
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First π-linker featuring mercapto and isocyano anchoring groups within the same molecule: synthesis, heterobimetallic complexation and self-assembly on Au(111)†
Jason C.
Applegate
a,
Monisola K.
Okeowo
a,
Nathan R.
Erickson
a,
Brad M.
Neal
a,
Cindy L.
Berrie
*a,
Nikolay N.
Gerasimchuk
*b and
Mikhail V.
Barybin
*a
aDepartment of Chemistry, The University of Kansas, 1251 Wescoe Hall Drive, Lawrence, KS 66045, USA. E-mail: mbarybin@ku.edu; cberrie@ku.edu
bDepartment of Chemistry, Missouri State University, 901 S. National Ave., Springfield, MO 65897, USA. E-mail: NNGerasimchuk@MissouriState.edu
First published on 20th November 2015
Abstract
Mercapto (–SH) and isocyano (–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) terminated conducting π-linkers are often employed in the ever-growing quest for organoelectronic materials. While such systems typically involve symmetric dimercapto or diisocyano anchoring of the organic bridge, this article introduces the chemistry of a linear azulenic π-linker equipped with one mercapto and one isocyano terminus. The 2-isocyano-6-mercaptoazulene platform was efficiently accessed from 2-amino-6-bromo-1,3-diethoxycarbonylazulene in four steps. The 2-NC end of this 2,6-azulenic motif was anchrored to the [Cr(CO)5] fragment prior to formation of its 6-SH terminus. Metalation of the 6-SH end of [(OC)5Cr(η1-2-isocyano-1,3-diethoxycarbonyl-6-mercaptoazulene)] (7) with Ph3PAuCl, under basic conditions, afforded X-ray structurally characterized heterobimetallic Cr0/AuI ensemble [(OC)5Cr(μ-η1:η1-2-isocyano-1,3-diethoxycarbonyl-6-azulenylthiolate)AuPPh3] (8). Analysis of the 13C NMR chemical shifts for the [(NC)Cr(CO)5] core in a series of the related complexes [(OC)5Cr(2-isocyano-6-X-1,3-diethoxy-carbonylazulene)] (X = –NC, Br, H, SH, SCH2CH2CO2CH2CH3, SAuPPh3) unveiled remarkably consistent inverse-linear correlations δ(13COtrans) vs. δ(13CN) and δ(13COcis) vs. δ(13CN) that appear to hold well beyond the above 2-isocyanoazulenic series to include complexes [(OC)5Cr(CNR)] containing strongly electron-withdrawing substituents R, such as CF3, CFClCF2Cl, C2F3, and C6F5. In addition to functioning as a sensitive 13C NMR handle, the essentially C4v-symmetric [(–NC)Cr(CO)5] moiety proved to be an informative, remote, νNC/νCO infrared reporter in probing chemisorption of 7 on the Au(111) surface.
C) terminated conducting π-linkers are often employed in the ever-growing quest for organoelectronic materials. While such systems typically involve symmetric dimercapto or diisocyano anchoring of the organic bridge, this article introduces the chemistry of a linear azulenic π-linker equipped with one mercapto and one isocyano terminus. The 2-isocyano-6-mercaptoazulene platform was efficiently accessed from 2-amino-6-bromo-1,3-diethoxycarbonylazulene in four steps. The 2-NC end of this 2,6-azulenic motif was anchrored to the [Cr(CO)5] fragment prior to formation of its 6-SH terminus. Metalation of the 6-SH end of [(OC)5Cr(η1-2-isocyano-1,3-diethoxycarbonyl-6-mercaptoazulene)] (7) with Ph3PAuCl, under basic conditions, afforded X-ray structurally characterized heterobimetallic Cr0/AuI ensemble [(OC)5Cr(μ-η1:η1-2-isocyano-1,3-diethoxycarbonyl-6-azulenylthiolate)AuPPh3] (8). Analysis of the 13C NMR chemical shifts for the [(NC)Cr(CO)5] core in a series of the related complexes [(OC)5Cr(2-isocyano-6-X-1,3-diethoxy-carbonylazulene)] (X = –NC, Br, H, SH, SCH2CH2CO2CH2CH3, SAuPPh3) unveiled remarkably consistent inverse-linear correlations δ(13COtrans) vs. δ(13CN) and δ(13COcis) vs. δ(13CN) that appear to hold well beyond the above 2-isocyanoazulenic series to include complexes [(OC)5Cr(CNR)] containing strongly electron-withdrawing substituents R, such as CF3, CFClCF2Cl, C2F3, and C6F5. In addition to functioning as a sensitive 13C NMR handle, the essentially C4v-symmetric [(–NC)Cr(CO)5] moiety proved to be an informative, remote, νNC/νCO infrared reporter in probing chemisorption of 7 on the Au(111) surface.
