
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRNA G-quadruplex formation in defined sequence in living cells detected by bimolecular fluorescence complementation†
Hong-he
Liu
,
Ke-wei
Zheng
,
Yi-de
He
,
Quan
Chen
,
Yu-hua
Hao
and
Zheng
Tan
*
State Key Laboratory of Membrane Biology, Institute of Zoology, Chinese Academy of Sciences, Beijing 100101, P. R. China
First published on 4th April 2016
Abstract
G-quadruplexes are implicated in many essential cellular processes and sequences with potential to form a G-quadruplex are widely present in DNA and RNA. However, it is difficult to know whether a sequence of interest naturally forms a G-quadruplex in living cells. Here we report the detection of a G-quadruplex in defined RNA sequences in living cells in a natural intracellular environment. A G-quadruplex forming sequence in a RNA transcript is tagged at proximity with an aptamer. The two structures are recognized respectively by two probe proteins each of which is fused with a split half of enhanced green fluorescent protein (eGFP). Simultaneous binding of the two proteins to RNA brings the two halves of eGFP into proximity, permitting them to reconstitute into a functional eGFP that emits fluorescence to signal the formation of a G-quadruplex in RNA. We show that a G-quadruplex can form in RNA and can be detected with sequence and structure specificity under both in vitro and in vivo conditions. The results, therefore, provide direct evidence for the formation of RNA G-quadruplexes in live cells and the method provides a useful tool to validate G-quadruplex formation in a specific sequence under a natural cellular condition.
Introduction
G-quadruplexes are four-stranded structures formed in guanine-rich nucleic acids. Because of their implication in physiological and pathological processes as well as their potential use as therapeutic targets,1–5 G-quadruplexes are gaining increasing attention from researchers in different disciplines in recent years. Sequences with potential to form intramolecular G-quadruplexes are extremely abundant in genomes and genes. For example, ∼370![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 of such sequences are present in the human genome.6 These numbers are expanded because certain non-canonical sequences are also capable of forming G-quadruplexes. For example, >700000 G-quadruplex-forming sequences have been identified in the human genome by high-throughput sequencing.7 Recently, we found that G-quadruplexes with an incomplete G-quartet layer can form in DNA, resulting in structures that respond to guanine derivatives in the environment.8 The amount of motifs with potential to form such structures is in the same order of those for the canonical G-quadruplexes in the human genome. In our earlier studies, we also found that a hybrid type of G-quadruplexes can form during transcription when a non-template DNA strand bears two or more guanine tracts (G-tract) downstream of the transcription start sites (TSS).9–12 This finding raised the number of potential G-quadruplex forming sites to >1.5 million in the human genome.9
000 of such sequences are present in the human genome.6 These numbers are expanded because certain non-canonical sequences are also capable of forming G-quadruplexes. For example, >700000 G-quadruplex-forming sequences have been identified in the human genome by high-throughput sequencing.7 Recently, we found that G-quadruplexes with an incomplete G-quartet layer can form in DNA, resulting in structures that respond to guanine derivatives in the environment.8 The amount of motifs with potential to form such structures is in the same order of those for the canonical G-quadruplexes in the human genome. In our earlier studies, we also found that a hybrid type of G-quadruplexes can form during transcription when a non-template DNA strand bears two or more guanine tracts (G-tract) downstream of the transcription start sites (TSS).9–12 This finding raised the number of potential G-quadruplex forming sites to >1.5 million in the human genome.9
Besides DNA, RNA also forms G-quadruplexes, for example, in the human telomere sequence,13–17 the most studied sequence in the G-quadruplex field. In humans, there are >100000 potential G-quadruplex-forming motifs in RNA.9 RNA G-quadruplexes form during transcription to stimulate mitochondrial transcription termination and primer formation.18 Their biological implications can be found in gene expression and telomere maintenance.19,20
Detection of G-quadruplex formation in cells is necessary for determining the function of G-quadruplexes. The progress made in the past few years on G-quadruplex-recognizing antibodies and ligands has resulted in the establishment of several detecting methods. In an early study, G-quadruplexes were detected in the telomeres of ciliated protozoa Stylonychia. The huge number of telomeres and their particular organization pattern in the macronucleus facilitated a direct visualization of G-quadruplexes by antibodies generated from ribosome display.21 More recently, G-quadruplex structures have been visualized by an antibody,22,23 and captured by chemical ligands24 in human cells, providing evidence for the existence of G-quadruplexes in cells. A common feature of these methods is that they detect G-quadruplexes in cells in a large set of different sequences without sequence identities and most of them involve disruption or fixation of cells.21–25 Therefore, they have an advantage of viewing an overall picture of G-quadruplex formation in cells. A potential G-quadruplex forming sequence may not necessarily form a G-quadruplex in cells. For a specific G-quadruplex forming sequence, it is still difficult to know if a G-quadruplex can form inside cells in the sequence. In this case, the detection of G-quadruplex formation in the sequence is essential to establish a connection between the structure and a cellular process.
