
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-luminescent switching of metal-free organic phosphors for luminometric detection of organic solvents†
Min Sang
Kwon
a,
Jake H.
Jordahl
b,
Andrew W.
Phillips
c,
Kyeongwoon
Chung
c,
Sunjong
Lee
f,
Johannes
Gierschner
 g,
Joerg
Lahann
abcd and
Jinsang
Kim
*abcde
g,
Joerg
Lahann
abcd and
Jinsang
Kim
*abcde
aDepartment of Materials Science and Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: jinsang@umich.edu
bDepartment of Chemical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA
cMacromolecular Science and Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA
dDepartment of Biointerface Institute, University of Michigan, Ann Arbor, Michigan 48109, USA
eDepartment of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, USA
fMaterials and Process R&BD Group, Korea Institute of Industrial Technology (KITECH), South Korea
gMadrid Institute for Advanced Studies – IMDEA Nanoscience, Calle Faraday 9, Ciudad Universitaria de Cantoblanco, 28049, Madrid, Spain
First published on 4th January 2016
Abstract
Metal-free organic phosphors can be an attractive smart optical sensing materials since, in such compounds, intersystem crossing (ISC) and the phosphorescence process are placed in subtle competition with fluorescence, internal conversion (IC), and non-radiative decay pathways. Here, we report a unique environment-dependent multi-luminescence switching behavior of metal-free organic phosphorescent materials. Through combined photophysical measurements and computational electronic structure analysis, we systematically investigated how physicochemical properties of organic solvents affect the photophysical pathways of the metal-free organic phosphors. By rationally adapting the finding into phosphor-doped electrospun polymer fibers, we developed a new luminometric sensory platform and achieved selective detection of eight different common organic solvents. The presented finding provides new possibilities for metal-free organic phosphors to be a novel class of smart optical sensory materials.
Introduction
Controlling the luminescent properties of organic materials in response to external stimuli such as light, temperature, pressure, magnetic or electric fields, and chemical analytes serve a crucial role in a wide range of applications including displays, optical devices, lasers, biological imaging probes, and sensors.1 Key material classes that can be used for such smart materials are fluorescent organic molecules and polymers and phosphorescent metal–organic complexes.2 The luminescent switching mechanisms in these materials are changes in electron and/or energy transfer,3 excimer/exciplex via charge transfer (CT) complex formation,4 and HOMO–LUMO gap alternation5 caused by a chemical reaction and the alteration of molecular stacking and/or conformation. Although much effort has been devoted to these material classes, development of a new material class having novel luminescent transduction mechanisms can significantly improve the desired properties and extend the potential application of smart luminescent materials.Metal-free organic phosphorescent materials are photophysically distinct from conventional fluorescent materials and phosphorescent organometallic complexes. Only certain classes of organic emitters having particular moieties (for example, aromatic carbonyls, nitrogen heterocycles, and aromatic sulfides)6 show room temperature phosphorescence (RTP) because spin–orbit coupling (SOC) is typically inefficient without heavy-metal atom. In such compounds, intersystem crossing (ISC) from S1 to Tn and the phosphorescence process is not fast enough (kP < 102 s−1) and, thereby, those processes are placed in subtle competition with fluorescence, internal conversion (IC), and non-radiative decay pathways (Scheme 1b). Therefore, the emission properties of metal-free organic phosphors are sensitively dependent on (1) the nature of excited electronic states (i.e. the nature of S1, Tn, and T1) and (2) radiationless deactivation pathways, which are susceptible to environmental conditions like the presence of oxygen, rigidity of matrices, and temperature.7 Despite the fact that these photophysical properties make metal-free organic phosphors attractive smart optical sensing materials, metal-free organic phosphorescent materials have been rarely explored for sensing except for recent oxygen sensors.8,9e
 | ||
| Scheme 1 Multi-luminescent switching of metal-free organic phosphors (a) chemical structures of metal-free organic phosphors and polymers. (b) A general Jablonski diagram of organic emitters. (c) Photoluminescence images of composite fiber mats before (left) and after exposure of polar (middle) or nonpolar (right) organic solvents. General rules for efficient RTP emitters are given in the gray box. | ||
Here, we report environment-responsive multi-luminescence switching behavior of metal-free organic phosphorescent materials. The phosphors described in Scheme 1a, Br6A and G1, exhibit strong green phosphorescence in specific polymer matrices while they show either blue fluorescence or no luminescence in solutions depending on solvent polarity (Scheme 1c). Through combined experimental analysis and quantum chemical (QC) calculations, we thoroughly investigate the photophysical pathways of metal-free organic phosphors to understand the observed multi-luminescence switching. Moreover, we develop a new sensory platform for luminometric detection of organic solvents based on the multi-luminescence switching behavior of the phosphors. Combinations of three different types of polymer composite fiber mats produce unique luminescence color patterns, which makes it possible to successfully differentiate eight different common organic solvents.