Introduction
Mercapto (–SH) and isocyano (–NC) substituents are among particularly popular anchoring groups in coordination and surface chemistry as they are well-known to provide stable junctions at metal/organic interfaces.1–3 Even though dimercapto- and diisocyano-functionalized molecular linkers have long been attracting interest of theorists4–9 and experimentalists10–15 in the quest for efficient organoelectronic materials,16–20 species containing both –SH and –NC functionalities in the same molecule are not presently known and constitute a formidable synthetic challenge. Indeed, a mercapto group is incompatible with reaction conditions commonly employed to form an isocyano substituent,21 whereas free organic isocyanides are unlikely to tolerate chemical environments typically involved in the syntheses of mercaptans (thiols).22–24 In the context of targeting isocyanothiols for bridging metal-based electron reservoirs, a potentially straightforward strategy to circumvent the above dilemma would be to anchor either the –NC or the –SH terminus of such a hypothetical linker prior to forming and tethering its other end. There is only one related example in the literature, albeit not involving a mercapto group per se but rather its disulfide surrogate.25,26 In their elegant approach to covalently bind nickel clusters to a gold surface via the 4-isocyanophenylthiolate bridge, Kubiak and coworkers attached both –NC ends of otherwise non-isolable 1,2-bis(4-isocyanophenyl)disulfide to trinuclear nickel clusters in the μ3,η1 fashion.25 The resulting salt, [{Ni3(μ3-I)(μ2-dppm)3(μ3,η1-CNC6H4S–)}2]2+(I−)2 (dppm = bis(diphenylphosphino)methane), underwent homolysis of its S–S moiety upon exposure to a gold surface to give rectifying, presumably ionic, monolayer films.26
Earlier this year, Ratner and van Dyck proposed a new paradigm for the design of efficient molecular rectifiers that involved two π-conjugated units asymmetrically anchored to metallic electrodes and separated by a decoupling bridge.27 Their intriguing theoretical study suggested mercapto and cyano (–CN) junctions for accommodating the asymmetric anchoring on the premises that the –SH and –CN termini would facilitate alignments of a linker's HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital), respectively, through Fermi level pinning.27 We note that, from a practical standpoint, mercapto/isocyano asymmetric anchoring would be worth considering as well, given that the isocyano group offers a substantially more stable junction within a wider range of organometallic platforms compared to its isomeric cyano congener.2
Herein, we introduce chemistry of the first, to the best of our knowledge, π-linker equipped with mercapto and isocyano anchoring groups. The linker's core is comprised of the non-alternant aromatic framework of azulene, a substitution-free molecular diode which has, among other unusual physico-chemical characteristics, complementary orbital density distributions within its Frontier molecular orbitals (Fig. 1).28
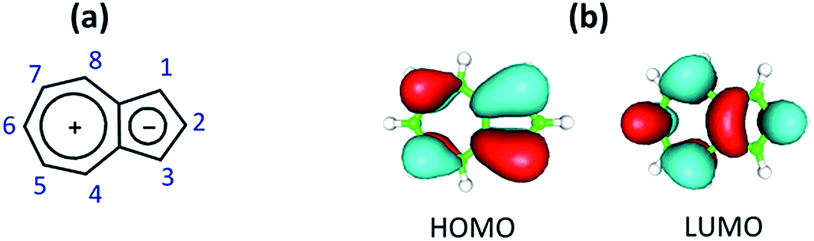 | ||
| Fig. 1 (a) Polar resonance form of azulene and C-atom numbering scheme of the azulenic scaffold; (b) Frontier molecular orbitals of azulene. | ||
Results and discussion
Recent synthetic breakthroughs in functionalization of the azulenic scaffold along its molecular axis have expanded the toolbox for developing low band-gap conducting and optoelectronic materials.29–33 The design of the title π-conjugated linker was influenced by and capitalized on our earlier studies involving 2,6-diisocyano- and 2,6-dimercapto-1,3-diethoxycarbonylazulenes, shown in Fig. 2 (compounds 1 and 2, respectively). As illustrated in Fig. 2, one can envision pursuing two hybrids of 1 and 2: 2-isocyano-6-mercapto-1,3-diethoxycarbonylazulene (3a) and 2-mercapto-6-isocyano-1,3-diethoxycarbonylazulene (3b). Among these two hybrids, 3a is particularly interesting because each substituent in its structure reinforces the molecular dipole of the azulenic framework. In fact, our Density Functional Theory (DFT) calculations suggest that the dipole moment of the “parent” azulene molecule should increase nearly 10-fold upon incorporation of all substituents to form 3a (Fig. 3). | ||
| Fig. 2 2,6-Diisocyano-1,3-diethoxycarbonylazulene (1),32 2,6-dimercapto-1,3-diethoxy-carbonylazulene (2),29 and their hypothetical hybrids 3a and 3b. | ||
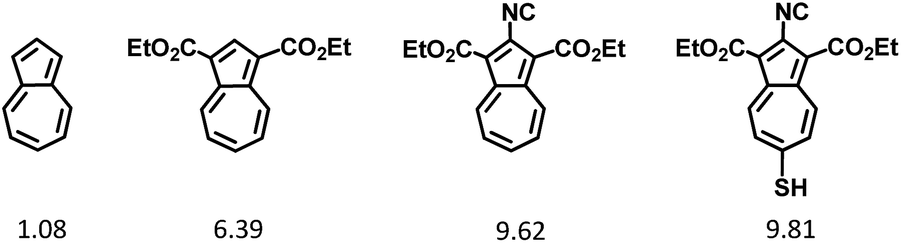 | ||
| Fig. 3 DFT-calculated electric dipole moments (in Debye) of azulene and its derivatives. | ||
Our synthetic approach to constructing and metalating 3a is shown in Scheme 1. Treating pink 2-formamido-6-bromo-1,3-diethoxycarbonylazulene32,34 with ethyl 3-mercapto-propionate in refluxing pyridine afforded persimmon-coloured thioether 4 in a high yield. Dehydrating the 2-formamido group of 4 cleanly provided peach-red 2-isocyanoazulene derivative 5. Unlike 1,2-bis(4-isocyanophenyl)disulphide (vide supra),255 is thermally and air-stable for practical purposes and can be stored under ambient conditions for at least a few weeks without spectroscopically (1H NMR, FTIR) detectable deterioration. Compound 5 reacted with Cr(CO)5(THF) via its 2-NC end to form orange Cr0 adduct 6. No product featuring the thioether S → Cr(CO)5 interaction35 was documented in this reaction. The [(–NC)Cr(CO)5] moiety of 6 tolerated the basic environment and subsequent acidification of the reaction mixture used to convert 6 into auburn organometallic thiol 7, which constitutes 3a with its 2-NC terminus anchored to the 16-e− [Cr(CO)5] fragment. Metalation of the 6-SH end of 7 with PPh3AuCl under basic conditions yielded orange-red crystals of heterobimetallic Cr0/AuI complex 8 after a simple workup.
