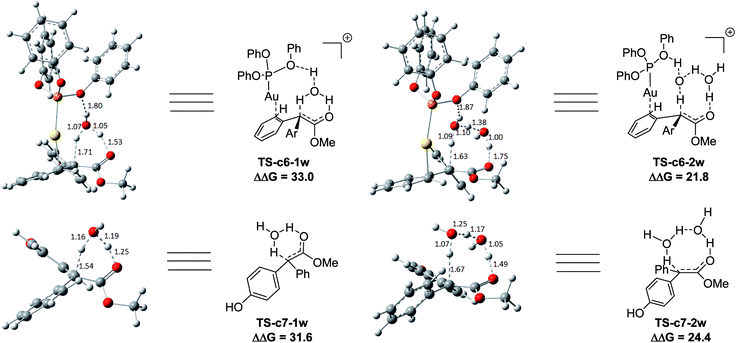 |
| | Fig. 3 The structures of TS-c6-1w, TS-c6-2w, TS-c7-1w and TS-c7-2w in the pathways of C–H functionalization of phenol catalysed by (PhO)3PAuSbF6. The values of the free energy barriers ΔΔG are in kcal mol−1. | |
Control experiments were carried out in our lab (Scheme 1) to demonstrate the proposed mechanisms of gold carbene insertion into the C–H bond of phenol. When 1 equivalent of D2O was added into the reaction system, product Pro-o8 was deuterated with the percentage of 22% (eqn (1)). The relatively low deuterium ratio might be attributed to the existence of trace amount of H2O19 that could also undertake the function of proton transfer. Additionally, it is well known that the hydrogen in the OH group of phenol could readily exchange with the deuterium in D2O, which leads to an increased H2O or DOH amount in solution. If a great number of H2O molecules participate in the process of the proton shuttle, the ratio of deuterated product Pro-o8 will definitely decrease. We also considered the possibility whether the phenols can serve as the proton shuttle. However, the calculated energy barrier between the transition state structure TS-c6-2p (ESI†) and Int-c5 for [1,3]-H shift is highly up to 36.1 kcal mol−1, which implies that it hardly occurs. Eqn (2) manifests that the direct proton exchange of Pro-o8 with D2O is impossible under the reaction conditions. More importantly, eqn (3) demonstrates that the reaction of 1 and deuterated phenol 2-5d gives rise to the product Pro-o8 without deuterium, which supports that direct [1,2]-H shift9 (TS*-c3) does indeed not occur in this case. The control experiments demonstrate that this kind of C–H bond insertion reactions governed by a gold carbene follows a novel mechanism involving a water assisting process, rather than previous commonly proposed direct [1,2]-H shift.5,8a,8c,9
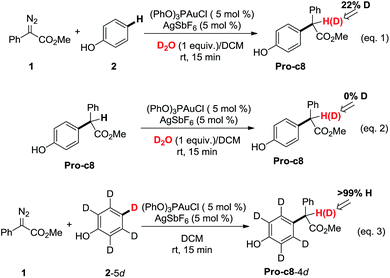 |
| | Scheme 1 Control experiments. | |
Proposed pathway of O–H insertion catalysed by (PhO)3PAuSbF6
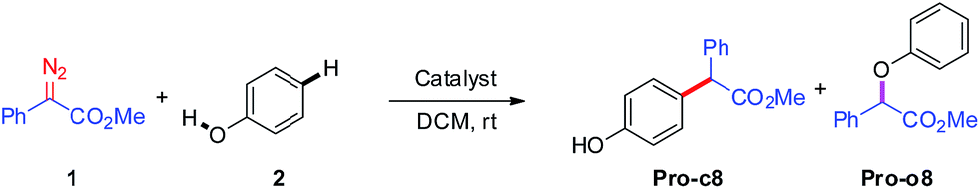
We next turn to address the question why the O–H insertion of phenol does not occur with (PhO)3PAuSbF6 as the catalyst. In Fig. 4, the calculated results show that oxonium ylide20Int-o2 formed through TS-o1 with a modest barrier. In the case of O–H bond insertion, a possible migration of complex LAu+ to the carbonyl group in Int-o2 is also taken into account, but the resulted structure Int*-o4 is quite high in energy. The oxonium ylide Int-o2 tends to isomerize to the more stable Int-o4viaTS-o3. The migration of the gold catalyst to the phenyl group in Int-o4 leads to the isomer Int-o5. It should be noted that Int*-o5 is quite energetically unstable, with the gold complex attached to the oxygen atom in the hydroxyl group. Similarly to the mechanism of C–H insertion in Fig. 1, the rearrangements of Int-o5 to Pro-o8 pass through the gold-associated or gold-free pathways via the specific water-assisted transition states. We also locate the two-water assisted transition states TS-o6-2w for the gold-associated pathway and TS-o7-2w for the gold-free pathway, respectively (Fig. 5). The activation barrier of TS-o6-2w (15.8 kcal mol−1) is relatively lower that of TS-o7-2w (18.4 kcal mol−1), indicating that the transformation of Int-o5 to Pro-o8 favours the gold-associated pathway viaTS-o6-2w rather than TS-o7-2w.
