
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePt(IV) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action†
Raji
Raveendran
a,
Jeremy Phillip
Braude
b,
Ezequiel
Wexselblatt
a,
Vojtech
Novohradsky
c,
Olga
Stuchlikova
cd,
Viktor
Brabec
c,
Valentina
Gandin
 *b and
Dan
Gibson
*a
*b and
Dan
Gibson
*a
aInstitute for Drug Research, School of Pharmacy, The Hebrew University, Jerusalem, 91120, Israel
bDipartimento di Scienze del Farmaco, Universita di Padova, Via Marzolo 5, 35131 Padova, Italy. E-mail: valentina.gandin@unipd.it
cInstitute of Biophysics, Academy of Sciences of the Czech Republic, v.v.i., Kralovopolska 135, CZ-61265 Brno, Czech Republic
dDepartment of Biophysics, Faculty of Science, Palacky University, 17. listopadu 12, CZ-77146 Olomouc, Czech Republic
First published on 15th January 2016
Abstract
Our study demonstrates that Pt(IV) derivative of cisplatin, with two axial PhB ligands, ctc-[Pt(NH3)2(PhB)2Cl2], is a very potent cytotoxic agent against many different human cancer cell lines and is up to 100 fold more potent than cisplatin, and significantly more potent than the Pt(IV) derivatives of cisplatin with either two hydroxido, two acetato or two valproato ligands. The high potency of this compound (and some others) is due to several factors including enhanced internalization, probably driven by “synergistic accumulation” of both the Pt moiety and the phenylbutyrate, that correlates with enhanced DNA binding and cytotoxicity. ctc-[Pt(NH3)2(PhB)2Cl2] inhibits 60–70% HDAC activity in cancer cells, at levels below the IC50 values of PhB, suggesting synergism between Pt and PhB. Mechanistically, ctc-[Pt(NH3)2(PhB)2Cl2] induces activation of caspases (3 and 9) triggering apoptotic signaling via the mitochondrial pathway. Data also suggest that the antiproliferative effect of ctc-[Pt(NH3)2(PhB)2Cl2] may not depend of p53. Pt(IV) derivatives of cisplatin with either two axial PhB or valproate ligands are more potent than their oxaliplatin analogs. ctc-[Pt(NH3)2(PhB)2Cl2] is significantly more potent than its valproate analog ctc-[Pt(NH3)2(VPA)2Cl2]. These compounds combine multiple effects such as efficient uptake of both Pt and PhB with DNA binding, HDAC inhibition and activation of caspases to effectively kill cancer cells.
Introduction
Despite the widespread clinical use of cisplatin, carboplatin and oxaliplatin, (Fig. 1), their drawbacks have prompted medicinal chemists to design novel platinum based drugs with improved therapeutic properties1–3 and clinicians to combine platinum drugs with other therapeutics in order to try and improve their clinical efficacy.4–6 | ||
| Fig. 1 The FDA approved platinum anticancer drugs (top row) and several HDAC inhibitors. | ||
Several reports describe the co-administration of platinum anticancer agents with inhibitors of histone deacetylases (HDACis), primarily with vorinostat and valproate (Fig. 1) that resulted in improved therapeutic profiles.7–9 HDACis are emerging as a new class of epigenetic anticancer drugs that can alter gene transcription and exert antitumor effects such as growth arrest, differentiation, apoptosis, and inhibition of tumor angiogenesis.10,11 The FDA approved in 2006 the first HDAC inhibitor – suberoylanilide hydroxamic acid (SAHA vorinostat) to treat the rare cancer cutaneous T-cell lymphoma (CTCL).12 Inhibition of HDAC results in hyperacetylated histones that cannot associate with the nuclear DNA leaving the DNA in an open form making it more susceptible to DNA damaging agents.
Platinum anticancer agents are believed to trigger the death of cancer cells by covalently binding to two adjacent guanines on the same strand of the nuclear DNA. The binding of the platinum to the DNA distorts the double helical structure of the DNA causing a kink towards the major groove. The cellular response to the distortion of the DNA determines the fate of the cancer cell and can lead to the death of the cancer cell.13–15
Thus, combining HDACis that expose the nuclear DNA with cytotoxic DNA damaging agents, such as platinum drugs, could, at least in theory, enhance the efficacy of the platinum drugs. Marmion and co-workers combined cis-[Pt(NH3)2] with the malonate derivatives of SAHA and belinostat and prepared trans-[Pt(py)(NH3)(VPA)2] in an attempt to capitalize on the potential synergistic effect of combining platinum complexes with HDACis (Fig. 2).16–18 Unfortunately these compounds, whose activity hinges on the intracellular aquation of the Pt(II) complexes to release the HDACi, were not as potent as might be expected.
 | ||
| Fig. 2 Platinum(II) complexes with HDAC inhibitor ligands. | ||
Rather than rely on aquation for the simultaneous release of the two antiproliferative agents inside the cancer cell, it is possible to harness the favorable chemical properties of the octahedral Pt(IV) complexes towards that end. Pt(IV) complexes are prepared from the square planar Pt(II) complexes by oxidatively adding two ligands in the axial positions. The d6 electronic configuration of the Pt(IV) complexes confers inertness to the compounds and retards unwanted interactions with nucleophiles prior to reaching the cancer cells. The prevailing assumption is that the Pt(IV) complexes are reduced inside the cell, simultaneously releasing the original cytotoxic Pt(II) drugs as well as the two axial ligands.19–22 Thus, attaching HDACis to the axial positions of the Pt(IV) derivatives of cisplatin or oxaliplatin should result in simultaneous release inside the cell of two antiproliferative agents that act by different mechanisms on different cellular targets.
Recent reports describe the preparation and biological properties of Pt(IV) derivatives of cisplatin or oxaliplatin with two axial valproate ligands (that are HDAC inhibitors).23–26 In both cases, these compounds are reported to be very potent cytotoxic agents against several types of cancer cell lines. Osella and co-workers concluded that the cytotoxicity of ctc-[Pt(NH3)2(VPA)2Cl2] is due solely to the antiproliferative action of the platinum and that the enhanced efficacy relative to cisplatin was only due to enhanced accumulation. They also stated that there cannot be a synergistic effect between VPA and cisplatin because the concentration of the VPA ligands of ctc-[Pt(NH3)2(VPA)2Cl2] that are released in the cells (∼μM levels), is too low to inhibit histone deacetylases, since the IC50 value of VPA is in the mM range.24
On the other hand, Shen and co-workers claim that the cytosolic ctc-[Pt(NH3)2(VPA)2Cl2] is reduced in the cells to yield cisplatin and VPA eliciting HDAC inhibitory effect and inducing cell cycle arrest at S phase and cell death through apoptosis in a time-dependent manner.23 Neither Osella nor Shen measured the inhibition of HDAC activity in the cancer cells.
Recently we demonstrated that Pt(IV) derivatives of cisplatin with axial VPA ligands inhibit HDAC activity in cells resulting in enhanced acetylation of histone 3, decondensation of heterochromatin and more efficient DNA platination by cytoplasmic platinum.26
We believe that the potency of these “dual action” prodrugs might be due to a combination of several cellular events rather than just inhibition of HDAC activity combined with DNA platination. With the purpose of shedding light on the mechanism of action of Pt(IV)–HDACi pro-drugs, and optimizing the choice of the platinum moiety and axial HDACi ligands, we describe the synthesis and biological activities of Pt(IV) derivatives of cisplatin, oxaliplatin and trans-[Pt(n-butylamine)(piperidino-piperidine)Cl2]+ with two different HDAC inhibitors – valproate (VPA) and 4-phenylbutyrate (PhB).
Results and discussion
Synthesis of the compounds
The twelve compounds synthesized for this study are depicted in Fig. 3. The symmetric compounds I, III, VI, VIII and IX–XII were synthesized following standard procedures.27 In brief, cisplatin or oxaliplatin were oxidized with H2O2 to yield ctc-[Pt(NH3)2(OH)2Cl2] (IX) or [Pt(DACH)(OH)2(ox)] (X) that were then reacted with an excess of the anhydrides of valproic acid, 4-phenylbutyric acid or acetic acid to yield the desired compounds.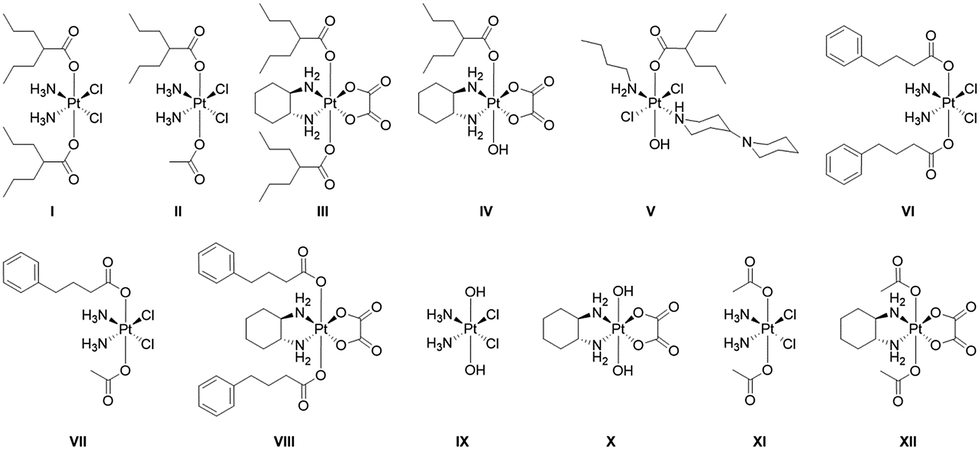 | ||
| Fig. 3 Platinum(IV) complexes with axial valproate or 4-PhB ligands studied in this work. | ||
The non-symmetric compound ctc-[Pt(NH3)2(OH)(OAc)Cl2] was prepared by oxidizing cisplatin in acetic acid.28 It was subsequently reacted with 1.5 equivalents of the appropriate anhydride in DMF to yield ctc-[Pt(NH3)2(L)(OAc)Cl2] where L = VPA, PhB.
trans-[Pt(n-butylamine)(piperidino-piperidine)Cl2]+ was prepared as described29 and was oxidized with H2O2 to yield ttt-[Pt(n-butylamine)(piperidino-piperidine)(OH)2Cl2]+ which was then reacted with valproic anhydride to yield ttt-[Pt(n-butylamine)(piperidino-piperidine)(VPA)(OH)Cl2]+.