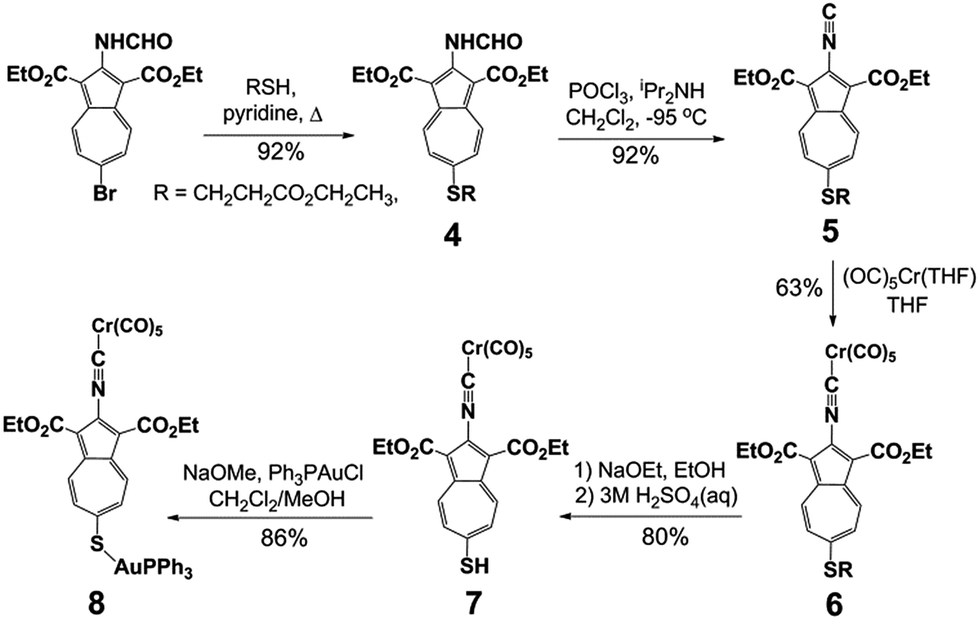 | ||
| Scheme 1 Synthesis and metalation of the 2-isocyano-6-mercaptoazulene motif. | ||
The solid-state structure of 8·¾CH2Cl2 features two very similar but crystallographically independent molecules of 8 in the asymmetric unit that are linked together via a weak Au⋯Au interaction36 of 3.2102(4) Å (Fig. 4, 5, S3 and S4†). The partially positively charged 7-membered ring of the highly polarizable azulenic moiety in each of these molecules of 8 undergoes donor–acceptor face-centred stacking37 with a Ph-ring of the other molecule's PPh3 ligand giving the intercentroid distances38 of 3.65 and 3.76 Å. Heterobimetallic complex 8 may be viewed as a hybrid of our X-ray structurally characterized mononuclear Cr0 and AuI adducts of 1 and 2, respectively, depicted in Fig. 6 (complexes 9 (ref. 32) and 10 (ref. 29)). While the S–Au–P unit in 10 is practically linear (ca. 177.4°),29,39 bending of the S–Au–P angle (ca. 166.5°) in 8 is undoubtedly a consequence of the Au⋯Au bonding reinforced further by the “aromatic donor-acceptor interactions”.37 The above structural perturbations do not significantly affect the Au–S–C angle in 8 compared to that in 10, which are ca. 107.8° 105.0°, respectively. Notably, the solid state structure of 10 exhibits neither aurophilic nor aromatic stacking interactions akin to those observed for 8.29
 | ||
| Fig. 4 ORTEP diagram (50% thermal ellipsoids) of the asymmetric unit of 8·¾CH2Cl2 emphasizing weak aurophilic interaction between two crystallographically independent molecules of 8. The disordered CH2Cl2 molecules or crystallization (Fig. S4†) and H-atoms are omitted for clarity. | ||
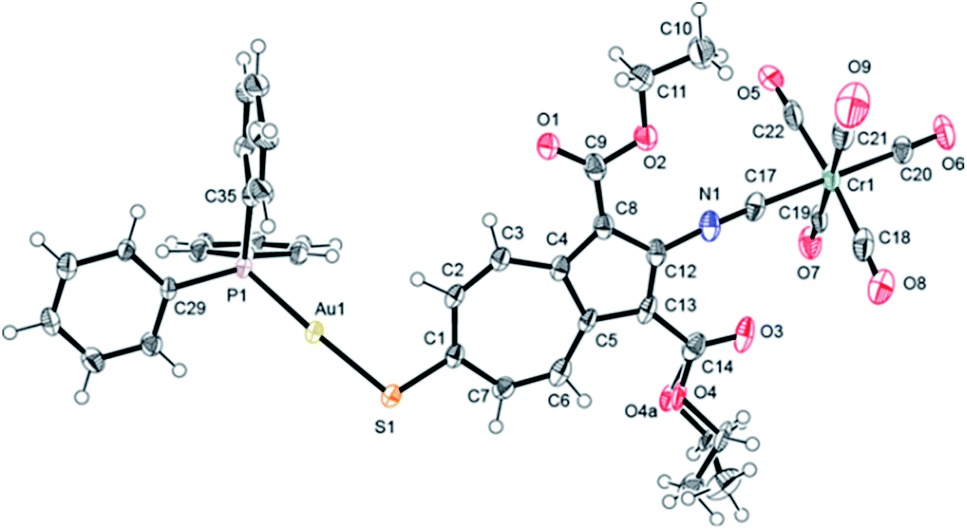 | ||
| Fig. 5 Molecular structure of one of the two crystallographically independent molecules of 8 (50% thermal ellipsoids). The OEt unit attached to C14 exhibits a positional disorder (Fig. S4†). Selected interatomic distances (Å) and angles (°): Au1–P1 2.268(1), Au1–S1 2.318(1), S1–C1 1.744(5), Cr1–C17 1.960(6), Cr1–C18 1.901(8), Cr1–C19 1.900(6), Cr1–C20 1.891(6), Cr1–C21 1.901(7), Cr1–C22 1.891(7), C17–N1 1.155(7), C18–O8 1.155(8), C19–O7 1.141(6), C20–O6 1.142(6), C21–O9 1.146(7), C22–O5 1.147(7), P1–Au1–S1 165.82(5), Au1–S1–C1 108.1(2), C12–N1–C17 172.6(6), Cr1–C17–N1 175.3(5). | ||
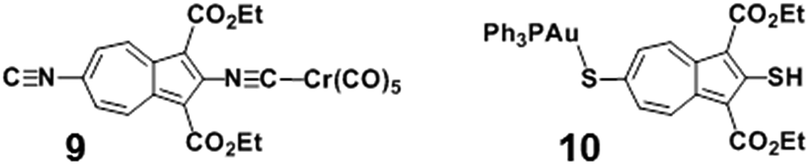 | ||
| Fig. 6 Previously X-ray structurally characterized mononuclear Cr0 and AuI complexes 9 (ref. 32) and 10 (ref. 29). | ||
The metric parameters for the octahedral [(–NC)Cr(CO)5] core in 8 are quite similar to those observed for 9 (ref. 32) and many other complexes (ArylNC)Cr(CO)5.39 Comparison of the Cr–CN and CN bond distances40 for 8 and 9 (Table 1) may hint that the 2-isocyanoazulene ligand in 8 has a somewhat higher σ-donor/π-acceptor ratio than that in 9, thereby reflecting the difference in electron-donating/withdrawing characteristics of –SAuPPh3versus –NC groups at position 6 of the azulenic scaffold. However, this suggestion should be taken cum grano salis as such subtle variations in d(Cr–CN) and d(CN) are statistically ambiguous, especially under the 3σ criterion. More drastic changes in the electronic nature of the isocyanide ligand's substituent do lead to significant alterations in the Cr–CN and CN bond lengths in (RNC)Cr(CO)5 as illustrated in Table 1 for R = tBu (ref. 42) and FCCF2.43
Compounds 4–8 are highly coloured substances. The lowest energy electronic absorption band for 5 occurs at 484 nm (ε = 1.55 × 103 M−1 cm−1) and is 259 cm−1 red-shifted compared to the S0 → S1 transition documented for 4 (Fig. S1†). This red shift arises from the greater electron-withdrawing influence of the 2-isocyano group in 5versus the 2-formamido group in 4 on the energy of the azulenic scaffold's LUMO (Fig. 1b).31,44 The UV-vis spectra of 6 and 7 are nearly identical and feature very intense absorption bands at 454 (ε = 3.1 × 104 M−1 cm−1) and 452 (ε = 2.6 × 104 M−1 cm−1), respectively, that have a substantial contribution from the dπ(Cr) → pπ*(CNAzulenyl) charge transfer (Fig. 7 and S1†). Our time-dependent DFT (TD-DFT) calculations for 7 suggest that the transition at 452 nm (TD-DFT: 416 nm) has 85% HOMO → LUMO character (Fig. 8a). Upon metalation of 7 to form 8, this band not only red-shifts to 469 nm (TD-DFT: 463 nm for 8a, the truncated model of 8 featuring OMe and PMe3 groups instead of OEt and PPh3, respectively, Fig. 8b) but also more than doubles in intensity (ε = 5.4 × 104 M−1 cm−1). This intensity gain is due to the addition of the n(S) → pπ*(CNAzulenyl) character to the HOMO → LUMO transition observed for 8 (cf. the 445 nm band for 10 in Fig. 7).29 As in the case of 9 and 10,29,32 the LUMOs of 7 and 8a constitute the π*-system of the azulenic moiety with contributions from both anchoring groups while their HOMOs involve the entire 2-isocyano-6-azulenylthiolate motif (Fig. 8).