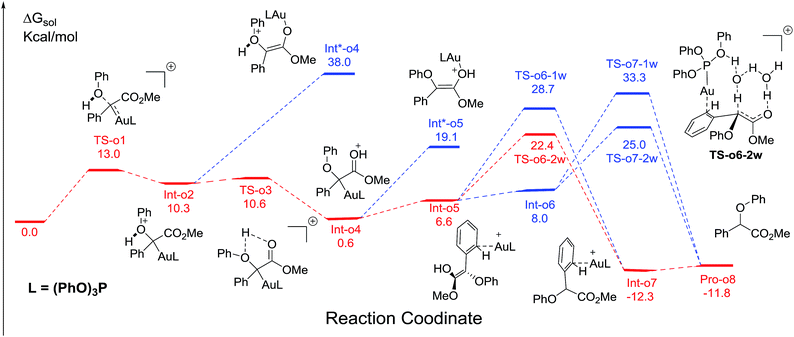 |
| | Fig. 4 The calculated free energy profiles ΔG and the corresponding structures of the intermediates and transition states along different possible O–H insertion pathways catalyzed by (PhO)3PAuSbF6. The reasonable pathway is shown in red and other possible pathways are in blue. All values of free energies are relative to the gold carbene and phenol reactants and in the units of kcal mol−1. | |
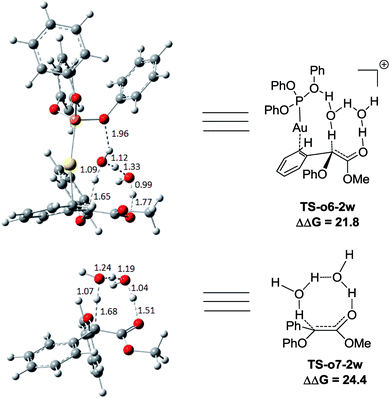 |
| | Fig. 5 The structures of TS-o6-2w and TS-o7-2w in the proposed O–H functionalization of phenols catalysed by (PhO)3PAuSbF6. The distances are in the units of angstroms and the values of free energy barriers ΔΔG are in kcal mol−1. | |
Rationale of the chemoselectivity
Comparing these two competitive reaction pathways in Fig. 1 and 4, it is revealed that both rearrangements from Int-c5 or Int-o5 to the final products are the rate-determining steps in the overall pathways, with almost the same total barriers of 21.8 kcal mol−1. Nevertheless, the DFT calculations reveal that the thermal stability of the intermediate of C–H insertion remarkably differs from that of O–H insertion. For example, the intermediate Int-c4 is more stable than Int-o4 by 9.1 kcal mol−1, with an exothermic energy of 8.5 kcal mol−1. As for the final products, the energy of Pro-c8 is 10.8 kcal mol−1 lower than that of the O–H insertion product Pro-o8. Besides, the endothermicity of TS-c6-2w is 13.3 kcal mol−1, whereas the corresponding energy of TS-o6-2w is up to 22.4 kcal mol−1, relative to the sum of the reactants. Such a large endothermicity of TS-o6-2w means that it is more difficult to pass through it to reach the product though collision in solution. The calculated reaction pathways in Fig. 1 and 4 reveal that the stability of intermediates plays an important role in chemoselectivity and accounts for the unique chemoselectivity toward C(sp2)–H insertion.