All compounds were characterized by 195Pt NMR spectroscopy and when possible by mass spectrometry. They were purified by preparative HPLC and their purity was ascertained by analytical HPLC and by elemental analysis. Of the compounds with HDAC inhibitors in the axial positions compound I was previously reported by Shen23 and Osella24 and compounds I–IV by us.25,26 Compounds V–VIII are reported here for the first time.
Cytotoxicity
Compounds I–XII, uncoordinated PhB as well as cisplatin and oxaliplatin were screened against seven different cancer cell lines representative of lung (A549), breast (MCF-7), pancreatic (BxPC3), kidney (A498), prostate (PC3) and colon (HCT-15) carcinoma, along with melanoma (A375). Compounds I–VII were also screened against cisplatin sensitive and resistant ovarian cancer cell line (A2780 and A2780cisR). The cytotoxicity parameters, in terms of IC50 (the median growth inhibitory concentration calculated from dose–survival curves) obtained after 72 h exposure, are reported in Table 1.| A2780 | A2780cisR | MCF-7 | A549 | HCT-15 | PC3 | A498 | BxPC3 | A375 | |
|---|---|---|---|---|---|---|---|---|---|
| a An average of three measurements. | |||||||||
| CDDP | 2.8 ± 0.2 | 13.8 ± 0.2 | 17.4 ± 0.2 | 8.35 ± 0.87 | 11.32 ± 1.06 | 2.25 ± 0.82 | 17.53 ± 1.21 | 11.36 ± 1.14 | 4.03 ± 0.95 |
| OXP | 0.52 ± 0.27 | 4.2 ± 1.6 | 3.36 ± 0.2 | 1.46 ± 0.31 | 1.15 ± 0.43 | 5.24 ± 0.86 | 7.94 ± 1.04 | 4.15 ± 0.93 | 6.30 ± 1.11 |
| I | 0.25 ± 0.09 | 0.30 ± 0.10 | 0.91 ± 0.30 | 1.12 ± 0.32 | 0.81 ± 0.12 | 2.13 ± 0.45 | 2.43 ± 0.72 | 1.13 ± 0.14 | 1.25 ± 0.25 |
| II | 1.02 ± 0.6 | 1.62 ± 0.30 | 5.74 ± 1.10 | 8.36 ± 1.14 | 6.36 ± 1.03 | 7.32 ± 1.24 | 4.21 ± 0.95 | 3.54 ± 0.66 | 2.17 ± 0.53 |
| III | 1.3 ± 0.2 | 8.8 ± 2.1 | 5.7 ± 0.9 | 20.5 ± 2.14 | 16.4 ± 1.63 | 10.1 ± 1.17 | 5.21 ± 1.10 | 14.4 ± 2.16 | 4.17 ± 0.87 |
| IV | 5.4 ± 0.2 | 16.1 ± 0.7 | 31.6 ± 2.1 | 60.3 ± 3.34 | 51.5 ± 3.26 | 53.2 ± 4.24 | 21.1 ± 3.07 | 19.8 ± 2.25 | 47.3 ± 2.53 |
| V | 1.18 ± 0.33 | 1.29 ± 0.29 | 8.80 ± 1.48 | 15.5 ± 1.13 | 14.4 ± 2.05 | 16.8 ± 1.41 | 8.64 ± 0.95 | 4.43 ± 1.94 | 12.4 ± 2.93 |
| VI | 0.14 ± 0.03 | 0.12 ± 0.03 | 0.18 ± 0.03 | 0.17 ± 0.04 | 0.31 ± 0.04 | 0.41 ± 0.06 | 0.43 ± 0.02 | 0.19 ± 0.01 | 0.16 ± 0.02 |
| VII | 0.65 ± 0.04 | 1.54 ± 0.27 | 3.43 ± 0.14 | 7.16 ± 1.11 | 5.17 ± 0.85 | 4.34 ± 1.25 | 8.42 ± 1.47 | 10.14 ± 1.69 | 6.25 ± 1.94 |
| VIII | 0.90 ± 0.08 | 1.96 ± 0.44 | 2.28 ± 0.46 | 19.63 ± 3.22 | 12.54 ± 1.13 | 6.35 ± 1.19 | 9.32 ± 1.07 | 7.64 ± 1.15 | 0.64 ± 0.13 |
| IX | — | — | 11.52 ± 3.71 | 22.52 ± 4.54 | 15.34 ± 3.23 | 2.98 ± 1.35 | 21.98 ± 0.85 | 6.23 ± 1.47 | 1.93 ± 0.75 |
| X | — | — | 19.88 ± 1.41 | 26.14 ± 2.89 | 18.89 ± 3.18 | 5.85 ± 1.08 | 24.36 ± 5.35 | 14.33 ± 2.09 | 6.85 ± 1.55 |
| XI | — | — | 39.52 ± 4.45 | 32.32 ± 4.25 | 40.23 ± 2.86 | 10.11 ± 2.02 | 20.12 ± 0.85 | 36.59 ± 3.39 | 29.69 ± 5.16 |
| XII | — | — | 46.58 ± 1.48 | 35.14 ± 4.25 | 48.28 ± 3.33 | 15.85 ± 2.87 | 22.45 ± 5.05 | 29.69 ± 5.16 | 19.59 ± 2.84 |
| PhB | — | — | 1735 ± 27 | 1989 ± 22 | 1214 ± 81 | 795 ± 18 | 925 ± 81 | 1523 ± 36 | 25.65 ± 2.89 |
Uncoordinated HDAC inhibitor ligands, VPA and PhB, displayed very low cytotoxic activities, with IC50 values in the millimolar range, in agreement with the literature.25,26,29 With two exceptions, the cytotoxic activity of mono- and bis-VPA Pt(IV) derivatives of cisplatin (I and II) was significantly higher than that elicited by cisplatin against all tested cell lines. Over the seven cancer cell lines, the average IC50 values (μM), were 1.4 for ctc-[Pt(NH3)2(VPA)2Cl2] (I), 5.4 for ctc-[Pt(NH3)2(VPA)(OAc)Cl2] (II), and 10.3 for cisplatin. To assess the impact that the VPA ligands might have on the potency we prepared the Pt(IV) derivatives of cisplatin with non-bioactive axial ligands; with two hydroxido axial ligands (IX) and with two acetato ligands (XI) and measured their cytotoxicity against five cancer cell lines (Table 1). The bis-VPA compound was significantly more potent than both compounds IX and XI in all cell lines having an average IC50 of 1.4 μM compared with 7.0 and 29.2 μM for IX and XI respectively. The mono-VPA compound (II) was more potent than the bis-OAc compound (XI) in all five cell lines but only somewhat more potent that the bis-OH (IX). Interestingly, the bis-OH (IX) was significantly more potent that the bis-OAc compound in all five cell lines. These results, similar to those reported for ctc-[Pt(NH3)2(VPA)2Cl2],23,24,26 clearly indicate that Pt(IV) derivatives of cisplatin with bioactive VPA ligands are significantly more potent than both the parent cisplatin and its Pt(IV) derivatives with two hydroxido or two acetato axial ligands. In particular, ctc-[Pt(NH3)2(VPA)2Cl2] (I) was about 19, 14 and 10 times more effective than cisplatin against breast (MCF-7), colon (HCT-15) and pancreatic (BxPC3) cancer cells, respectively. When one VPA ligand of ctc-[Pt(NH3)2(VPA)2Cl2] was replaced with an acetato to give ctc-[Pt(NH3)2(VPA)(OAc)Cl2] (II), there was a 1.7–7.9 fold reduction in the potency compared to ctc-[Pt(NH3)2(VPA)2Cl2]. Comparing the averages over the seven cell lines, the bis-VPA derivative of cisplatin (I) was 3.9 fold more potent than the mono-VPA monoacetato analog (II). Yet, with the exception of the PC3 cells, ctc-[Pt(NH3)2(VPA)(OAc)Cl2] was as or more potent than cisplatin in all tested cell lines.