 | ||
| Fig. 7 UV-vis spectra of 7, 8, and 10 (ref. 29) in CH2Cl2 at 25 °C. | ||
 | ||
| Fig. 8 DFT-calculated Frontier MO's of (a) 7 and (b) 8a, a truncated model of 8. | ||
Whilst considering 13C NMR signatures of the [(NC)Cr(CO)5] core in 6, 7, 8, and 9, we noticed that they were predictably sensitive to the nature of the substituent at position 6 of the azulenic scaffold. To further validate this initial observation, we expanded the above family of four related complexes [(OC)5Cr(2-isocyano-6-X-1,3-diethoxycarbonylazulene)] (X = SCH2CH2CO2CH2CH3, SH, SAuPPh3, NC) to include species with X = H (11) and Br (12). The top six rows in Table 2 contain 13C NMR data pertaining to the [(NC)Cr(CO)5] moiety in this series of six 2-isocyanoazulenic adducts. All of these 13C NMR measurements were performed for samples dissolved in CDCl3.
| Compound | δ(13CN) | δ(13COtrans) | δ(13COcis) |
|---|---|---|---|
| a All spectra were recorded in CDCl3. b Ref. 32. c Ref. 47. d Ref. 43. e Ref. 48. | |||
|
178.55 | 217.26 | 214.91 |

|
181.72 | 216.85 | 214.68 |

|
182.27 | 216.77 | 214.65 |

|
183.36 | 216.69 | 214.60 |
|
184.42 | 216.52 | 214.47 |

|
186.6 | 216.3 | 214.4 |
| F5C6–NCCr(CO)5c | 193.8 | 214.6 | 213.3 |
F2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) {F}C–NCCr(CO)5d {F}C–NCCr(CO)5d |
199.3 | 214.2 | 213.0 |
| ClF2C{ClF}C–NCCr(CO)5d | 208.2 | 212.0 | 212.0 |
| F3C–NCCr(CO)5e | 211.1 | 211.5 | 211.7 |
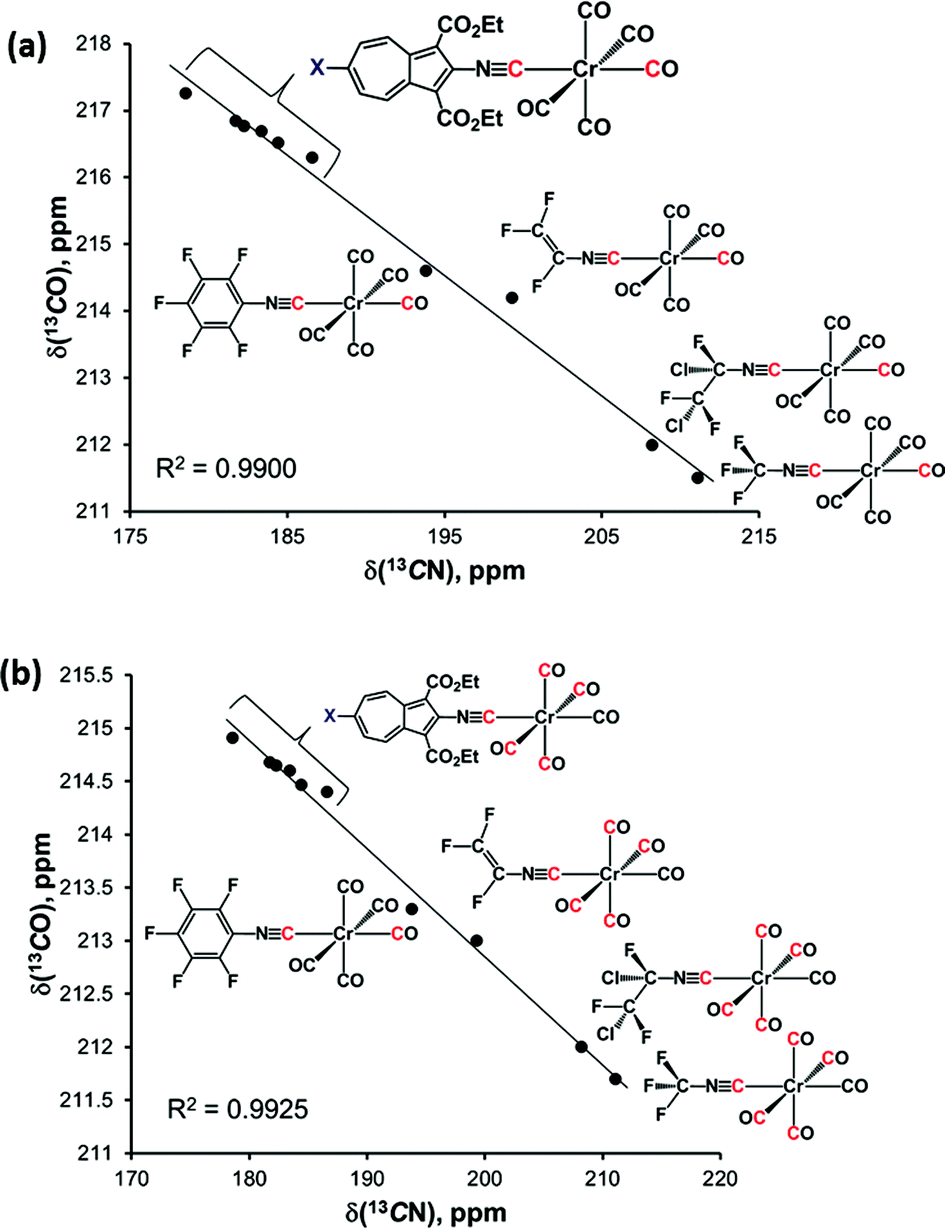
From Table 2 it is evident that as the net electron-releasing ability of X decreases (SAuPPh3 > SCH2CH2CO2CH2CH3 > SH > H > Br > NC), the δ(13CN) value for the isocyano carbon resonance increases in the range spanning ca. 8 ppm, thereby signifying gradual drop in the σ-donor/π-acceptor ratio of the 2-isocyano-6-X-azulene ligand. Concomitantly, both δ(13COtrans) and δ(13COcis) values decrease, albeit in tighter chemical shift ranges (∼1.0 and ∼0.5 ppm, respectively), indicating reduction in the electron richness of the Cr-centre. Even though the 13C chemical shifts of terminal CO and CNR ligands in low-valent complexes are influenced considerably by the paramagnetic shielding term, σpara, which reflects the degree of π-backbonding,40,45,46 it is more appropriate to interpret Δδ(13CN) and Δδ(13CO) as a combined σ-donor/π-acceptor effect.