In this work, we attempted to detect G-quadruplex structures in defined G-quadruplex forming sequences in RNA transcripts in living bacteria cells under a natural cellular condition by adapting the bimolecular fluorescence complementation (BiFC) technique.26 By transformation with appropriate plasmids, RNAs containing a G-quadruplex-forming sequence and an aptamer tag side-by-side were expressed in E. coli cells. In the meantime, two probe proteins that were fused respectively with two split halves of an enhanced green fluorescent protein (eGFP) were also expressed by transformation with another plasmid. When the two fusion proteins bound simultaneously to the aptamer and G-quadruplex on the RNA, the formation of the G-quadruplex in the RNA was reported by the fluorescence generated by the complementation of the two split halves of the eGFP brought together by the two probe proteins. By this method, we demonstrate that G-quadruplexes can form in RNA in living cells which can be detected with both sequence and structure specificity.
Results
Design of BiFC for detection of RNA G-quadruplex
We constructed several plasmids (ESI, Fig. S1A†) that could express RNAs containing a REX(A) aptamer at the 5′ side of three G-quadruplex forming motifs, TERC(Q), G4T(Q), and PITX(Q), respectively (Fig. 1) (RNA motifs are presented in italics and probe proteins in normal font here and thereafter). We examined G-quadruplex formation in the RNA sequences, TERC(Q), G4T(Q), or PITX(Q), using CD spectroscopy. Their CD spectra all displayed a positive peak at 265 and a negative peak at 240 nm (ESI, Fig. S2†) which are characteristic of parallel G-quadruplexes.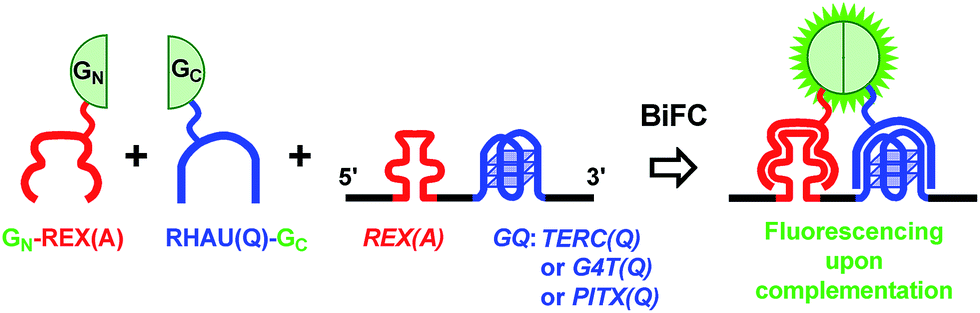 | ||
| Fig. 1 Schematic illustration of detection of a RNA G-quadruplex by the BiFC technique. An aptamer-binding protein REX(A) is fused with the N-terminal half (GN) and a G-quadruplex-binding protein RHAU(Q) is fused with the C-terminal half (GC) of an eGFP, respectively. When the GN-REX(A) and RHAU(Q)-GC bind to their targets REX(A) and G-quadruplex (GQ) on the same RNA, the two split halves of the eGFP are brought into close vicinity with each other such that they can reconstitute into an active eGFP, resulting in fluorescence emission. “A” and “Q” in parentheses indicate that a probe fusion protein is intended to target an aptamer or G-quadruplex, or that a RNA motif forms an aptamer or G-quadruplex, respectively. | ||