The behavior of the Pt(IV) derivatives of oxaliplatin with VPA axial ligands was very different than that observed for the Pt(IV) derivatives of cisplatin with VPA ligands. The antiproliferative potencies of compounds III, [Pt(DACH)(VPA)2(ox)] and IV, [Pt(DACH)(VPA)(OH)(ox)], were significantly lower than those of the parent Pt(II) complex, oxaliplatin. The average IC50 values (μM), over the seven cell lines, were 4.2, 10.9 and 40.7 for oxaliplatin, III and IV, respectively. Compound III was significantly more potent than the bis-acetato derivative of oxaliplatin (XII) with average IC50 values of 10.2 and 33.2 μM respectively and only somewhat more potent than the bis-hydroxido derivative of oxaliplatin (X) in four out of the five cell lines having an average IC50 of 10.2 μM compared with 13.2 for X. Again, the bis-hydroxido derivative of oxaliplatin was significantly more potent than its bis-acetato analog.
The trans-platinum derivative, trans-[Pt(n-butylamine)(VPA)(OH)(piperidino-piperidine)Cl2]+ (V), which possesses one VPA ligand in an axial position, elicited an average IC50 value over the seven tested cell lines (11.5 μM) only slightly higher than that of cisplatin. However, it retained a better in vitro antitumor potency with respect to cisplatin against four out of the seven tested cell lines (MCF-7, A549, A498 and BxPC3 cells).
Compounds VI and VII, the Pt(IV) derivatives of cisplatin with either two or one 4-phenylbutyrate (PhB) ligands in the axial positions, showed significant antiproliferative activity against all tested cell lines. Like with mono- and bis-VPA cisplatin derivatives, the bis-PhB cisplatin derivative, (VI), was significantly more potent than cisplatin, with average IC50 values (μM) of 0.26 (0.18–0.43) and 10.32 (2.25–17.53) respectively while the mono-PhB (VII) was only slightly more potent than cisplatin over the 7 cell lines having average IC50 values (μM) of 6.42 and 10.32 respectively. Remarkably, the bis-PhB derivative, ctc-[Pt(NH3)2(PhB)2Cl2] (VI) was nearly 100-, 60- and 50-fold more potent than cisplatin against MCF-7, BxPC3 and A549 cells, respectively. As in the case of ctc-[Pt(NH3)2(VPA)2Cl2] and ctc-[Pt(NH3)2(VPA)(OAc)Cl2], the symmetric ctc-[Pt(NH3)2(PhB)2Cl2] was considerably more effective than ctc-[Pt(NH3)2(PhB)(OAc)Cl2]. Notably, the bis-PhB derivative (VI) elicited IC50 values 4.6–53 fold lower than those obtained with the non-symmetric mono-PhB monoacetato complex (VII) and, on the average, about 24-fold more potent than cisplatin. Yet, ctc-[Pt(NH3)2(PhB)(OAc)Cl2] was more potent than cisplatin in five out of the seven cells lines by factors of 2.1–9. Thus, by replacing a PhB in ctc-[Pt(NH3)2(PhB)2Cl2] with an acetato ligand significantly diminished the potency of the resulting Pt(IV) complex.
Comparing the cytotoxicity results of I and VI, reveals that replacing both VPA ligands with PhB in the Pt(IV) derivatives of cisplatin increased the potency of the complex (on the average there is nearly a 5-fold improvement in potency). Conversely, no such effect was observed with the non-symmetric compounds and ctc-[Pt(NH3)2(PhB)(OAc)Cl2] and ctc-[Pt(NH3)2(VPA)(OAc)Cl2], elicited similar cytotoxicity profiles.
The Pt(IV) derivative of oxaliplatin with two PhB, [Pt(DACH)(PhB)2(ox)] was significantly (4–40 fold) less potent than its cisplatin analog (VI) and had similar potency to [Pt(DACH)(VPA)2(ox)] (III).
Furthermore, Pt(IV) complexes (I–VIII) were also tested for their ability to overcome acquired resistance by testing their in vitro cytotoxicity on a pair of human ovarian adenocarcinoma cell lines which have been selected for sensitivity/resistance to cisplatin (cancer cells A2780/A2780cisR). Both Pt(IV) derivatives of cisplatin with VPA axial ligands (I and II) exhibited similar potency towards the sensitive and the resistant ovarian cancer cells A2780 and A2780cisR. Compound I was 11 and 46 fold more potent than cisplatin against A2780 and A2780cisR cells, respectively, while compound II was 2.7 and 8.5 fold more potent than cisplatin. The resistance factors (R.F.: the ratio between IC50 values calculated for the resistant cells and those obtained with the sensitive ones) calculated for I and II were of 1.2 and 1.6, respectively, indicating their ability to circumvent acquired resistance to cisplatin. Similarly, the bis-PhB cisplatin Pt(IV) derivative (VI) was roughly 115 times more effective than cisplatin against the resistant ovarian cancer cell line A2780cisR, having a resistance factor of 0.85, thus attesting to the absence of cross-resistance with cisplatin.
The Pt(IV) derivatives of oxaliplatin with either VPA or PhB axial ligands as well as the Pt(IV) derivative of cisplatin with one PhB showed cross-resistance with cisplatin in the A2780cisR cells (R.F. 6.8, 3.0, 2.4 and 2.2 for III, IV, VII and VIII, respectively).
Cellular accumulation and binding to nuclear DNA
With the aim of identifying a possible correlation between cytotoxic activity and cellular accumulation, the cellular Pt content was measured in MCF-7 cells treated for 24 h with equi-Pt concentrations (5 μM) of the Pt(IV) compounds. The cellular platinum levels were quantified by means of GF-AAS analysis, and the results expressed as pg Pt per 106 cells, are shown in Fig. 4. | ||
| Fig. 4 (a) Cellular and nuclear accumulation of equi-Pt doses (5 μM) incubated with MCF-7 cells for 24 h. (b) Platination levels of nuclear DNA following a 24 h incubation of 5 μM of the complexes with MCF-7 cells. | ||
The levels of cell associated platinum strictly depend on the structures of the Pt(IV) complexes. With the exception of compound IV, all the Pt(IV) complexes with HDAC inhibitors as axial ligands accumulated in cells more efficiently compared to the dihydroxido or diacetato Pt(IV) derivatives (IX–XII) and interestingly, the Pt(IV) derivatives of cisplatin and oxaliplatin with two axial hydroxido ligands accumulated more efficiently than their analogs with two axial acetato ligands. Among the Pt(IV) complexes with HDAC inhibitors as axial ligands, the accumulation pattern was VI > I > II ≈ III ≈ VII ≈ VIII > V > IV. Overall, uptake results suggest that the Pt(IV) derivatives of cisplatin with two HDACis in the axial positions accumulated most efficiently, and the bis-PhB derivative (VI) accumulated somewhat more efficiently than its bis-VPA counterpart (I). Interestingly, the cellular accumulation of Pt(IV) derivatives of cisplatin with one acetato and either one VPA or one PhB (II and VII) and the Pt(IV) derivatives of oxaliplatin with either two VPA or two PhB ligands (III and VIII) displayed similar accumulation. In this cell line, the transplatinum complex with one VPA ligand also accumulated very efficiently. The same pattern of cellular accumulation was obtained employing A375 melanoma cells (not shown).
We measured the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of compounds VI and VIII to see if a correlation exists between lipophilicity and cell association. To quantify the hydrophobicity of the platinum complexes, partition coefficients (logP) values for octanol/water partition were measured using the shake-flask method. The logP values for cisplatin, compounds I, II, III, VI and VIII are shown in Table 2.
P of compounds VI and VIII to see if a correlation exists between lipophilicity and cell association. To quantify the hydrophobicity of the platinum complexes, partition coefficients (logP) values for octanol/water partition were measured using the shake-flask method. The logP values for cisplatin, compounds I, II, III, VI and VIII are shown in Table 2.
P (octanol/water) values obtained by shake flask methoda
| Compound | Formula | logP |
|---|---|---|
| a Results are expressed as the mean ± SD from three independent experiments. | ||
| Cisplatin26 | −2.25 ± 0.04 | |
| I | ctc-[Pt(NH3)2(VPA)2Cl2]26 | 0.25 ± 0.03 |
| II | ctc-[Pt(NH3)2(Ac)(VPA)Cl2]26 | 0.17 ± 0.02 |
| III | [Pt(DACH)(VPA)2(ox)]25 | 0.37 ± 0.08 |
| IV | [Pt(DACH)(VPA)(OH)(ox)]25 | 0.10 ± 0.05 |
| VI | ctc-[Pt(NH3)2(PhB)2Cl2] | −0.73 ± 0.05 |
| VIII | [Pt(DACH)(PhB)2(ox)] | 0.25 ± 0.02 |
Interestingly, there is no correlation between lipophilicity and cellular association. The highest accumulation is observed for compound VI followed by compound I. Although compounds I and VIII have the same logP values the former has much higher accumulation than the latter and compound III with the highest logP (0.37) does not accumulate as well as compounds VI and I.
Platination of nuclear DNA is a critical step in triggering the death of cancer cells by Pt based drugs. Since co-administration with HDAC inhibitors can facilitate binding of platinum drugs to nuclear DNA by increasing the exposed area of nuclear DNA, we also measured the nuclear accumulation of the complexes as well as the amount of platinum bound to nuclear DNA, to see whether either of them correlates with cytotoxicity. Cells were treated for 24 h with 5 μM of the tested complexes and the nuclei and nuclear DNA were isolated and the amount of Pt in each sample was determined by means of GF-AAS analysis. The results expressed as pg Pt per 106 cells for nuclear Pt content are depicted in Fig. 4a and the DNA platination levels expressed as pg Pt μg−1 DNA appear in Fig. 4b.