Closer examination of the 13C NMR data in the top six rows of Table 2 unveiled remarkably consistent inverse-linear relationships δ(13COtrans) vs. δ(13CN) and δ(13COcis) vs. δ(13CN), as illustrated in Fig. 9. This figure also confirms that remote modulation the Cr-centre's electron richness mediated by the 2,6-azulenic framework affects the trans-CO ligand to a greater extent than the cis-CO's of the [(NC)Cr(CO)5] moiety. Would the trends depicted in Fig. 9 hold beyond the 2-isocyanoazulenic series? To address this question, we considered (RNC)Cr(CO)5 species containing strongly electron-withdrawing substituents R, for which 13C NMR data acquired in the same solvent (CDCl3) were available (bottom four rows in Table 2). The expanded δ(13COtrans) vs. δ(13CN) and δ(13COcis) vs. δ(13CN) plots that, in addition to the 2-isocyanoazulenic complexes, include (OC)5Cr(CNR) with R = C6F5,47 C2F3,43 CFClCF2Cl,43 and CF3 (ref. 48) are shown in Fig. 10, which again demonstrates excellent inverse-linear correlations now spanning substantially wider Δδ(13CN) and Δδ(13CO) windows.
 | ||
| Fig. 9 (a) Plot of δ(13COtrans) vs. δ(13CN) chemical shifts (in CDCl3) in the 13C NMR spectra of 6, 7, 8, 9, 11, and 12; (b) plot of δ(13COcis) vs. δ(13CN) chemical shifts (in CDCl3) in the 13C NMR spectra of 6, 7, 8, 9, 11, and 12. | ||
 | ||
| Fig. 10 (a) Plot of δ(13COtrans) vs. δ(13CN) chemical shifts (in CDCl3) in the 13C NMR spectra of all compounds from Table 2; (b) plot of δ(13COcis) vs. δ(13CN) chemical shifts (in CDCl3) in the 13C NMR spectra of all compounds from Table 2. | ||
The above δ(13CO)/δ(13CN) NMR analysis serves as a convenient tool for quantifying even subtle electronic influence of a CNR ligand's substituent R. In this regard, it offers a simple alternative to the well-established method involving correlation the carbonyl 13C chemical shifts with the corresponding CO force constants (kCO) for complexes (RNC)Cr(CO)5.40,49,50 Unfortunately, changes in kCO due to mild electronic perturbations of the R group are often not clearly discernible.40,51 Determining the values of kCO's under the C4v symmetry for complexes (RNC)5Cr(CO)5 using the Cotton–Kraihanzel (C–K) approximation52 is a straightforward but somewhat tedious task that carries fundamental limitations52–54 and relies on the availability of the complete νCO vibrational profile52 (ΓνCO = 2A1 + B1 + E, e.g., Fig. 11 (ref. 55 and 56) and Table S17†). In the IR spectra of LM(CO)5 species, the lower energy νCO(A1) band is often obscured by the intense νCO(E) band,40,51–54 which compromises the accuracy of experimental determination of this νCO(A1) value (vide infra).
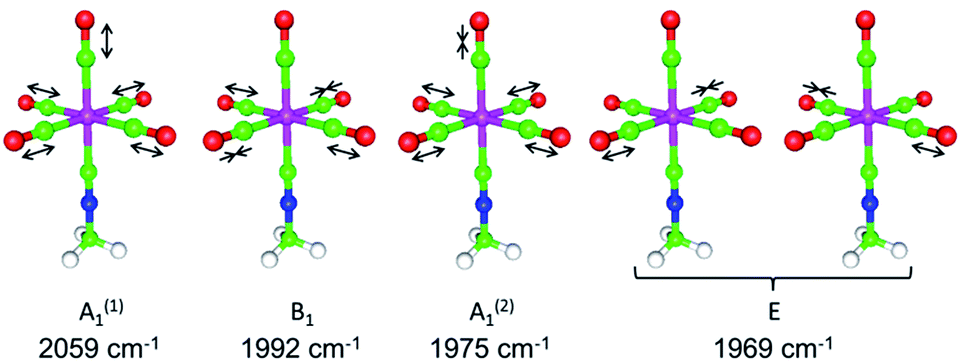 | ||
| Fig. 11 DFT-calculated νCO vibrational profile for (MeNC)Cr(CO)5 in the gas phase.55,56 | ||
Similar to the trend in δ(13CN) for the (RNC)Cr(CO)5 adducts in Table 2, the 13C NMR resonance for the terminal C-atom in the available uncoordinated 2-isocyanoazulenes moves upfield upon increasing electron-donating power of the substituent X at the azulenic 6-position (δ = 179.9,32 178.0, 177.5, 176.3 ppm in CDCl3 for X = C, Br, H, SCH2CH2CO2CH2CH3, respectively). Yet, the νNC stretching frequency for these free 2-isocyano-azulenes (2126 ± 1 cm−1 in CH2Cl2) is insensitive to the nature of the group X. However, upon proceeding from 8 to (6, 7, 11) to 12 to 9, the νNC band undergoes a small red shift (Table 3), thereby suggesting decrease in the σ-donor/π-acid ratio of the isocyanide ligand, especially when 8 is compared to 9 and 12.