In order to further understand the chemoselectivity, the reactions of phenol with α-phenyldiazoester catalysed by (PhO)3PAuSbF6 at different temperatures were also carried out. The ratios of Pro-c8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pro-o8 decreased from >20:1 to 2.5:1 along with lowering the reaction temperature from rt to −40 °C (Fig. 6; for details, see ESI†). These results indicated that the C–H functionalization process was more favored at high temperature than at low temperature. According to the free energy showed in Fig. 1 and 4, Int-c4 and Int-o4 should be the key intermediates for C–H functionalization and O–H insertion. As shown in Fig. 6, Int-c4 was more stable via a higher barrier whilst Int-o4 was more unstable via a lower barrier. That is, the formation of Int-c4 was the process of thermodynamic control, leading to C–H functionalization. In contrast, the generation of Int-o4 underwent kinetic control, leading to O–H insertion. The control experiments at different temperatures and DFT calculations are in excellent agreement.
:Pro-o8 decreased from >20:1 to 2.5:1 along with lowering the reaction temperature from rt to −40 °C (Fig. 6; for details, see ESI†). These results indicated that the C–H functionalization process was more favored at high temperature than at low temperature. According to the free energy showed in Fig. 1 and 4, Int-c4 and Int-o4 should be the key intermediates for C–H functionalization and O–H insertion. As shown in Fig. 6, Int-c4 was more stable via a higher barrier whilst Int-o4 was more unstable via a lower barrier. That is, the formation of Int-c4 was the process of thermodynamic control, leading to C–H functionalization. In contrast, the generation of Int-o4 underwent kinetic control, leading to O–H insertion. The control experiments at different temperatures and DFT calculations are in excellent agreement.
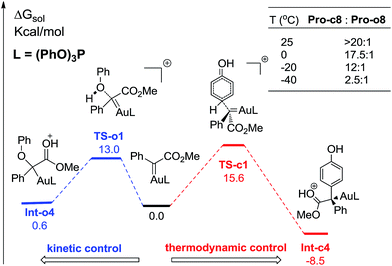 |
| | Fig. 6 The reaction temperature on the chemoselectivity and the competitive pathways for the formation of two key intermediates. All values of free energies are relative to the gold carbene and phenol reactants and in the units of kcal mol−1. | |
Two competitive pathways catalyzed by Ph3PAuSbF6
Additionally, the mechanisms of C–H and O–H insertions of phenol catalyzed by Ph3PAuSbF6 are also shown in Fig. 7, with the corresponding barriers listed in Table 2. On the one hand, compared to the corresponding pathways in Fig. 1 and 4, we found that the ligand Ph3P has a remarkable electronic effect on the change of dynamics barriers of electrophilic addition to intermediates Int-o12 and Int-c12. For C–H insertion, the addition barrier viaTS-c9 was enhanced to be 18.2 kcal mol−1, 2.6 kcal mol−1 higher than that of TS-c1. In contrast, the barrier of O–H insertion viaTS-o9 decreases from 13.0 to 11.8 kcal mol−1. It is no doubt that the change of the barriers of addition is disadvantageous to C–H insertion but advantageous to O–H insertion. On the other hand, the calculated barrier of hydrogen transfer viaTS-o15-2w in O–H insertion is 18.0 kcal mol−1, relatively lower than the barrier of 22.0 kcal mol−1 for C–H insertion. Therefore, the reaction of C–H insertion shown in Fig. 7 is significantly slowed down in comparison to the case when it is catalysed by (PhO)3PAuSbF6 in Fig. 1. Product of C–H insertion is still more stable than the counterpart of O–H insertion in terms of thermodynamics according to the current DFT calculations. Considering both the dynamic and thermodynamic effects, the calculated pathways (Fig. 7) could account for the low chemoselectivity for Pro-c8 and Pro-o8 catalysed by Ph3PAuSbF6 that has been observed in our experiments (Table 1, entry 7).