Results and discussions
The phosphors, Br6A and G1, show strong green RTP in specific amorphous polymer matrices, i.e. isotactic poly(methyl methacrylate) (iPMMA) and poly(vinyl alcohol) (PVA), at ambient conditions (Scheme 1c). As we reported previously, this is due to the combined characteristic molecular features of the phosphors and of the host polymer matrices.9 The phosphors promote ISC via SOC by the creation of a nπ* type triplet (Tn) state through the benzaldehyde moiety as well as by the intra- and intermolecular heavy atom effect of bromine through direct participation in the relevant frontier π/π*-type MOs, providing efficient solid-state phosphorescence (see below for details). The rigidity, high triplet energy, and oxygen barrier properties of host polymer matrices can greatly suppress the radiationless deactivation processes of triplet excitons, resulting in efficient RTP in host–guest polymer composites (Table S1†).8–10The phosphors (Br6A and G1) also show interesting solvent-dependent photophysical properties in solution. In polar solvents, e.g. methanol (MeOH) and water, the molecules show strong blue fluorescence. In other solvents like chloroform (CHCl3), fluorescence is effectively switched off (Fig. 1). No phosphorescence is observed under gated conditions at room temperature in their deoxygenated solutions. On the other hand, cooling the phosphor solutions to 77 K or introducing them into solid matrices of iPMMA or PVA, result in noticeable phosphorescence (Scheme 1c and Table S1†).9 Photophysical analysis, combined with time-dependent density functional theory (TD-DFT) calculations, were conducted to elucidate the process.11 Chloroform solution of Br6A showed detectable fluorescence so that we could quantify fluorescence quantum efficiency (QY) and lifetime and we thus successfully compared with the values in methanol solution of Br6A (Table 1), allowing for the quantitative analysis of the observed multi-luminescence switching. However, unfortunately, it is technically very difficult to quantify these values in some non-polar solvents such as ether since the molecules are barely fluorescent as one can see in Fig. 1.
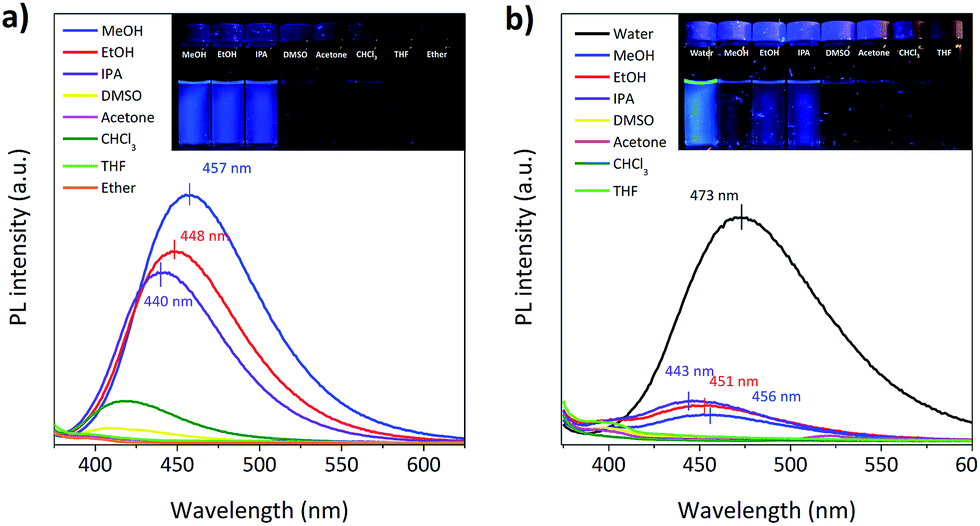 | ||
| Fig. 1 Fluorescence spectra of (a) Br6A and (b) G1 in various solvents are presented. Inset shows the fluorescence images of luminophores in solution under a 365 nm handheld UV lamp. | ||