As with whole cell accumulation experiments, with the exception of compound IV, all the Pt(IV) complexes with HDAC inhibitors as axial ligands were more effective in platinating DNA compared to the dihydroxido and diacetato Pt(IV) derivatives (IX–XII). In particular, the highest nuclear Pt accumulation and DNA platination levels were observed with compound VI whereas the lowest values were detected for complex IV. These data corroborate the hypothesis that coordination of HDAC inhibitors in axial positions facilitates binding of the Pt(II) moiety to DNA. Furthermore, for all tested complexes the intranuclear accumulation and DNA platination profiles were comparable, suggesting that nuclear internalization correlated with DNA-platination.
HDAC inhibition
The complexes described here were designed specifically to release either VPA or PhB inside the cells in order to inhibit HDAC activity and pave the way for more efficient DNA platination. Thus, in addition to measuring the levels of DNA platination, we also measured the ability of these pro-drugs to inhibit HDAC activity in cells.MCF-7 cells were incubated for 24 h with cytotoxic IC50 concentrations of compounds I–XII and their HDAC activity was determined in cells and, for comparison, the HDAC inhibitory effect was also assayed in isolated nuclei of MCF-7 treated cells (Fig. 5a).
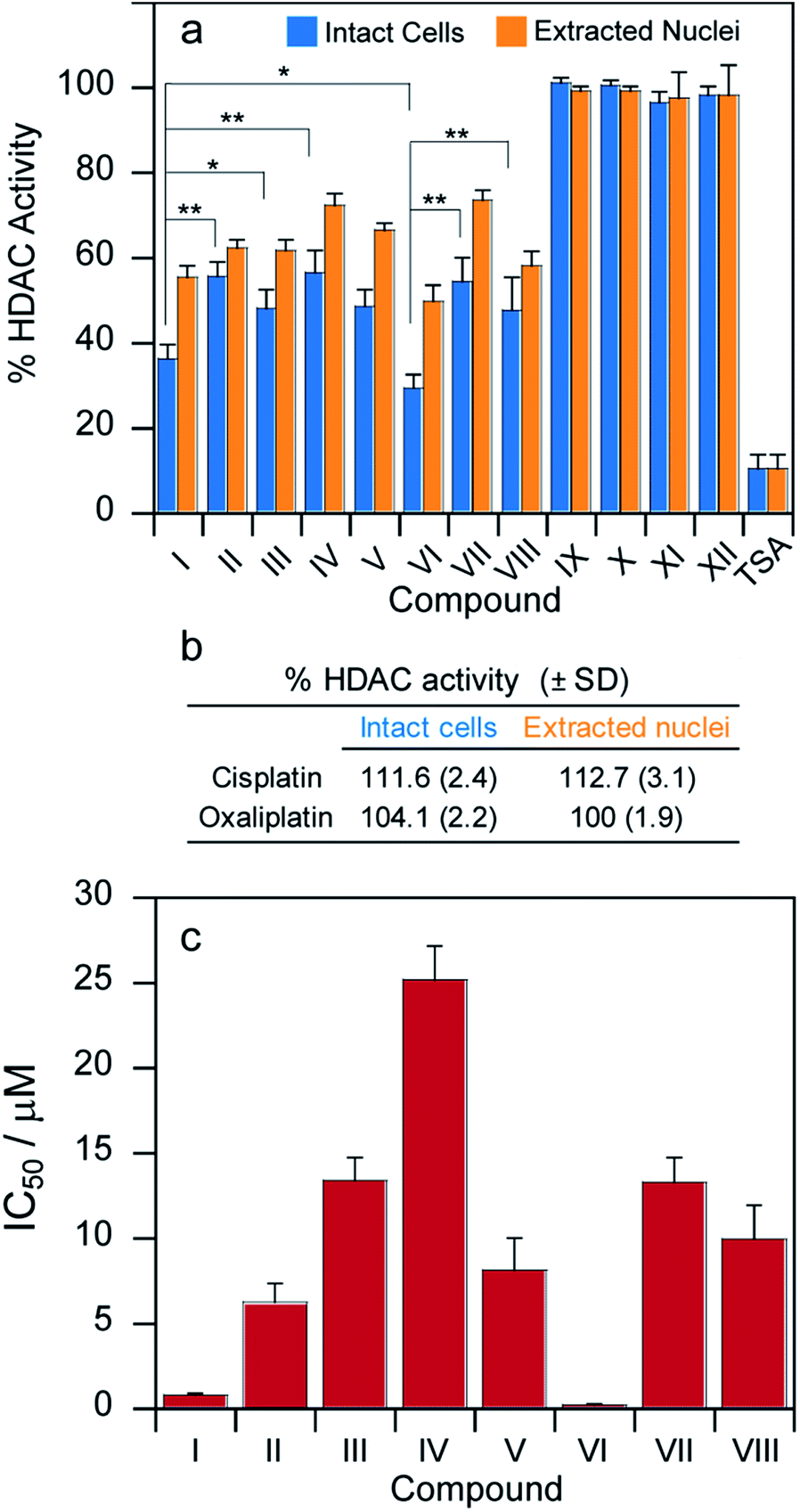 | ||
| Fig. 5 (a) MCF-7 cells were incubated for 24 h with IC50 concentrations of tested complexes. The HDAC activity was determined in cells (light blue bars) or in nuclear extracts (orange bars), *P < 0.05, **P < 0.01. (b) MCF-7 cells were incubated for 24 h with IC50 concentrations of either cisplatin or oxaliplatin and HDAC activity was determined. (c) MCF-7 cells were incubated for 24 h with increasing concentrations of tested complexes. HDAC IC50 values were calculated by 4 PL model (P < 0.05). | ||
In both experiments, the pattern of HDAC inhibition was very similar; resembling those obtained from cytotoxicity and platination assays. As expected, cisplatin, oxaliplatin as well as Pt(IV) complexes IX–XII lacking the HDAC inhibitors in the axial position were not effective in modifying HDAC activity (Fig. 5a and b). Compounds I and VI are significantly more potent HDAC inhibitors than the other derivatives. More importantly, their IC50 values (μM) calculated in term of cellular HDAC inhibitory activity were three orders of magnitude lower than those reported for free VPA or PhB, respectively. As a general consideration, the bis-HDACi compounds were more efficient than the corresponding mono-HDACi. However, all the complexes were significantly less potent HDAC inhibitors compared with trichostatin A (TSA).30
In addition to measuring the inhibition of HDAC activity at cytotoxic IC50 concentrations, we incubated the MCF-7 cells for 24 h with increasing concentrations of tested complexes and measured the HDAC activity for each compound. The HDAC inhibitory IC50 values were calculated by 4-PL model (Fig. 5c). The IC50 values are all in the μM range. Compounds I and VI are significantly more potent inhibitors of cellular HDAC activity than the other derivatives. More importantly, the IC50 values (μM) calculated in terms of cellular HDAC inhibitory activity were three orders of magnitude lower than those reported for VPA or PhB for all the complexes.