Table 2 The calculated free energetic spans ΔΔG of hydrogen transfer through transition states (TSs) assisted by one water molecule and two water molecules in Fig. 6. The values of ΔΔG are in the units of kcal mol−1
| TSs |
TS-c14-2w
|
TS-c15-2w
|
TS-o14-2w
|
TS-o15-2w
|
| ΔΔG |
24.1 |
22.0 |
20.5 |
18.0 |
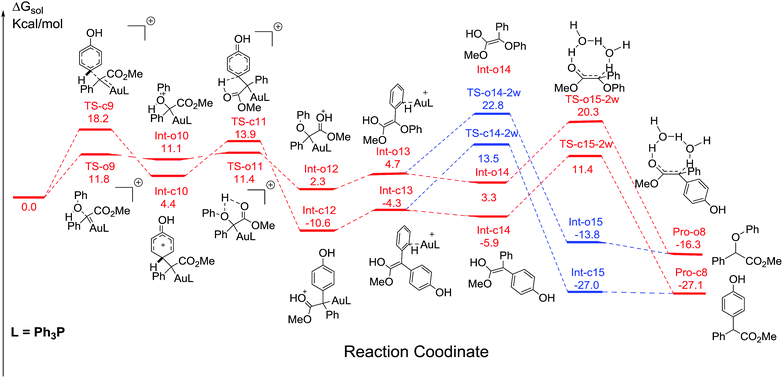 |
| | Fig. 7 The competitive pathways of C–H and O–H insertion catalyzed by Ph3PAuSbF6. The two reasonable pathways are shown in red and the other possible pathways in blue. All values of free energies are relative to the gold carbene and phenol reactants and in the units of kcal mol−1. | |
Conclusions
In summary, the origins of the unique chemoselectivity of C–H bond functionalization of phenol with diazo compounds catalyzed by (PhO)3PAuSbF6 are due to the high thermodynamic stability of intermediates and products, as well as the modest energy barrier of the rate-determining step. It is proposed for the first time that the reactions of gold carbenes with phenol pass through the specific pathways involving the formation of stable enols. The hydrogen transfers in the rearrangements from enol to final products follow a novel reaction pathway with two water molecules serving as a proton shuttle rather than the previous commonly proposed direct [1,2]-H shift. The DFT calculations also reveal that the oxygen atoms in the ligand (PhO)3P play an important role in stabilizing the transition states of hydrogen transfer. The observed ligand effects of metal complexes on the reaction pathways are well rationalized based on the results of the present DFT calculations. The novel mechanistic insights delineated above will refine the model of gold catalyst in carbene chemistry and contribute to the future development of this field as well as the rational designing in chemo- and site-selective organic transformation.
Acknowledgements
Financial support by the 973 Program (2011CB808600), Shanghai Pujiang Program (14PJ1403100), National Natural Science Foundation of China (21572065, 21433004, 21473056, 21425205), Changjiang Scholars and Innovative Research Team in University (PCSIRT) is greatly appreciated. We acknowledge the support of the NYU-ECNU Center for Computational Chemistry at NYU Shanghai.
Notes and references
-
Synthetic and Natural Phenols, ed. J. H. P. Tyman, Elsevier, 1996 Search PubMed.
- For recent highlights, see:
(a) D.-G. Yu, F. de Azambuja and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 7710 CrossRef CAS;
(b) P. MacLellan, Nat. Chem., 2014, 6, 375 CrossRef; for selected examples, see:
(c) R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112 CrossRef CAS;
(d) R. B. Bedford and M. E. Limmert, J. Org. Chem., 2003, 68, 8669 CrossRef CAS PubMed;
(e) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109 CrossRef CAS PubMed;
(f) R. Zhu, J. Wei and Z.-J. Shi, Chem. Sci., 2013, 4, 3706 RSC;
(g) Z. Huang, L. Jin, Y. Feng, P. Peng, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7151 CrossRef CAS;
(h) Z. Wu, F. Luo, S. Chen, Z. Li, H. Xiang and X. Zhou, Chem. Commun., 2013, 49, 7653 RSC;
(i) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567 CrossRef CAS PubMed;
(j) A. Seoane, N. Casanova, N. Quiñones, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2014, 136, 834 CrossRef CAS;
(k) Y. E. Lee, T. Cao, C. Torruellas and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 6782 CrossRef CAS PubMed;
(l) C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 9303 CrossRef CAS PubMed;
(m) K. V. N. Esguerra, Y. Fall, L. Petitjean and J.-P. Lumb, J. Am. Chem. Soc., 2014, 136, 7662 CrossRef CAS;
(n) Y. Kuninobu, T. Matsuki and K. Takai, J. Am. Chem. Soc., 2009, 131, 9914 CrossRef CAS PubMed;
(o) C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458 CrossRef CAS PubMed.