| System | ε m/M−1 cm−1 | λ abs/nm | λ em/nm | Φ F 17 | τ F 17/ns | k nF/ns−1 | k F/ns−1 | f | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Absa | SBb | QCc | ||||||||
| a From f = 4.319 × 10−9 [M−1 cm2]∫εm(ν)dν. b From the simplified Strickler–Berg relation.12,13 c From QC calculations (TD-DFT). d In vacuum. | ||||||||||
| MeOH | 5812 | 352 | 457 | 0.128 | 2.4 | 0.363 | 0.053 | 0.09 | 0.111 | S1: 0.16, S2: 0 |
| CHCl3 | 5490 | 355 | 419 | 0.005 | 0.5 | 1.990 | 0.010 | 0.09 | 0.017 | S1: 0.00, S2: 0.13d |
The main characteristics of the photophysical processes were analyzed by UV-vis absorption and steady-state and time-resolved photoluminescence (PL) spectroscopy (Table 1 and Fig. 1 and S1 in the ESI†). In MeOH, the radiative rate (kF) from S1 to S0 is 0.053 ns−1, while in CHCl3 it is only 0.010 ns−1. Thus, the oscillator strength of the emitting state (f) must be significantly different in the two solvents. The oscillation strength is accessible via the Strickler–Berg equation,12,13 which relates f with kF, giving fMeOH = 0.11, whereas fCHCl3 = 0.017. On the other hand, f of the main absorption extracted by integration of the absorption spectrum is f = 0.09 (Table 1). This compares well with the photophysical data in MeOH, and thus indicates that in this case emission originates from an allowed transition from S0 to S1 which gives rise to the main absorption. Contrary, in CHCl3, an essentially dark S1 state has to be proposed. Thus, from the experimental data we suggest a solvent-dependent state-switching mechanism.
The alternative deactivation pathways for fluorescence (knF) are internal conversion (kIC) and ISC (kISC to the triplet manifold with consecutive phosphorescence, kP, or ISC to S0, kTS, or quenching processes to S0, kq); knF is considerably slower by a factor of six in MeOH (0.363 ns−1) compared to CHCl3 (1.990 ns−1), see Table 1. Considering that no phosphorescence is observed at room temperature in neither solvents, at least one of the radiationless deactivation pathways (kIC, kISC, kTS, kq), must be very fast relative to kF under these conditions (Scheme 1a).