Another plasmid (ESI, Fig. S1B†) was constructed to express two probe proteins, GN-REX(A) and RHAU(Q)-GC (ESI, Fig. S3†), that could bind the RNA aptamer and G-quadruplexes, respectively. The REX(A) aptamer in the RNA was recognized by GN-REX(A),27 a REX protein fused with the N-terminal half of eGFP (GN). REX is a human T-cell leukemia virus type 1 (HTLV-1) encoded protein with high binding affinity to the REX(A) aptamer.27 On the other hand, the G-quadruplexes were recognized by RHAU(Q)-GC, a RHAU protein fused with the C-terminal half of eGFP (GC). RHAU is a human DEAH-box RNA helicase that has been reported to bind the intramolecular RNA G-quadruplex in TERC and PITX in vitro and in cells.28–30 When both probe fusion proteins bound to their targets on the same RNA, the two eGFP fragments would be brought into close proximity to form a functional eGFP that could emit fluorescence to signal the presence of a G-quadruplex in the sequence (Fig. 1). If a G-quadruplex was not present on the RNA, its recognizing protein would not bind RNA. In this case, the RNA-mediated complementation would not take place.
In vitro RNA target recognition by probe proteins
We first used an electrophoretic mobility shift assay (EMSA) to test if the probe proteins could recognize their intended targets. RNAs containing TERC(Q), PITX(Q), G4T(Q), and their mutants, TERC(Qm), PITX(Qm), G4T(Qm), were incubated with each individual probe protein and the RNA/protein complexes were resolved on a native gel. The results in Fig. 2A show that both the GN-REX(A) (lanes 2–4, 9–11) and RHAU(Q)-GC (lanes 5–7) protein bound their targets in a concentration-dependent manner. The binding of RHAU(Q)-GC to the TERC(Q) motif was specific to G-quadruplex because when the TERC(Q) motif in the RNA was mutated to TERC(Qm) to disable the formation of a G-quadruplex, no binding was detected (lanes 12–14). When the wild type REX(A)–TERC(Q) RNA was incubated with both GN-REX(A) and RHAU(Q)-GC, an additional band appeared (lane 15), demonstrating that the