- For selected reviews, see:
(a)
Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, ed. M. P. Doyle, M. A. McKervey and T. Ye, Wiley-VCH, New York, 1998 Search PubMed;
(b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS;
(c) H. M. L. Davies and Y.-J. Lian, Acc. Chem. Res., 2012, 45, 923 CrossRef CAS;
(d) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC;
(e) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379 CrossRef PubMed;
(f) Z. Liu and J. Wang, J. Org. Chem., 2013, 78, 10024 CrossRef CAS PubMed.
- For reviews, see:
(a) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC;
(b) X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed;
(c) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed;
(d) D. J. Miller and C. J. Moody, Tetrahedron, 1995, 51, 10811 CrossRef CAS. For selected examples, see:
(e) P. Yates, J. Am. Chem. Soc., 1952, 74, 5376 CrossRef CAS;
(f) Z. Zhu and J. H. Espenson, J. Am. Chem. Soc., 1996, 118, 9901 CrossRef CAS;
(g) Z. Qu, W. Shi and J. Wang, J. Org. Chem., 2004, 69, 217 CrossRef CAS PubMed;
(h) Z. Zhang, Y. Liu, L. Ling, Y. Li, Y. Dong, M. Gong, X. Zhao, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 4330 CrossRef CAS PubMed;
(i) J. R. Sosa, A. A. Tudjarian and T. G. Minehan, Org. Lett., 2008, 10, 5091 CrossRef CAS;
(j) K. J. Kilpin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328 RSC;
(k) R. A. Maurya, C. P. Park, J. H. Lee and D.-P. Kim, Angew. Chem., Int. Ed., 2011, 50, 5952 CrossRef CAS;
(l) T. Osako, D. Panichakul and Y. Uozumi, Org. Lett., 2012, 14, 194 CrossRef CAS;
(m) K. B. S. Magar and Y. R. Lee, Org. Lett., 2013, 15, 4288 CrossRef PubMed. For asymmetric version, see:
(n) T. C. Maier and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 4594 CrossRef CAS PubMed;
(o) C. Chen, S.-F. Zhu, B. Liu, L.-X. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 12616 CrossRef CAS;
(p) X.-L. Xie, S.-F. Zhu, J.-X. Guo, Y. Cai and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 2978 CrossRef CAS.
-
(a) Z. Yu, B. Ma, M. Chen, H.-H. Wu, L. Liu and J. Zhang, J. Am. Chem. Soc., 2014, 136, 6904 CrossRef CAS PubMed;
(b) Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 9817 CrossRef CAS PubMed.
- For selected recent accounts and general reviews, see:
(a) A. Corma, A. Leyva-Perez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS;
(b) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS PubMed;
(c) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS;
(d) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358 RSC;
(e) B.-L. Lu, L. Dai and M. Shi, Chem. Soc. Rev., 2012, 41, 3318 RSC;
(f) D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462 CrossRef CAS;
(g) Y.-W. Sun, Q. Xu and M. Shi, Beilstein J. Org. Chem., 2013, 9, 2224 CrossRef PubMed;
(h) F. López and J. L. Mascareñas, Beilstein J. Org. Chem., 2013, 9, 2250 CrossRef PubMed;
(i) G. Cera and M. Bandini, Isr. J. Chem., 2013, 53, 848 CrossRef CAS;
(j) W. Yang and A. S. K. Hashmi, Chem. Soc. Rev., 2014, 43, 2941 RSC;
(k) F. López and J. Mascareñas, Chem. Soc. Rev., 2014, 43, 2904 RSC;
(l) C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902 CrossRef CAS PubMed;
(m) A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864 CrossRef CAS;
(n) L. Zhang, Acc. Chem. Res., 2014, 47, 877 CrossRef CAS;
(o) F. López and J. L. Mascareñas, Beilstein J. Org. Chem., 2011, 7, 1075 CrossRef;
(p) D. Qian and J. Zhang, Chem. Rec., 2014, 14, 280 CrossRef CAS PubMed;
(q) D. Qian and J. Zhang, Chem. Soc. Rev., 2015, 44, 677 RSC.