In order to understand the solvent-dependent state-switching and the impact on radiationless deactivation, we performed TD-DFT calculations.14 In vacuum, the S1 state, calculated at 3.66 eV (339 nm), is of nπ* nature (Fig. 2 and Table S2 in the ESI†) and is thus forbidden (f = 0),15 whereas the bright S2 state of ππ* nature state is calculated at 3.77 eV (329 nm) with f = 0.13. In MeOH, the nπ* character is essentially unchanged (3.78 eV; 328 nm), while the ππ* state is significantly stabilized (3.61 eV; 343 nm) and becomes the emissive state, which is in agreement with experiment. It should be noted, however, that although TD-DFT accurately predicts the qualitative behavior, i.e. the state-switching by a solvent effect, it does not capture all details. In fact, state-switching is predicted already for CHCl3.16
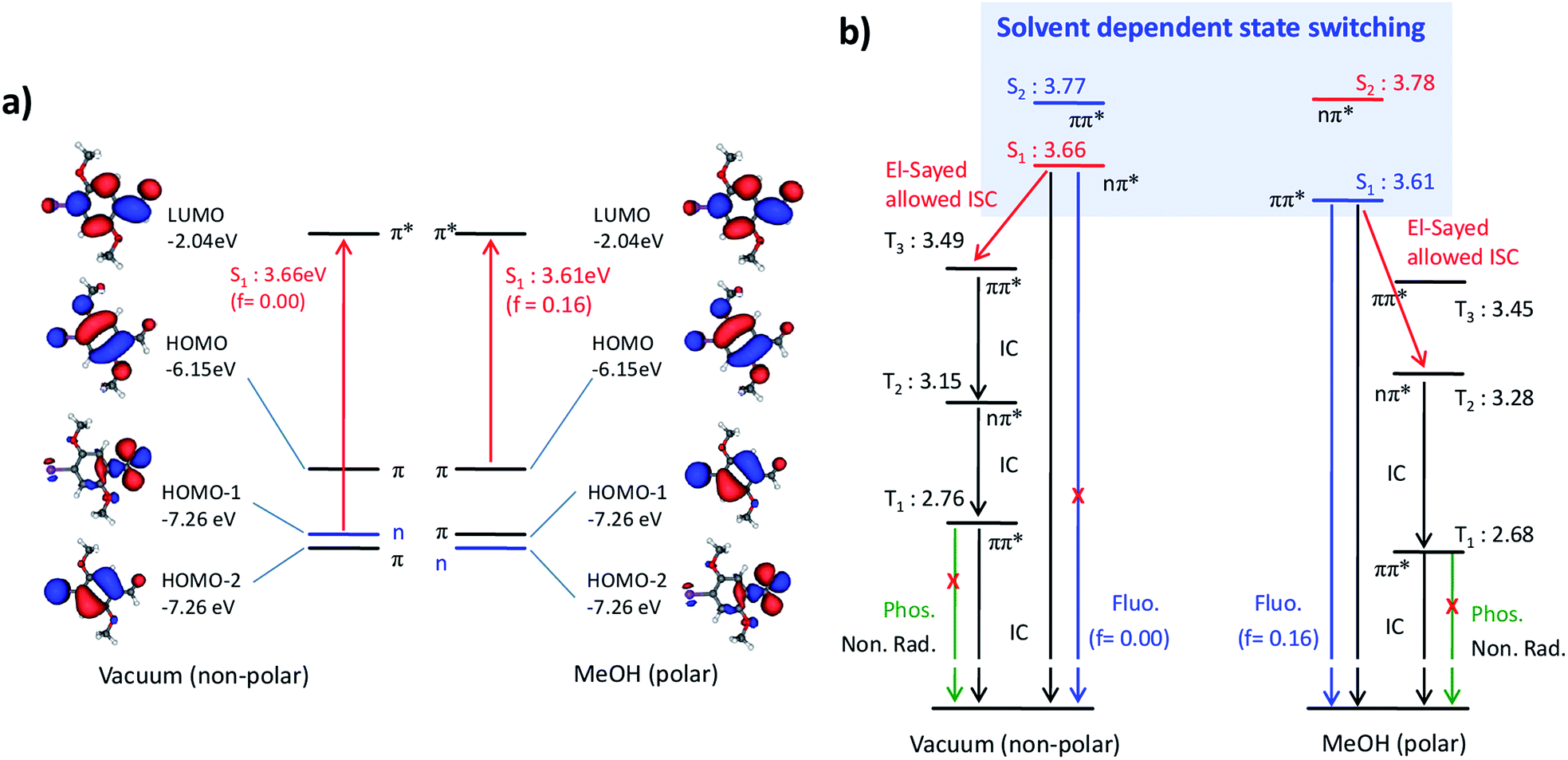 | ||
| Fig. 2 (a) Equilibrium structure and frontier MOs of Br6A. (b) Term diagrams of Br6A in vacuum and methanol as calculated by TD-DFT; nπ*-type states are indicated, while the other states are all of ππ* character. Energies are given in eV. | ||
Radiationless processes cannot be drawn from QC calculations as long as the excited state hypersurfaces are not known in detail, which are not properly accessible with TD-DFT. However, some basic statements can be made. Firstly, the decrease in knF in polar vs. non-polar solvents cannot be due to ISC from T1 to S0 because the nature of T1 is the same in both cases, and the relative change in the state energy is small (Table 1 and Fig. 2b). Although a solvent-dependent change of the IC pathway (S1 → S0) cannot be excluded since the nature of S1 changes, the peculiar solvent-dependent state ordering suggests an alternative explanation. In MeOH, ISC from S1(ππ*) to the energetically close T3(ππ*) state is El-Sayed forbidden and is only promoted by SOC through the heavy-atom effect of bromine, while ISC in vacuum is El-Sayed allowed, i.e. a transition from S1(nπ*) to T3(ππ*). The T2(nπ*) state on the other hand, which could serve for an El-Sayed allowed ISC process in MeOH, is significantly lower in energy and is thus much less efficient for ISC compared to the situation in vacuum. Finally, the phosphorescence process in Br6A is only promoted by the heavy atom effect, and is not El-Sayed allowed, e.g. as in benzophenone.5,6 Thus, alternative deactivation pathways, i.e. ISC and quenching processes from T1 to S0, can effectively compete with phosphorescence at ambient conditions (Scheme 1a and Fig. 2b).