two proteins could simultaneously bind to the RNA. The simultaneous binding also relied on the presence of a G-quadruplex because the extra band disappeared when the mutated TERC(Qm) motif was used (lane 16). For the REX(A)–G4T(Q) and REX(A)–PITX(Q) RNAs, similar results were also obtained. The binding of RHAU(Q)-GC to the G4T(Q) (Fig. 2B, lanes 5–7) and PITX(Q) (Fig. 2C, lanes 5–7) also depended on the presence of a G-quadruplex structure and mutation in the REX(A)–G4T(Qm) and REX(A)–PITX(Qm) abolished the binding (Fig. 2B and C, lanes 12–14).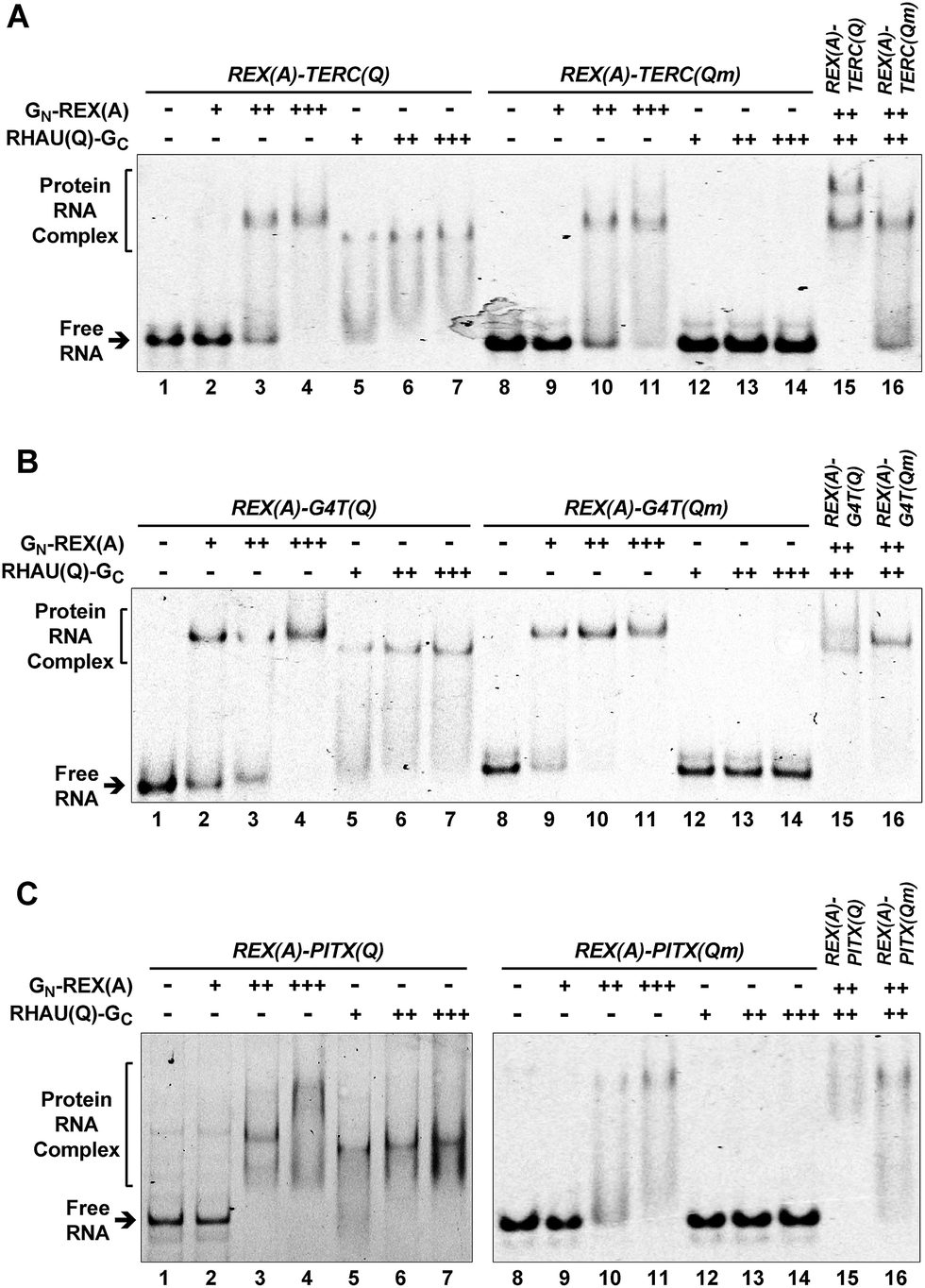 | ||
| Fig. 2 Specific probe protein binding to the intended RNA target revealed by an electrophoretic mobility shift assay (EMSA). (A) REX(A)–TERC(Q), (B) REX(A)–G4T(Q), (C) REX(A)–PITX(Q) RNA and their G-quadruplex mutants, REX(A)–TERC(Qm), REX(A)–G4T(Qm), and REX(A)–PITX(Qm) were incubated with either protein GN-REX(A) or RHAU(Q)-GC or both in K+ solution before being resolved by native gel electrophoresis. The –, +, ++, and +++ indicate protein concentrations of 0, 125, 500, and 1250 nM, respectively, for GN-REX(A) and of 0, 250, 1000, and 2500 nM, respectively, for RHAU(Q)-GC. “Qm” in parenthesis indicates the mutated G-quadruplex sequence in this and the rest of the figures. | ||
The G-quadruplex is stabilized by monovalent cations in the order of K+ > Na+ > Li+.31 Our aforementioned EMSA was carried out in K+ solution to promote formation of a G-quadruplex in the RNAs. To further verify the structural specificity of RHAU(Q)-GC, we performed the same experiments with K+ being replaced by Li+. As is shown in Fig. S5,† this substitution dramatically reduced the binding of RHAU(Q)-GC to the G-quadruplex targets. For example, a majority of the REX(A)–TERC(Q) RNAs remained free in Li+ solution whereas they became fully shifted in K+ solution in the presence of >1 μM of RHAU(Q)-GC (Fig. 2A, lanes 6–7 versus Fig. S5A, lanes 6–7†). This phenomenon also hold true for REX(A)–G4T(Q) RNA (lanes 6–7 in Fig. 2Bversus in ESI, Fig. S5B†). Collectively, the results obtained with mutations in the RNAs (Fig. 2) and wild-type RNA in Li+ solution (Fig. S5†) confirmed that the binding of RHAU(Q)-GC to the RNAs was G-quadruplex specific.