-
(a) E. Nakamura, N. Yoshikai and M. Yamanaka, J. Am. Chem. Soc., 2002, 124, 7181 CrossRef CAS;
(b) A. A. C. Braga, F. Maseras, J. Urbano, A. Caballero, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2006, 25, 5292 CrossRef CAS.
- For gold-catalyzed carbene transfer from diazo compounds, see:
(a) M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284 CrossRef CAS;
(b) M. R. Fructos, P. de Frémont, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2006, 25, 2237 CrossRef CAS;
(c) I. Rivilla, B. P. Gõmez-Emeterio, M. R. Fructos, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2011, 30, 2855 CrossRef CAS;
(d) E. López, G. Lonzi and L. A. López, Organometallics, 2014, 33, 5924 CrossRef;
(e) J. Barluenga, G. Lonzi, M. Tomás and L. A. López, Chem.–Eur. J., 2013, 19, 1573 CrossRef CAS PubMed;
(f) J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 11916 CrossRef CAS;
(g) J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 13314 CrossRef CAS PubMed;
(h) Z.-Y. Cao, X. Wang, C. Tan, X.-L. Zhao, J. Zhou and K. Ding, J. Am. Chem. Soc., 2013, 135, 8197 CrossRef CAS PubMed;
(i) V. V. Pagar, A. M. Jadhav and R. S. Liu, J. Am. Chem. Soc., 2011, 133, 20728 CrossRef CAS;
(j) A. M. Jadhav, V. V. Pagar and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 11809 CrossRef CAS PubMed;
(k) S. K. Pawar, C.-D. Wang, S. Bhunia, A. M. Jadhav and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 7559 CrossRef CAS;
(l) D. Zhang, G. Xu, D. Ding, C. Zhu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 11070 CrossRef CAS PubMed;
(m) G. Xu, C. Zhu, W. Gu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 883 CrossRef CAS;
(n) Y. Zhou, B. G. Trewyn, R. J. Angelici and L. K. Woo, J. Am. Chem. Soc., 2009, 131, 11734 CrossRef CAS PubMed;
(o) G. Lonzi and L. A. López, Adv. Synth. Catal., 2013, 355, 1948 CrossRef CAS;
(p) A. Prieto, M. R. Fructos, M. M. Díaz-Requejo, P. J. Pérez, P. Pérez-Galán, N. Delpont and A. M. Echavarren, Tetrahedron, 2009, 65, 1790 CrossRef CAS.
- A. Padwa, D. J. Austin, A. T. Price, M. A. Semones, M. P. Doyle, M. N. Protopopova, W. R. Winchester and A. Tran, J. Am. Chem. Soc., 1993, 115, 8669 CrossRef CAS.
- Y. Liang, H. Zhou and Z.-X. Yu, J. Am. Chem. Soc., 2009, 131, 17783 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
-
(a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS;
(b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS.
-
(a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS;
(b) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Chem. Phys., 2001, 22, 976 CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
-
R. Dennington, T. Keithm and J. Millam, GaussView, Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- In order to rule out the pathway via the Büchner reaction, more control experiments were also carried out in our lab. Please see Section 3 in the ESI† for more details.
- Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS.
- For a nice experiment example of O–H insertion via oxonium ylides, see: X. Zhang, H. Huang, X. Guo, X. Guan, L. Yang and W. Hu, Angew. Chem., Int. Ed., 2008, 47, 6647 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Data for new compounds, experimental procedures and theoretical studies on mechanisms. See DOI: 10.1039/c5sc04319k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a