Further studies
To gain more insights into the mode of action of the newly synthesized Pt(IV) derivatives, compounds I and VI were chosen for further experiments. In particular, we evaluated and compared their capacity to induce apoptosis in human breast MCF-7 cells. Nuclear DNA fragmentation and apoptosome complex formation are critical steps in the apoptotic process.31 DNA fragmentation tests performed on MCF-7 cells treated for 12 or 24 h with IC50 concentrations of I and VI showed the ability of both Pt(IV) derivatives to increase mono- and oligo-nucleosome formation, to a very similar extent, in a time-dependent manner (Fig. 6a). Notably, after a 24 h treatment, I and VI induced respectively a 1.5 and 2 times higher nucleosome formation compared with cisplatin. Interestingly, both complexes containing HDAC inhibitors in the axial position were also more effective than Pt(IV) derivatives of cisplatin containing biologically inactive ligands, namely IX and X. Apoptotic cell death induction was also confirmed through a Hoechst 33342 staining. As depicted in Fig. 6b, MCF-7 cells treated with IC50 concentrations of I and VI displayed, pyknotic nuclei, chromatin condensation and fragmentation characteristics, typical of apoptosis.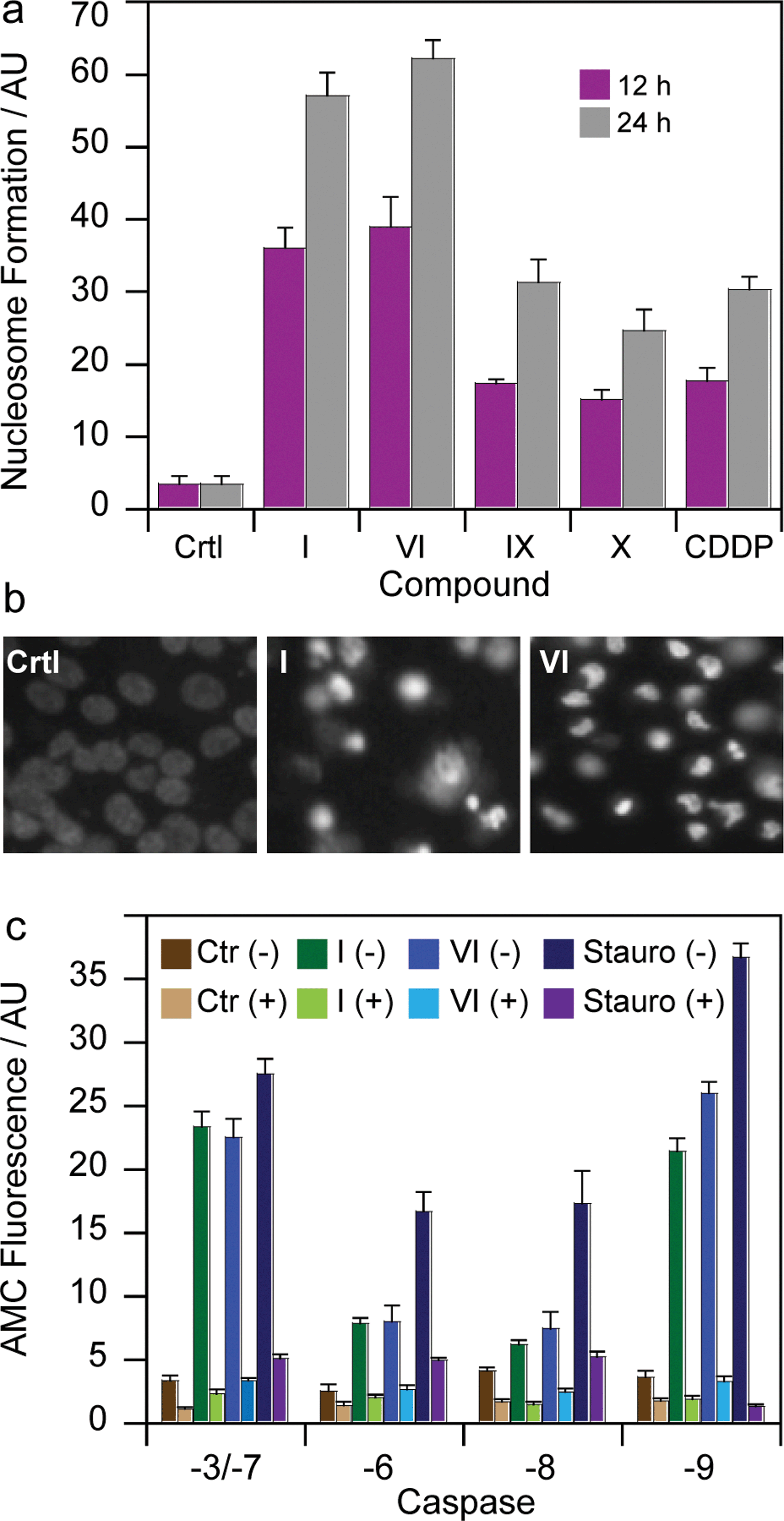 | ||
| Fig. 6 (a) Nuclear DNA fragmentation. MCF-7 cells were treated for 12 or 24 h with IC50 of I, VI or cisplatin (CDDP). Quantitative estimation of DNA fragmentation was obtained with an ELISA test. Data are the means of five independent experiments. Error bars indicate SD. (b) MCF-7 cells were treated with IC50 of I and VI for 72 h and stained with the fluorescent dye Hoechst 33258. (c) Caspase activity. MCF-7 cells incubated for 24 h with IC50 of I, VI or staurosporine ± broad-spectrum caspase inhibitor zVAD, and processed for caspase-3/-7, -6, -8, -9 activity. Data are the means of at least three independent experiments. Error bars indicate SD. | ||
To assess the mechanism of the triggered apoptotic process, the activity of the two initiator caspases (-8 and -9), and the downstream effectors (-3/-7 and -6) was determined in MCF-7 treated cultures. Staurosporine, an alkaloid inducing apoptosis through caspase-3/-7 and -9 activation, and z-VADfmk a pan caspase inhibitor, were used as positive and negative controls, respectively. As shown in Fig. 6c, both derivatives I and VI provoked a substantial activation of all caspases. Notably, after 24 h of incubation with IC50 concentrations of I and VI, the cleavage of caspases-3/-7 and -9 was higher with respect to activation of caspases -6 and -8. Caspase-3/-7 and -9 cleavage reached values similar to that exerted by the well-known caspase-dependent apoptosis inducer staurosporine.32
Interestingly, pre-treatment of cancer cells with zVAD, a cell-permeable pan-caspase inhibitor, strongly decreases caspase activation induced by Pt(IV) derivatives, thus confirming the role played by these proteases in cell death caused by the Pt(IV) complexes. Since caspases -3/-7 and -9 are mainly involved in intrinsic apoptosis pathway, whereas caspase -6 and -8 are extrinsic caspase executors, these data suggest that I and VI act predominantly by induction of apoptotic signaling via the mitochondrial pathway. Further support comes from studies in the myeloma cell lines demonstrating that PhB treatment induced activation of caspases -3, -7, and -9 accompanied by cleavage of their substrates and internucleosomal DNA degradation.33
Based on these findings, we thought of interest to evaluate the effect induced by Pt(IV) derivatives on mitochondria membrane potential (ΔΨ, MMP). MMP depletion generally precedes mitochondrial-driven apoptotic cell death.34 By treating MCF-7 cells with IC50 concentrations of I and VI for 24 and 48 h, the fluorescence intensity decreases with increasing exposure times, attesting to a time-dependent increase in percentage of cells with depleted MMP (Fig. 7a).
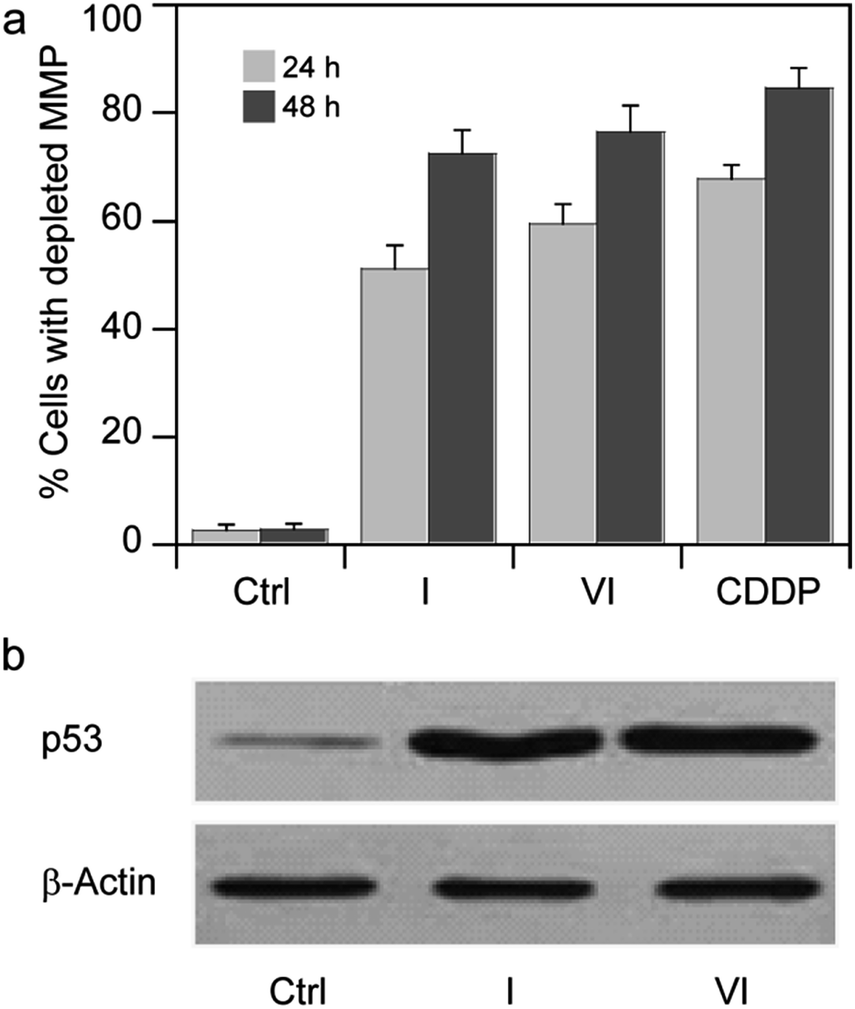 | ||
| Fig. 7 (a) Effects of I or VI on cellular mitochondrial membrane potential. MCF-7 cells were treated for 24 or 48 h with IC50 of I or VI. The percentage of cells with hypopolarized mitochondrial membrane potential was determined by Mito-ID® Membrane Potential Kit. Data are the means of five independent experiments. Error bars indicate SD. (b) MCF-7 cells were treated with IC50 of I or VI for 24 h. The amount of p53 was detected by Western blotting analysis as described in the ESI.† | ||
Satraplatin, ctc-[Pt(NH3)(c-hexylamine)(OAc)2Cl2], and other Pt(IV) derivatives have been recently shown to induce cell death in human cancer cells via p53 and p21 induction.35,36