Fig. 12a shows the FTIR spectrum of thiol 7 in CH2Cl2. In addition to the characteristic νS–H and νNC bands at 2583 and 2140 cm−1, respectively, it features a typical pattern in the νCO stretching region for a LM(CO)5 species.57 The band at 2049 cm−1 corresponds to the νCO mode A1(1) where all five CO ligands vibrate in-phase (cf.Fig. 11). The very weak band at 2000 cm−1 is due to the νCO vibration of B1-symmetry, which is IR-forbidden under the strict C4v symmetry but gains slight intensity because of minor deviations of the structure from the idealized C4v geometry. The intense νCO band at 1958 cm−1 chiefly represents the doubly degenerate vibration of E-symmetry. This νCO(E) band obscures the remaining IR-active νCO mode A1(2). Interestingly, perturbations of the local C4v symmetry in 7 through crystal packing interactions in the solid state are sufficient to split the E-mode into two separate νCO peaks while unmasking the original A1(2) mode (Fig. 12b).
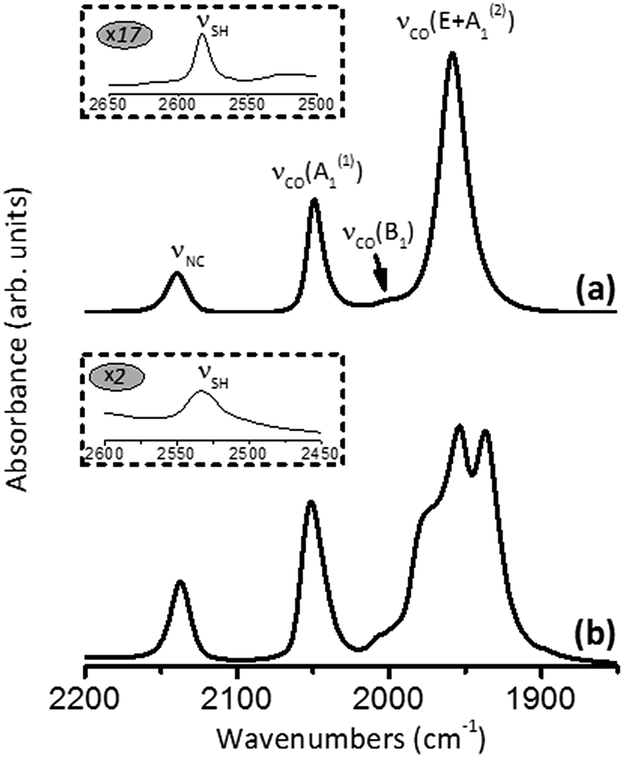 | ||
| Fig. 12 FTIR spectra of 7 in (a) CH2Cl2 and (b) KBr. | ||
Exposing ca. 1 × 1 cm2 gold substrates to a 2 mM solution of 7 in CHCl3 without protection from air and ambient lighting reproducibly afforded self-assembled monolayer (SAM) films of 7 on the Au(111) surface. This chemisorption process is presumably accompanied by formation of the thiolate junction and the release of H2.8,17,58 The reflection absorption infrared (RAIR) spectrum of the SAM of 7 on Au(111) is shown in Fig. 13a. In addition to the νNC absorption at 2135 cm−1, it features two νCO bands. The νCO region in this RAIR spectrum, however, is quite different from that in Fig. 11a in terms of peak intensities and energies. The lowest energy intense νCO band in the solution IR spectrum of 7, which is primarily attributed to the νCO mode of E symmetry, practically vanishes upon the SAM formation, while simultaneously uncovering the hidden A1(2) band of much lower intensity. This observation implies approximately parallel orientation of the cis-CO ligands with respect to the gold surface. Indeed, surface IR selection rules59 dictate that only vibrations contributing to dipole changes perpendicular to the surface are IR-active. Consequently, any vibrations occurring nearly parallel to the surface would have low IR intensity. Given that the C–N–C unit in 7 is expected to be essentially linear, the appearance of the RAIR spectrum in Fig. 13a suggests upright orientation (i.e., straight C–S–Ausurface angle) of the molecules in the SAMs of 7.
The “hollow-linear” coordination of organic thiolates in their SAMs on Au(111), akin to that depicted in Fig. 13b, has been predicted to accommodate the strongest S–Au interaction and induce S → Au(111) charge transfer via S(3p)–Au π-bonding.60,61 In the context of the chemistry presented herein, this means that the gold surface would effectively function as an electron-withdrawing “substituent”, thus, enhancing π-acidity of the 2-isocyanoazulene ligand and, in turn, decreasing electron richness of the [Cr(CO)5] unit. The A1(1) and A1(2)νCO bands at 2058 and 1995 cm−1 in the RAIR spectrum in Fig. 11 both exhibit significant blue shifts compared to the corresponding νCO peaks in the solution FTIR spectrum of 7 (2049 and 1958 cm−1, respectively, Fig. 10a). The magnitudes of these shifts appear to be too high, especially in the case of the A1(2) mode, to be attributed solely to differences in intermolecular interactions within the SAM vs. solution of 7. The larger change in energy of the νCO A1(2) mode compared to that of the A1(1) mode upon chemisorption of 7 stems from the greater contribution of the trans-CO stretch to the former.62
 | ||
| Fig. 13 (a) RAIR spectra of 7 adsorbed on Au(111); (b) the “hollow-linear” coordination mode of 7 adsorbed on Au(111). | ||
The tilt angle of the aromatic moiety in SAMs of benzenoid mercaptoarenes on Au(111) can be highly variable.17 We have recently shown that 2-mercaptoazulene and several of its derivatives form monolayer films on Au(111) with approximately upright assembly of the azulenylthiolate constituents.63 Our optical ellipsometry measurements on multiple SAM samples of 7 provided consistent SAM thickness values that nicely corroborate the monolayer nature of these films and upright orientation of the molecules on the gold surface (Table 4). In terms of their composition, the SAMs of 7 and 9 on Au(111) differ only in the surface anchoring group (thiolate vs. isocyanide) and appear to exhibit essentially identical thicknesses.64 Notably, neither RAIR spectroscopic nor ellipsometric data collected for the SAMs of 7 on Au(111) would be consistent with the “on-top-bent”59 or any other adsorption models of 7 invoking a bent C–S–Ausurface geometry. The ellipsometric measurements on SAM films formed from our recently reported29 6-mercapto-1,3-diethoxycarbonyl-azulene and 6-mercapto-2-chloro-1,3-diethoxycarbonylazulene also corroborate that these 6-mercaptoazulenes self-assemble on Au(111) surfaces in the upright fashion (Table 4).