We prepared polymer composites to study the feasibility of an array-based selective detection of common organic solvents. Array-based detection is a potentially powerful method for the detection of various chemical analytes.18,19 Since sensor arrays produce a distinct pattern of responses based on cross-reactive sensor elements, they may provide a characteristic fingerprint for analytes having similar chemical and physical properties. We selected Br6A–iPMMA, G1–PVA80, and G1–PVA100 polymer composites as cross-reactive sensor elements (Fig. 1a and 3). Phosphors act as a signaling unit in the detection platform due to their environment-dependent multi-luminescence. Polymer matrices offer a rigid environment for efficient solid-state phosphorescence of embedded phosphors, serve as an oxygen barrier, and confer additional selectivity through distinct solubility toward different organic solvents. Once the embedded phosphors are exposed to a solvent, they can undergo a distinct luminometric transition: (1) maintain green phosphorescence, (2) change from green phosphorescence to blue fluorescence or (3) become non-luminescent. To prepare polymer composites, electro-spinning was chosen because it allows rapid and cost-effective fabrication of fibrous polymer composites with large surface areas, resulting in fast and reproducible differentiation of solvents.
 | ||
| Fig. 3 (a) SEM images of the Br6A–iPMMA (upper), G1–PVA80 (middle), and G1–PVA100 electrospun polymer fiber mats. (b) Photographs of the Br6A–iPMMA (upper), G1–PVA80 (middle), and G1–PVA100 electrospun polymer fiber mats before (CTRL) and after exposure to various organic solvents under a 365 nm hand-held UV lamp. | ||
Electrospun fiber mats were fabricated from three different viscous polymer solutions containing the phosphors, Br6A or G1. Polymer solutions were pumped through a syringe needle charged to a high voltage. The resultant microfibers were collected on a high speed rotating drum collector covered with aluminum foil. Scanning electron microscope (SEM) images confirm the successful formation of microfibers by the electrospinning method (Fig. 3a). The detailed fabrication conditions of the polymer composite fibers are given in the ESI.†
The resulting composite fiber mats show strong green phosphorescence at ambient conditions, which corresponds well with conventional drop-cast films in terms of emission wavelength and phosphorescence intensity (Fig. 3b). This implies that electrospinning is an efficient method to make phosphorescent polymer fiber composites. Pipette drops (ca. 50 μl) of organic solvents were placed onto the three different fiber mats and the resulting luminescence color change was monitored after 5 min. The polymer mats show distinctively different luminometric responses upon exposure to the organic solvents. For example, the Br6A–iPMMA composite polymer mats displayed the green phosphorescence to blue fluorescence transition when it was exposed to polar solvents like MeOH, ethanol (EtOH), and isopropanol (IPA). Alcoholic solvents can dissolve the polymer matrix and therefore efficiently break the halogen bonded luminophore networks, resulting in phosphorescence quenching and blue fluorescence emission, like in solution. Solvents with medium polarity such as dimethyl sulfoxide (DMSO), acetone, CHCl3, and tetrahydrofuran (THF) can also dissolve the polymer matrix and thus induce the transition from green phosphorescence to non-luminescence. Since water and ether cannot dissolve the polymer matrix, the polymer mats maintain their original green phosphorescence emission properties. G1–PVA80 and G1–PVA100 also show their own characteristic luminescence patterns in response to solvents because the polymers have different solubility parameters in the solvents. THF and IPA dissolved PVA80 well and, thus, phosphorescence was efficiently quenched, while phosphorescence was maintained in PVA100 for both solvents. Based on the combination/patterns of responses, we successfully differentiated eight different solvents by simply monitoring the emission color patterns.