The promotion of apoptosis by the platinum complexes may be connected with the induction of tumor suppressor p53, which plays a pro-apoptotic role.37–39 To investigate the possible involvement of p53 pathway in the mechanism of action of compounds I and VI, MCF-7 cells were treated for 24 h with I and VI. As depicted in Fig. 7b, treatment resulted in an increase in p53 activation. In addition, the effect of these platinum complexes on the viability of human colon carcinoma cells expressing p53 (HCT116 p53+/+) or non-expressing p53 (HCT116 p53−/−) was investigated. The cells were treated with I or VI for 72 h as described in the ESI.† The IC50 values and DNA content in the treated cells were determined as described in previously published articles.37–39 The data showed that compound I significantly reduced the number of cells in a dose-dependent manner, being only slightly more effective in p53-proficient cells compared to the p53-knockout cells (Table 3 and Fig. 8a). These results suggest that transcription factor p53 does not play a marked role in the mechanism of action of compound I. The efficacy of compound VI in killing p53+/+ and p53−/− cells was not too different (Table 3 and Fig. 8b). It significantly reduced the number of cells in a dose-dependent manner, being equally effective in p53-proficient and p53-knockout cells, indicating that compound VI operates via a pathway that is p53-independent. In this regard, the mechanism of action of compounds I and VI appears to be fundamentally different from that of cisplatin, which was shown to be more effective in p53-wild-type over the p53-mutant tumor cells.40
| HCT-116+/+ | HCT-116−/− | |
|---|---|---|
| a Results are expressed as the mean ± SD from three independent experiments. | ||
| I | 0.46 ± 0.07 | 1.4 ± 0.2 |
| VI | 0.22 ± 0.03 | 0.34 ± 0.08 |
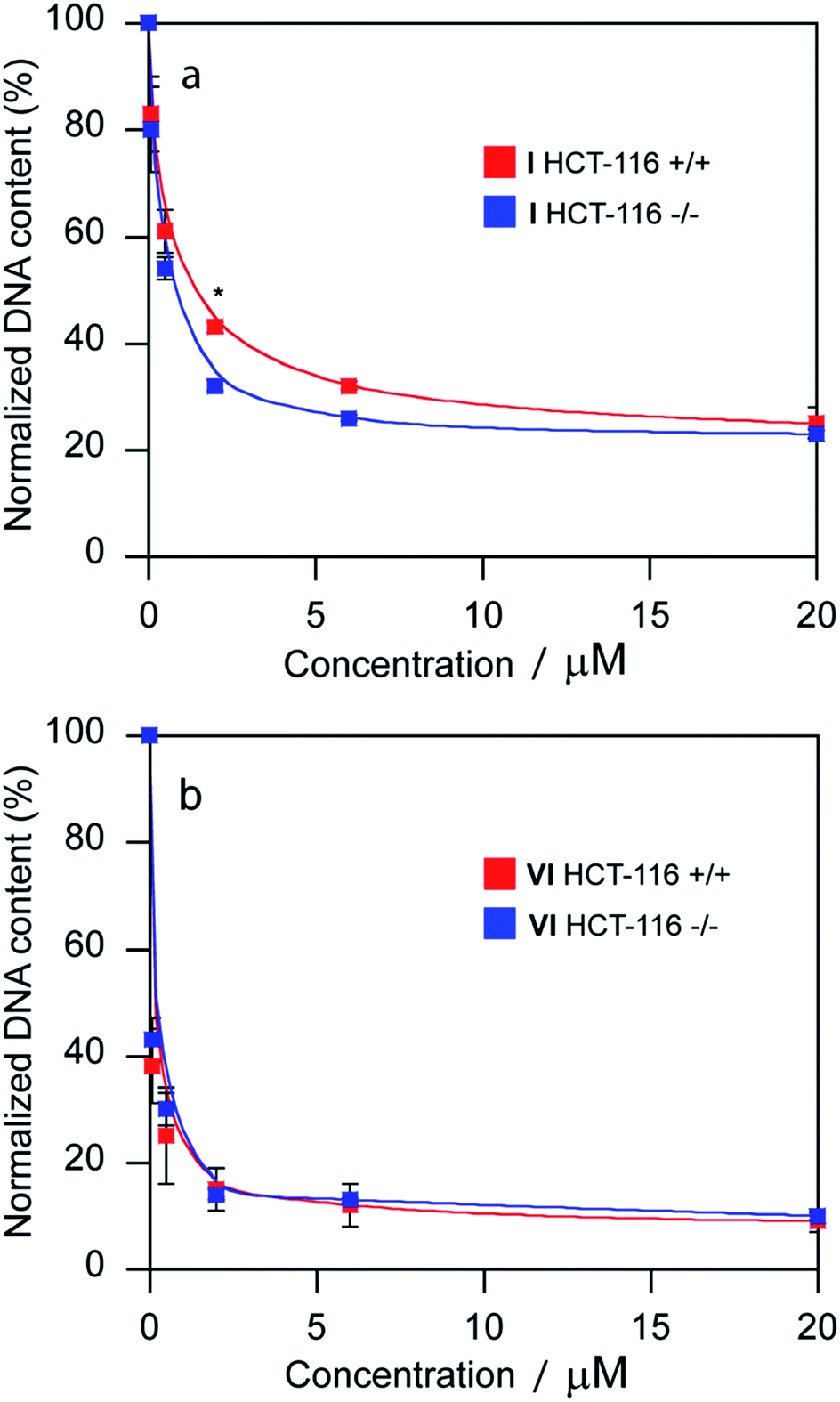 | ||
| Fig. 8 Sensitivity of HCT-116 cells [parental cells (p53+/+) and its p53-null derivative (p53−/−)] to the treatment with compounds I and VI. DNA quantification of total cells after treatment with compound I (a) and VI (b) assessed as a measure of fluorescence of Sybr Green I. DNA contents of control, untreated cells were taken as 100%. The error bars represent the SDs, experiments were done in triplicate. (*) Denotes significant (P < 0.05) difference from p53-wild-type (p53+/+) cells. | ||
Discussion
The hypothesis underlying this work was that by combining in the same molecular entity DNA damaging agents with pharmacophores that can facilitate platination of nuclear DNA we might be able to obtain new compounds with improved ability to kill cancer cells. Towards this end, we designed Pt(IV) derivatives of cisplatin and oxaliplatin that have one or two HDACis as axial ligands, such that the conjugation of hydrophilic Pt(II) complexes with lipophilic anionic HDACis will yield neutral conjugates that will enhance cellular accumulation of both the Pt moiety as well as the HDCAis and that once inside the cell these pro-drugs will be activated by reduction simultaneously releasing the Pt drug and the HDACis. Another goal was to study how the type of platinum moiety (cisplatin vs. oxaliplatin) and the nature of the HDACi (VPA vs. PhB) affect the various parameters associated with cytotoxicity (cellular and nuclear accumulation, DNA platination, HDAC inhibition etc.). To investigate these goals, we synthesized eight Pt(IV) complexes with HDACis in their axial position and compared their cytotoxicity to the parent Pt drugs (cisplatin or oxaliplatin), to the HDACis (VPA of PhB) and to the Pt(IV) derivatives of cisplatin and oxaliplatin with non-bioactive axial ligands (IX–XII).Screened against nine different cancer cell lines the eight Pt(IV) compounds elicited different responses. The Pt(IV) derivatives of cisplatin with two VPA or PhB were significantly more potent than cisplatin (13 and 50 fold respectively) while the VPA or PhB derivatives of oxaliplatin were only approximately as potent as oxaliplatin. Interestingly, although oxaliplatin was more effective than cisplatin over the nine cell lines (4.9 vs. 9.9 μM), the bis-VPA derivative of oxaliplatin was approximately 8.5 fold less potent than the bis-VPA derivative of cisplatin. Similarly, the bis-PhB derivative of cisplatin is nearly 20 fold more potent than its oxaliplatin analog. Even though it is less potent than oxaliplatin, cisplatin seems to be more effective than oxaliplatin in these types of Pt(IV) pro-drugs. Comparing the contribution of VPA and PhB to the cytotoxicity we see that for the Pt(IV) derivatives of cisplatin the bis-PhB derivative (VI) is approximately five fold more potent than it bis-VPA analog (I) and the bis-PhB derivative of oxaliplatin has similar potency to the bis-VPA derivative. So it seems like PhB is somewhat more effective than VPA at eliciting cytotoxicity.
Taken together the cytotoxicity results confirm that the conjugation of either two VPA or two PhB to the axial positions of cisplatin yields very effective derivatives, endowed with cytotoxic potency extraordinarily superior to those of free VPA or PhB or to the Pt(IV) derivatives of cisplatin or oxaliplatin with non-bioactive ligands.