| Mercaptoazulene derivative | D obs | D calc |
|---|---|---|
| a Average of five measurements at different spots on multiple SAM samples. b Calculated from the X-ray structural data for 8, 6-mercapto-1,3-diethoxycarbonylazulene (ref. 29), and 6-mercapto-2-chloro-1,3-diethoxy-carbonylazulene (ref. 29), as well as by assuming straight C–S–Ausurface angle and the Au(111)–S distance of 2.45 Å (ref. 58). | ||
| 7 | 18.3 ± 2.7 | 17.1 |
| 6-Mercapto-1,3-diethoxycarbonylazulene | 12.8 ± 1.9 | 13.3 |
| 6-Mercapto-2-chloro-1,3-diethoxycarbonylazulene | 14.6 ± 1.9 | 13.3 |
Conclusions
The asymmetric nonbenzenoid aromatic framework of azulene proved to be a convenient platform for accessing the first π-linker terminated with both mercapto and isocyano junction moieties. Anchoring the 2-isocyano end of this linker was an important prerequisite to successfully installing its 6-mercapto terminus. The 13C NMR signatures of the octahedral [(–NC)Cr(CO)5] core in related complexes 6, 7, 8, 9, 11, and 12 provided a sensitive spectroscopic handle for tuning electron richness of the Cr0-centre through mediation by the 2,6-azulenic framework. Moreover, the remarkably consistent inverse-linear trends δ(13COtrans)/δ(13CN) and δ(13COcis)/δ(13CN) for a wide spectrum of complexes (RNC)Cr(CO)5 offer a simple and more accurate alternative to the δ(13CO)/kCO strategy in quantifying electronic influence of the substituent R in isocyanide ligands. This 13C NMR approach utilizes feedback from the entire [(–NC)Cr(CO)5] unit rather than focusing on the [Cr(CO)5] fragment in the δ(13CO)/kCO method. In addition, the C4v-symmetric [(–CN)Cr(CO)5] moiety served as a distinctly informative νNC/νCO infrared reporter for probing self-assembly of the 6-mercaptoazulenic motif on the Au(111) surface. We hope that the chemistry of the 2-isocyano-6-mercaptoazulenic platform introduced herein will facilitate further development and experimental validation of the emerging concept of asymmetric anchoring relevant to the design of organic electronics materials. Efforts to access and isolate completely free (i.e., unmetalated) 3a are currently in progress.
Acknowledgements
This work was funded by the US National Science Foundation through grant # CHE-1214102 to MVB. The NMR instrumentation used in this study was purchased through NIH Shared Instrumentation grants S10RR024664 and S10OD016360, as well as NSF MRI grant CHE 0320648.Notes and references
-
Functional Supramolecular Architectures: For Organic Electronics and Nanotechnology, ed. P. Samori and F. Cacialli, Wiley-VCH, Weinheim, 2011, vol. 1–2 Search PubMed
.
-
M. V. Barybin, J. J. Meyers Jr and B. M. Neal, in Isocyanide Chemistry - Applications in Synthesis and Material Science, ed. Nenajdenko V., Wiley-VCH, Weinheim, 2012, pp. 493–529 Search PubMed
-
M. Lasar and R. J. Angelici, in Modern Surface Organometallic Chemistry, ed. J.-M. Basset, R. Psaro, D. Roberto and R. Ugo, Wiley-VCH, Weinheim, 2009, pp. 513–556 Search PubMed
- M. Bürkle, J. K. Viljas, D. Vonlanthen, A. Mishchenko, G. Schön, M. Mayor, T. Wandlowski and F. Pauly, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 075417 CrossRef
- H. Häkkinen, Nat. Chem., 2012, 4, 443 CrossRef PubMed
- C. Morari, G.-M. Rignanese and S. Melinte, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 115428 CrossRef
- Y. Li, D. Lu, S. A. Swanson, J. C. Scott and G. Galli, J. Phys. Chem. C, 2008, 112, 6413 CAS
- C. Chu, J. A. Ayres, D. M. Stefanescu, B. R. Walker, C. B. Gorman and G. N. Parsons, J. Phys. Chem. C, 2007, 111, 8080 CAS
- R. B. Pontes, A. R. Rocha, S. Sanvito, A. Fazzio and A. J. R. da Silva, ACS Nano, 2011, 5, 795 CrossRef CAS PubMed
- J. Kestell, R. Abuflaha, M. Garvey and W. T. Tysoe, J. Phys. Chem. C, 2015, 119, 23042 CAS
- C. Bruot, J. Hihath and N. Tao, Nat. Nanotechnol., 2012, 7, 35 CrossRef CAS PubMed
- Y. Kim, T. Pietsch, A. Erbe, W. Belzig and E. Scheer, Nano Lett., 2011, 11, 3734 CrossRef CAS PubMed
- B. Kim, S. H. Choi, X. Y. Zhu and C. D. Frisbie, J. Am. Chem. Soc., 2011, 133, 19864 CrossRef CAS PubMed
- B. Kim, J. M. Beebe, Y. Jun, X.-Y. Zhu and C. D. Frisbie, J. Am. Chem. Soc., 2006, 128, 4970 CrossRef CAS PubMed
- K. L. Murphy, W. T. Tysoe and D. W. Bennett, Langmuir, 2004, 20, 1732 CrossRef CAS
- C. A. Mirkin and M. A. Ratner, Annu. Rev. Phys. Chem., 1992, 43, 719 CrossRef CAS
- J. C. Love, L. A. Esttoff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS PubMed
- B. K. Pathem, S. A. Claridge, Y. B. Zheng and P. S. Weis, Annu. Rev. Phys. Chem., 2013, 64, 605 CrossRef CAS PubMed
- D. K. James and J. M. Tour, Chem. Mater., 2004, 16, 4423 CrossRef CAS
- S. H. Choi, B. Kim and C. D. Frisbie, Science, 2008, 320, 1482 CrossRef CAS PubMed
-
I. Ugi, Isonitrile Chemistry, Academic Press, New York, 1971 Search PubMed
-
R. J. Cremlyn, An Introduction to Organosulfur Chemistry, John Wiley & Sons LTD, 1996 Search PubMed