More interestingly, MeOH and EtOH, which show exactly the same emission color patterns, were further distinguished by response time. Much faster emission color change (from green phosphorescence to blue fluorescence) was observed in MeOH compared to EtOH (Movie S1 in the ESI†). We attribute this difference in response time to the different solubility of iPMMA for MeOH and EtOH.
Conclusions
In conclusion, we report a unique environment-dependent multi-luminescence switching of metal-free organic phosphorescent materials by controlling competing photophysical pathways. Through combined photophysical and electronic structure analysis, we reveal that the observed multi-luminescence switching is due to subtle changes in the photophysical pathways of the metal-free organic phosphors, e.g. solvent-dependent state-switching. By rationally adapting the finding, we developed a new optical sensory platform, the phosphor-doped electrospun polymer fibers, for the luminometric detection of common organic solvents, and achieved selective detection of eight different common organic solvents. Moreover, MeOH and EtOH were successfully distinguished by emission color change response time. We believe that our results provide new possibilities for metal-free organic phosphors to be a novel class of smart optical sensory materials.Acknowledgements
This work was supported by the National Science Foundation (DMREF DMR 1435965) and a Korea Institute of Industrial Technology Grant. The work in Madrid was supported by the Spanish Science Ministry (project MultiCrom, grant no. CTQ2014-58801), and by the Campus of International Excellence (CEI) UAM + CSIC.Notes and references
- (a) M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759 CrossRef CAS PubMed; (b) T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685 CrossRef CAS PubMed; (c) A. Kishimura, T. Yamahita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546 CrossRef CAS PubMed; (d) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS PubMed; (e) J. Gierschner and S. Y. Park, J. Mater. Chem. C, 2013, 1, 5818 RSC; (f) K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. Kim, S. Takayama, J. Gierschner, A. J. Matzger and J. Kim, ACS Cent. Sci., 2015, 2, 94 CrossRef; (g) M. S. Kwon, G. Jang, D. Bilby, B. Milián-Medina, J. Gierschner, T. S. Lee and J. Kim, RSC Adv., 2014, 4, 46488 RSC.
- (a) R. Yerushalmi, A. Scherz, M. E. van der Boom and H. Kraatz, J. Mater. Chem., 2005, 15, 4480 RSC; (b) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS; (c) J. Hu and S. Liu, Macromolecules, 2010, 43, 8315 CrossRef CAS.
- (a) R. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327 RSC; (b) M. S. Kwon, J. Gierschner, S. J. Yoon and S. Y. Park, Adv. Mater., 2012, 24, 5487 CrossRef CAS PubMed; (c) M. S. Kwon, J. Gierschner, J. Seo and S. Y. Park, J. Mater. Chem. C, 2014, 2, 2552 RSC.
- (a) S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed; (b) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175 CrossRef CAS; (c) J. Kim, D. T. McQuade, S. K. McHugh and T. M. Swager, Angew. Chem., Int. Ed., 2000, 39, 3868 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319 CrossRef CAS.
- (a) S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199 CrossRef CAS; (b) C. A. Parker and T. A. Joyce, Chem. Commun., 1968, 749 RSC; (c) M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8 CrossRef CAS; (d) P. Avouris, W. M. Gelbart and M. A. El-Sayed, Chem. Rev., 1977, 77, 793 CrossRef CAS.
- Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061 RSC.
- (a) G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747 CrossRef CAS PubMed; (b) P. Lehner, C. Staudinger, S. M. Borisov and I. Klimant, Nat. Commun., 2014, 5, 4460 CAS.