A necessary step in the cascade of events leading to the death of the cancer cell is the cellular internalization of the complexes. Consistent with our hypothesis we found that with one exception (IV), the HDACi ligands significantly increase the cellular association (compared with the complexes with the non-bioactive ligands) and that the bis-HDACi derivatives of cisplatin (I and VI) accumulated more efficiently than the bis-HDACi derivatives of oxaliplatin (III and VIII). Interestingly, as mentioned above, we did not find any correlation between cellular accumulation and hydrophobicity as characterized by logP values. Noteworthy, we found a correlation between the cell-associated Pt levels (following 24 h incubation with equi-Pt concentrations) and the IC50 values. These results are in agreement with reports that higher cellular accumulation correlates with improved cytotoxicity.41,42 Unlike cisplatin that is reported to accumulate in cells by both passive diffusion and via the CTR1 receptor, Pt(IV) derivatives are reported to accumulate only by passive diffusion43 and thus greater lipophilicity should increase cellular accumulation.44 We recently reported that the mono- and bis-VPA derivatives of cisplatin accumulate primarily in the cell membrane.26 The same general correlation patterns were observed between potency and Pt accumulation in the nucleus and platination of nuclear DNA. The most potent compounds (I and VI) were also the most abundant in the nucleus and displayed the highest levels of DNA platination. It seems like there is a correlation between the Pt levels associated with whole cells and their levels in the nucleus and bound to DNA. To assess whether the bioactive VPA or PhB ligands enhanced DNA platination compared to the non-bioactive OAc ligands we used the data from Fig. 4 to estimate the percentages of DNA platination relative to the total amount of Pt associated with the cells. We estimated that platinum content of DNA fraction isolated from MCF-7 cells expressed as a percentage of the total platinum accumulated in the cells was 18% for ctc-[Pt(NH3)2(VPA)2Cl2] (I), 16% for ctc-[Pt(NH3)2(PhB)2Cl2] (VI) and only 5% for ctc-[Pt(NH3)2(OAc)2Cl2] (XI). Thus, the platinum content bound to DNA obtained from MCF-7 cells treated with I and VI was approximately 36 and 41-fold greater, respectively, than that obtained from the cells treated with XI. One of the hypotheses behind the design of such pro-drugs was the hope to increase platination of nuclear DNA by inhibition of HDAC activity that would lead to decondensed chromatin that affords higher accessibility to platination. We recently corroborated this hypothesis and showed that Pt(IV) derivatives with VPA axial ligands downregulate the expression of HDACs, enhance acetylation of histone H3 and decondense heterochromatin.26 These effects were ascribed to the intracellular activity of VPA presumed to be released by reduction in the cell. Moreover, considering the total Pt inside the cells (not including the fraction trapped in the membrane), a considerably higher fraction of Pt from the Pt(IV)–VPA conjugates is bound to DNA compared with conjugates with biologically inactive ligands suggesting a synergistic effect brought about by the valproate.26 In the current study, we observed a correlation between the ability to inhibit HDAC activity in cells (IC50 values for HDAC inhibition) and the cytotoxicity IC50 values. Again, compounds I and VI showed the highest inhibitory effects while compound IV was least effective. Notably, the complexes inhibit 50% cellular HDAC activity at μM extracellular concentrations while mM levels of free VPA or PhB are necessary to obtain the same effect.
One of the most intriguing questions when studying “dual action” platinum agents is whether the high potency is due primarily to a synergistic effect between the HDAC inhibitors and the Pt complexes as proposed by some authors,23 or is it merely due to the enhanced cellular accumulation of cisplatin as suggested by others.24 It is generally accepted that lipophilic ligands enhance the cellular uptake of Pt complexes and that higher accumulation usually results in enhanced cytotoxicity. Thus by attaching the lipophilic VPA or PhB ligands to the Pt pro-drug we expect to enhance the potency of the Pt moiety by virtue of higher cellular accumulation. At neutral pH, both valproic acid and 4-phenylbutyric acid are monoanionic and therefore do not efficiently accumulate in the cancer cells. Attempts to increase the cellular accumulation of valproate include neutralization of the charge by esterifying the carboxylate and forming for instance valproyl ester-valpramide of acyclovir.45 Ligation of VPA or PhB to the axial positions of the Pt(IV) complex converts them to neutral metalloesters thereby facilitating their cellular uptake. Conjugation of VPA or PhB to the Pt(IV) results in compounds endowed with “synergistic accumulation” where cisplatin and VPA (or PhB) are prodrugs of each other and the conjugation affords a significant enhancement in intracellular accumulation of both relative to either cisplatin or free VPA (or PhB).
Due partly to inefficient internalization into the cells, extracellular VPA or PhB are not very potent inhibitors of the cellular activity of HDAC and hence their IC50 values (in the mM range) do not provide any indication to the levels of intracellular VPA (or PhB) necessary to efficiently inhibit HDAC activity. The in vitro inhibitory potency of VPA or PhB might provide an indication to the cellular concentrations of VPA or PhB necessary to attain effective inhibition of HDACs. The in vitro IC50 values for PhB range from 64 to 260 μM for class I HDACs (HDAC1, HDAC2, HDAC3 and HDAC8) and were greater than 2000 μM for class IIa HDACs (HDAC4, HDAC5, HDAC7 and HDAC9) and 240 μM for HDAC6. For VPA the in vitro IC50 values for HDAC inhibition are 39 to 161 μM for class I and greater than 2000 μM for all the rest.46
The cellular concentrations of PhB or VPA released by compounds I and VI (in the low or sub-μM range) are well below the in vitro IC50 values for HDAC inhibition yet we observed 60–70% HDAC inhibition in cells for I and VI (Fig. 5a). Also, we obtained IC50 values for HDAC inhibition for compounds I and VI that are in the low μM (Fig. 5c). Together, this suggests that it might be due to a synergistic effect with the platinum.
These compounds are very potent and have the ability to induce an apoptotic cell death that encompass an increase of p53 activity, and that seems independent to p53 status. It is well known that the outcome of cancer chemotherapy strongly depends on the tumor p53 status,47 and tumor cells with mutated or deleted p53 tend to be less responsive to several common chemotherapeutics and radiation therapy.48 Hence, therapies that do not depend on functional p53 are clinically preferable, and can be a valid alternative strategy. Thus, Pt(IV)–HDACi complexes are very promising tools for the treatment of resistant neoplasia.
When we and others (Shen and Osella) conjugated VPA or PhB to Pt(IV), the rationale was to obtain “dual action” of HDAC inhibition (leading to higher accessibility to nuclear DNA) and thereby enabling more effective DNA platination that would trigger apoptosis. This however may have been an oversimplified view, since both VPA and PhB are not potent enough HDAC inhibitors to inhibit effectively HDACs at sub-μM levels. Thus, it might be reasonable to assume that the enhanced cytotoxic effects may arise from synergistic interactions between the platinum moieties and several cellular processes involving the VPA or PhB in addition to HDAC inhibition. VPA is engaged in cellular processes other than HDAC inhibition.26 PhB was reported to activate human peroxisome proliferator activated receptors, which are ligand-activated transcription factors that up-regulate the expression of several genes that code for lipid-metabolizing enzymes49 resulting in decreased conversion of mevalonic acid to farnesyl PPi,50 decreased cholesterol production, decreased protein prenylation,51,52 and decreased activation of the p21ras target p42MAPK/ERK2. It was suggested that PhB might have therapeutic utility as a cisplatin sensitizer in head and neck cancer by inhibiting the FA/BRCA pathway through the down regulation of BRCA1 as well as by an FA/BRCA-independent mechanism.53 PhB inhibits DNA damage-induced homologous recombination likely by mediating changes in chromatin acetylation. The combination of sodium phenylbutyrate with genotoxic agents can lead to different cell fates depending on the type of DNA damage inflicted.54
Thus, it seems that the impressive cytotoxicity displayed by the Pt(IV) derivatives of cisplatin with axial PhB ligands probably begins with the synergistic accumulation that facilitates high uptake of both cisplatin and the PhB (or VPA) and continues with the combined intracellular actions of cisplatin and PhB (or VPA) that include HDAC inhibition, triggering apoptosis and depleting MMP. Clearly, both intracellular PhB and VPA are involved in many other cellular processes which may or may not be involved in increasing the potency of the Pt(IV) compounds.
Conclusions
Our study demonstrates that Pt(IV) derivatives of cisplatin, with two axial PhB ligands are very potent cytotoxic agents against many different cancer cell lines and are significantly more potent than cisplatin or the Pt(IV) derivatives of cisplatin with either two hydroxido, two acetato or two valproato ligands. Conjugation of the anionic and lipophilic HDACis to Pt(IV) results in synergistic cellular accumulation that correlates with enhanced DNA binding and cytotoxicity. The results of this study suggest that Pt(IV) derivatives of cisplatin with two axial HDACis are superior to those of oxaliplatin despite the higher potency of oxaliplatin compared with cisplatin.Although designed to enhance DNA platination by inhibiting cellular HDAC activity, once inside the cell PhB or VPA can affect many cellular processes in addition to some HDAC inhibition.
Therefore, we cannot attribute the enhanced cytotoxicity to one specific cellular event and we believe that the “dual action” complexes, such as these, really are “multi-action” pro-drugs that once they get into the cells can trigger many different events that together lead to the death of the cancer cells.
Acknowledgements
VN, OS and VB were supported by the Czech Science Foundation (Grant 14-21053S). The research of OS was also supported by Palacky University in Olomouc (IGAPrF2015 025). The research of VG was supported by the University of Padova (Grants 60A04-0443, 60A04-3189 and 60A04-4015/15). This research was supported by the ISRAEL SCIENCE FOUNDATION (grant No. 1611/14) to DG.References
- J. Zhang, D. Liu, Y. Li, J. Sun, L. Wang and A. Zang, Mini-Rev. Med. Chem., 2009, 9, 1357–1366 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- A. G. Quiroga, Curr. Top. Med. Chem., 2011, 11, 2613–2622 CrossRef CAS.
- A. Ardizzoni, G. Antonelli, F. Grossi, L. Tixi, M. Cafferata and R. Rosso, Ann. Oncol., 1999, 10(suppl. 5), S13–S17 CrossRef PubMed.
- C. P. Belani, Semin. Oncol., 2002, 29, 4–9 CAS.