- C. M. Rayner, Contemp. Org. Synth., 1996, 3, 499 RSC
- I. V. Koval', Russ. J. Org. Chem., 2005, 41, 631 CrossRef
- G. M. Ferrence, J. I. Henderson, D. G. Kurth, D. A. Morgenstern, T. Bein and C. P. Kubiak, Langmuir, 1996, 12, 3075 CrossRef CAS
- J. I. Henderson, S. Feng, G. M. Ferrence, T. Bein and C. P. Kubiak, Inorg. Chim. Acta, 1996, 242, 115 CrossRef CAS
- C. van Dyck and M. A. Ratner, Nano Lett., 2015, 15, 1577 CrossRef CAS PubMed
- S. Vosskötter, P. Konieczny, C. M. Marian and R. Weinkauf, Phys. Chem. Chem. Phys., 2015, 17, 23573 RSC
- K. J. Scheetz, A. D. Spaeth, A. S. Vorushilov, D. R. Powell, V. W. Day and M. V. Barybin, Chem. Sci., 2013, 4, 4267 RSC
- M. Koch, O. Blacque and K. Venkatesan, J. Mater. Chem. C, 2013, 1, 7400 RSC
- T. R. Maher, A. D. Spaeth, B. M. Neal, C. L. Berrie, W. H. Thompson, V. W. Day and M. V. Barybin, J. Am. Chem. Soc., 2010, 132, 15924 CrossRef CAS PubMed
- T. C. Holovics, R. E. Robinson, E. C. Weintrob, M. Toriyama, G. H. Lushington and M. V. Barybin, J. Am. Chem. Soc., 2006, 128, 2300 CrossRef CAS PubMed
- M. V. Barybin, M. H. Chisholm, N. S. Dalal, T. H. Holovics, N. J. Patmore, R. E. Robinson and D. J. Zipse, J. Am. Chem. Soc., 2005, 127, 15182 CrossRef CAS PubMed
- This compound is accessible in three high-yielding steps from commercially available 2-chlorotropone and ethyl cyanoacetate: T. Nozoe, S. Seto, S. Matsumura and Y. Murase, Bull. Chem. Soc. Jpn., 1962, 35, 1179 CrossRef CAS
- H. G. Raubenheimer, M. W. Esterhuysen, G. Frenking, A. Y. Timoshkin, C. Esterhuysen and U. E. I. Horvath, Dalton Trans., 2006, 4580 RSC
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC
- Average value for two crystallographically unique molecules.
- A. E. Carpenter, C. C. Mokhtarzadeh, D. S. Ripatti, I. Havrylyuk, R. Kamezawa, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Inorg. Chem., 2015, 54, 2936 CrossRef CAS PubMed
- The differences in Cr–COtrans, COtrans, average Cr–COcis, and average COcis bond lengths in 8 and 9 are insignificant.
- W. Sattler and G. Parkin, Chem. Commun., 2009, 7566 RSC
- D. Lentz and D. Preugschat, Chem. Commun., 1992, 1523 RSC
- An EWG at position 2 of the azulenic framework stabilizes the LUMO without significantly affecting the HOMO: S. V. Shevyakov, H. Li, R. Muthyala, A. E. Asato, J. C. Croney, D. M. Jameson and R. S. H. Liu, J. Phys. Chem. A, 2003, 107, 3295 CrossRef CAS
- M. V. Barybin, W. W. Brennessel, B. E. Kucera, M. E. Minyaev, V. J. Sussman, V. G. Young Jr and J. E. Ellis, J. Am. Chem. Soc., 2007, 129, 1141 CrossRef CAS PubMed
- L. J. Todd and J. R. Wilkinson, J. Organomet. Chem., 1974, 77, 1 CrossRef CAS
- D. Lentz, M. Anibarro, D. Preugschat and G. Bertrand, J. Fluorine Chem., 1998, 89, 73 CrossRef CAS
- D. Lentz, Chem. Ber., 1984, 117, 415 CrossRef CAS
- O. A. Gansow, B. Y. Kimura, G. R. Dobson and R. A. Brown, J. Am. Chem. Soc., 1971, 93, 5922 CrossRef CAS
- D. L. Cronin, J. R. Wilkinson and L. J. Todd, J. Magn. Reson., 1975, 17, 353 CAS
- R. J. Dennenberg and D. J. Darensbourg, Inorg. Chem., 1972, 11, 72 CrossRef CAS
- F. A. Cotton and C. S. Kraihanzel, J. Am. Chem. Soc., 1962, 84, 4432 CrossRef CAS
- R. B. King and M. S. Saran, Inorg. Chem., 1972, 13, 74 CrossRef
- D. Karakaş and C. Kaya, J. Organomet. Chem., 2001, 640, 37 CrossRef
- Calculations performed at the BP86/TZVP level. For a similar analysis at the BP86/DZP level originally reported by King, et al., see: J. Wang, G. Li, Q. Li, Y. Xie and R. B. King, Polyhedron, 2012, 47, 165 CrossRef CAS
-
Cf. the experimentally observed IR-active νCO's of 2071 (A1(1)) and 1964 (A1(2) + E) cm−1 for (MeNC)Cr(CO)5 in n-hexanes: J. A. Connor, E. M. Jones, G. K. McEwen, M. K. Lloyd and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 1972, 1247 Search PubMed
- M. Y. Darensbourg, Prog. Inorg. Chem., 1985, 33, 221 CrossRef CAS
- A. Cossaro, R. Mazzarello, R. Rousseau, L. Casalis, A. Verdini, A. Kohlmeyer, L. Floreano, S. Scandolo, A. Morgante, M. L. Klein and G. Scoles, Science, 2008, 321, 943 CrossRef CAS PubMed
- H. A. Pearce and N. Sheppard, Surf. Sci., 1976, 59, 205 CrossRef CAS
- M. Tachibana, K. Yoshizawa, A. Ogawa, H. Fujimoto and R. Hoffmann, J. Phys. Chem. B, 2002, 106, 12727 CrossRef CAS
- H. Sellers, A. Ultman, Y. Shnidman and J. E. Eilers, NATO ASI Ser., Ser. B, 1992, 283, 441 CAS
- M. S. Davies, R. S. Armstrong and M. J. Aroney, Chimika Chronika, New Series, 1995, 24, 233 CAS
- B. M. Neal, A. S. Vorushilov, A. M. DeLaRosa, R. E. Robinson, C. L. Berrie and M. V. Barybin, Chem. Commun., 2011, 47, 10803 RSC
- D. L. DuBose, R. E. Robinson, T. C. Holovics, D. R. Moody, E. C. Weintrob, C. L. Berrie and M. V. Barybin, Langmuir, 2006, 22, 4599 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic and analytical data, details of the crystallographic and computational studies. CCDC 1410785. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04017e |
| This journal is © The Royal Society of Chemistry 2016 |