- (a) O. Bolton, K. Lee, H. Kim, K. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CrossRef CAS PubMed; (b) D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325 CrossRef CAS PubMed; (c) M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., Int. Ed., 2014, 53, 11177 CrossRef CAS; (d) O. Bolton, D. Lee, J. Jung and J. Kim, Chem. Mater., 2014, 26, 6644 CrossRef CAS; (e) D. Lee, J. Jung, D. Bilby, M. S. Kwon, J. Yun and J. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 2993 CrossRef CAS; (f) M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef CAS PubMed.
- (a) M. Bixon and J. Jortner, J. Chem. Phys., 1968, 48, 715 CrossRef CAS; (b) W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090 CrossRef CAS; (c) S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3368 CrossRef; (d) S. Reineke and M. A. Baldo, Sci. Rep., 2013, 4, 3797 Search PubMed; (e) J. Xu, A. Takai, Y. Kobayashi and M. Takeuchi, Chem. Commun., 2013, 49, 8447 RSC; (f) D. Chaudhuri, E. Sigmund, A. Meyer, L. Rock, P. Klemm, S. Lautenschlager, A. Schmid, S. R. Yost, T. V. Voorhis, S. Bange, S. Hoger and J. M. Lupton, Angew. Chem., Int. Ed., 2013, 52, 13449 CrossRef CAS PubMed; (g) M. Koch, K. Perumal, O. Blacque, J. A. Garg, R. Saiganesh, S. Kabilan, K. K. Balasubramanian and K. Venkatesan, Angew. Chem., Int. Ed., 2014, 53, 6378 CrossRef CAS.
- Limited here to Br6A, since we had shown that G1 had similar photophysical properties in solution.
- (a) S. J. Strickler and R. A. Berg, Chem. Phys., 1962, 37, 814 CAS; (b) J. P. Vikesland and S. J. Strickler, J. Phys. Chem., 1974, 60, 664 CrossRef.
- According to Strickler–Berg, the rate constant kF can be calculated from the oscillator strength of the transition responsible for the emission process (in the same solvent with refractive index n) and the absorption and emission maxima vabs, vem (in cm−1). In a simplified form this reads:
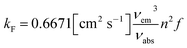 .
. - Since these are very subtle effects, this requires the correct geometry, a TD method to properly describe the energies of allowed and forbidden states, and finally an accurate description of solvent effects. Geometry optimization was carried out with DFT, employing the BH and HLYP functional with a 6-311G* basis set, as implemented in the Gaussian09 program package (M. J. Frisch et al., Gaussian, Inc., Wallingford, CT 2009, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009). The functional was shown to properly reproduce torsional angles, which turned out to be the most delicate issue in Br6A with respect to state ordering. Extensive screening of the potential hypersurface gave a planar stable equilibrium geometry (absence of negative frequencies). The TD part was done by employing the B3LYP functional (6-311G* basis set). Solvent effects were taken into account by the implicit polarizable continuum model (PCM).
- Zero oscillator strength of the nπ* state is obtained in the calculations; the increase in the experimental value (f = 0.053) is ascribed to Herzberg–Teller coupling through out-of-plane torsional modes. Indeed, our QC calculations on distorted Br6A structures give considerable oscillator strength to the nπ* transition.
- The qualitative picture does not further change by geometry relaxation, i.e. optimization of the excited states.
- O. Bolton, The directed heavy atom effect: a design principle for metal-free organic phosphors, Doctoral dissertation, University of Michigan, 2010, pp. 46–47.
- (a) N. A. Rakow and K. S. Suslick, Nature, 2000, 406, 710 CrossRef CAS PubMed; (b) J. Yoon, S. K. Chae and J. M. Kim, J. Am. Chem. Soc., 2007, 129, 3038 CrossRef CAS; (c) S. H. Lim, L. Feng, J. W. Kemling, C. J. Musto and K. S. Suslick, Nat. Chem., 2009, 1, 562 CrossRef CAS.
- J. Lee, H. T. Chang, H. An, S. Ahn, J. Shim and J. M. Kim, Nat. Commun., 2013, 4, 2461 Search PubMed.

Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03986j |
| This journal is © The Royal Society of Chemistry 2016 |