- Y. Itoyama, M. Kochi, S. Yamashiro, K. Yoshizato, J. Kuratsu and Y. Ushio, Neurol. Med. Chir., 1993, 33, 28–31 CrossRef CAS PubMed.
- H. V. Diyabalanage, M. L. Granda and J. M. Hooker, Cancer Lett., 2013, 329, 1–8 CrossRef CAS PubMed.
- W. J. Huang, Y. A. Tang, M. Y. Chen, Y. J. Wang, F. H. Hu, T. W. Wang, S. W. Chao, H. W. Chiu, Y. L. Yeh, H. Y. Chang, H. F. Juan, P. Lin and Y. C. Wang, Cancer Lett., 2014, 346, 84–93 CrossRef CAS PubMed.
- K. L. Jin, J. Y. Park, E. J. Noh, K. L. Hoe, J. H. Lee, J. H. Kim and J. H. Nam, J. Gynecol. Oncol., 2010, 21, 262–268 CrossRef CAS PubMed.
- P. W. Atadja, Prog. Drug Res., 2011, 67, 175–195 CAS.
- M. J. Lee, Y. S. Kim, S. Kummar, G. Giaccone and J. B. Trepel, Curr. Opin. Oncol., 2008, 20, 639–649 CrossRef CAS PubMed.
- B. S. Mann, J. R. Johnson, M. H. Cohen, R. Justice and R. Pazdur, Oncologist, 2007, 12, 1247–1252 CrossRef CAS PubMed.
- L. R. Kelland, Drug Resist. Updates, 2000, 3, 139–141 CrossRef CAS PubMed.
- M. A. Fuertes, J. Castilla, C. Alonso and J. M. Perez, Curr. Med. Chem., 2003, 10, 257–266 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- D. Griffith, M. P. Morgan and C. J. Marmion, Chem. Commun., 2009, 6735–6737 RSC.
- D. M. Griffith, B. Duff, K. Y. Suponitsky, K. Kavanagh, M. P. Morgan, D. Egan and C. J. Marmion, J. Inorg. Biochem., 2011, 105, 793–799 CrossRef CAS PubMed.
- V. Brabec, D. M. Griffith, A. Kisova, H. Kostrhunova, L. Zerzankova, C. J. Marmion and J. Kasparkova, Mol. Pharm., 2012, 9, 1990–1999 CrossRef CAS PubMed.
- C. M. Giandomenico, M. J. Abrams, B. A. Murrer, J. F. Vollano, M. I. Rheinheimer, S. B. Wyer, G. E. Bossard and J. D. Higgins, Inorg. Chem., 1995, 34, 1015–1021 CrossRef CAS PubMed.
- M. D. Hall and T. W. Hambley, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- C. F. Chin, D. Y. Wong, R. Jothibasu and W. H. Ang, Curr. Top. Med. Chem., 2011, 11, 2602–2612 CrossRef CAS PubMed.
- E. Wexselblatt and D. Gibson, J. Inorg. Biochem., 2012, 117, 220–229 CrossRef CAS PubMed.
- J. Yang, X. Sun, W. Mao, M. Sui, J. Tang and Y. Shen, Mol. Pharm., 2012, 9, 2793–2800 CrossRef CAS PubMed.
- M. Alessio, I. Zanellato, I. Bonarrigo, E. Gabano, M. Ravera and D. Osella, J. Inorg. Biochem., 2013, 129, 52–57 CrossRef CAS PubMed.
- V. Novohradsky, L. Zerzankova, J. Stepankova, O. Vrana, R. Raveendran, D. Gibson, J. Kasparkova and V. Brabec, J. Inorg. Biochem., 2014, 140, 72–79 CrossRef CAS PubMed.
- V. Novohradsky, L. Zerzankova, J. Stepankova, O. Vrana, R. Raveendran, D. Gibson, J. Kasparkova and V. Brabec, Biochem. Pharmacol., 2015, 95, 133–144 CrossRef CAS PubMed.
- J. J. Wilson and S. J. Lippard, Chem. Rev., 2014, 114, 4470–4495 CrossRef CAS PubMed.
- J. Z. Zhang, P. Bonnitcha, E. Wexselblatt, A. V. Klein, Y. Najajreh, D. Gibson and T. W. Hambley, Chem.–Eur. J., 2013, 19, 1672–1676 CrossRef CAS PubMed.
- Y. Najajreh, E. Khazanov, S. Jawbry, Y. Ardeli-Tzaraf, J. M. Perez, J. Kasparkova, V. Brabec, Y. Barenholz and D. Gibson, J. Med. Chem., 2006, 49, 4665–4673 CrossRef CAS PubMed.
- R. Furumai, Y. Komatsu, N. Nishino, S. Khochbin, M. Yoshida and S. Horinouchi, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 87–92 CrossRef CAS.
- S. J. Riedl and G. S. Salvesen, Nat. Rev. Mol. Cell Biol., 2007, 8, 405–413 CrossRef CAS PubMed.
- T. M. Caserta, A. N. Smith, A. D. Gultice, M. A. Reedy and T. L. Brown, Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties, Apoptosis, 2003, 8, 345–352 CrossRef CAS PubMed.
- T. E. Witzig, M. Timm, M. Stenson, P. A. Svingen and S. H. Kaufmann, Clin. Cancer Res., 2000, 6, 681–692 CAS.
- L. A. Gillies and T. Kuwana, J. Cell. Biochem., 2014, 115, 632–640 CrossRef CAS PubMed.
- M. Kalimutho, A. Minutolo, S. Grelli, G. Federici and S. Bernardini, Acta Pharmacol. Sin., 2011, 32, 1387–1396 CrossRef CAS PubMed.
- V. Pichler, P. Heffeter, S. M. Valiahdi, C. R. Kowol, A. Egger, W. Berger, M. A. Jakupec, M. Galanski and B. K. Keppler, J. Med. Chem., 2012, 55, 11052–11061 CrossRef CAS PubMed.
- J. D. Amaral, J. M. Xavier, C. J. Steer and C. M. Rodrigues, Discov. Med., 2010, 9, 145–152 Search PubMed.
- C. F. Chin, Q. Tian, M. I. Setyawati, W. Fang, E. S. Tan, D. T. Leong and W. H. Ang, J. Med. Chem., 2012, 55, 7571–7582 CrossRef CAS PubMed.
- J. Pracharova, T. Saltarella, T. Radosova Muchova, S. Scintilla, V. Novohradsky, O. Novakova, F. P. Intini, C. Pacifico, G. Natile, P. Ilik, V. Brabec and J. Kasparkova, J. Med. Chem., 2015, 58, 847–859 CrossRef CAS PubMed.
- P. M. O'Connor, J. Jackman, I. Bae, T. G. Myers, S. Fan, M. Mutoh, D. A. Scudiero, A. Monks, E. A. Sausville, J. N. Weinstein, S. Friend, A. J. Fornace Jr and K. W. Kohn, Cancer Res., 1997, 57, 4285–4300 Search PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- S. P. Oldfield, M. D. Hall and J. A. Platts, J. Med. Chem., 2007, 50, 5227–5237 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, I. Zanellato, I. Bonarrigo, M. Alessio, F. Arnesano, A. Galliani, G. Natile and D. Osella, J. Inorg. Biochem., 2015, 150, 1–8 CrossRef CAS PubMed.
- E. Gabano, M. Ravera, D. Colangelo and D. Osella, Curr. Chem. Biol., 2007, 1, 278–289 CAS.
- N. Tarasenko, S. M. Cutts, D. R. Phillips, G. Berkovitch-Luria, E. Bardugo-Nissim, M. Weitman, A. Nudelman and A. Rephaeli, Biochem. Pharmacol., 2014, 88, 158–168 CrossRef CAS PubMed.
- D. M. Fass, R. Shah, B. Ghosh, K. Hennig, S. Norton, W. N. Zhao, S. A. Reis, P. S. Klein, R. Mazitschek, R. L. Maglathlin, T. A. Lewis and S. J. Haggarty, ACS Med. Chem. Lett., 2011, 2, 39–42 CrossRef CAS PubMed.
- T. Soussi and C. Beroud, Nat. Rev. Cancer, 2001, 1, 233–240 CrossRef CAS PubMed.
- M. Wade, Y. C. Li and G. M. Wahl, Nat. Rev. Cancer, 2013, 13, 83–96 CrossRef CAS PubMed.
- T. Pineau, W. R. Hudgins, L. Liu, L. C. Chen, T. Sher, F. J. Gonzalez and D. Samid, Biochem. Pharmacol., 1996, 52, 659–667 CrossRef CAS PubMed.
- C. J. Marshall, Science, 1993, 259, 1865–1866 CAS.
- D. Samid, Z. Ram, W. R. Hudgins, S. Shack, L. Liu, S. Walbridge, E. H. Oldfield and C. E. Myers, Cancer Res., 1994, 54, 891–895 CAS.
- W. R. Hudgins, S. Shack, C. E. Myers and D. Samid, Biochem. Pharmacol., 1995, 50, 1273–1279 CrossRef CAS PubMed.
- K. Burkitt and M. Ljungman, Mol. Cancer, 2008, 7, 24 CrossRef PubMed.
- G. S. Kaiser, S. M. Germann, T. Westergaard and M. Lisby, Mutat. Res., 2011, 713, 64–75 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04205d |
| This journal is © The Royal Society of Chemistry 2016 |