In vitro fluorescence generation upon target binding
The capability of GN-REX(A) and RHAU(Q)-GC to simultaneously bind their targets in a RNA molecule provides a basis for the two proteins to form a BiFC complex. The two proteins did not show fluorescence emission when they were mixed together in the absence of a RNA target (Fig. 3A). When the RNA carrying a REX(A) aptamer and a TERC(Q) G-quadruplex was added, fluorescence was detected that increased over time (Fig. 3B). Mutation in TERC(Qm) prevented the generation of fluorescence because of the lack of a G-quadruplex. Similar results were obtained using the other two aptamer/G-quadruplex pairs (Fig. 3C and D). These results indicated that complementation took place when both GN-REX(A) and RHAU(Q)-GC bound to their targets in the RNA. They also demonstrated that the fluorescence signal could serve as an indication of G-quadruplex formation in RNA.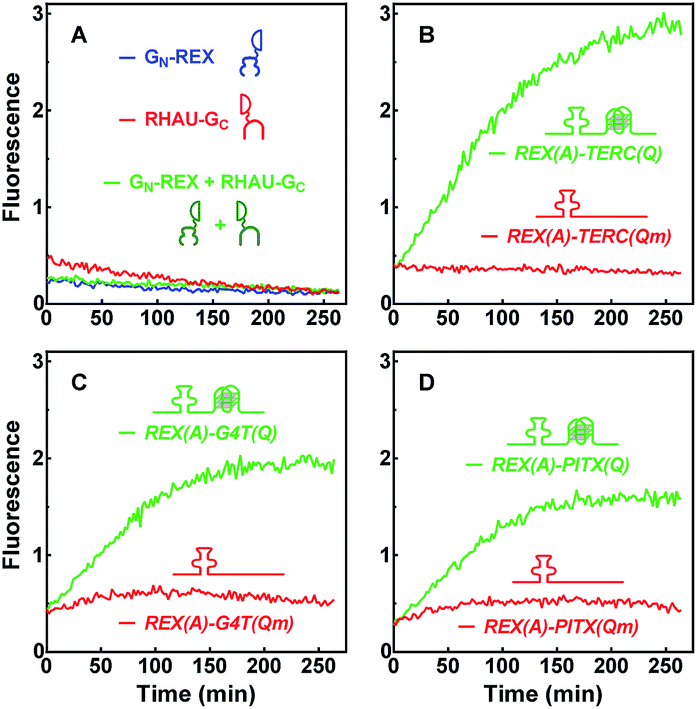 | ||
| Fig. 3 Fluorescence emission as a result of complementation between probe proteins upon binding to their RNA targets. Two probe proteins, GN-REX(A) and RHAU(Q)-GC were incubated individually or together in (A) the absence or (B–D) together in the presence of (B) REX(A)–TERC(Q) and REX(A)–TERC(Qm), or (C) REX(A)–G4T(Q) and REX(A)–G4T(Qm), or (D) REX(A)–PITX(Q) and REX(A)–PITX(Qm) RNAs. Fluorescence was monitored over time after sample mixing. | ||
Detection of G-quadruplexes in living E. coli
Using the fluorescence signal of the GN-REX(A) and RHAU(Q)-GC proteins generated upon binding to their targets, we detected G-quadruplex formation in RNA in living E. coli cells. After expression of the two proteins and RNA targets (Fig. S1A and B†) were induced by addition of IPTG, the E. coli cells were analyzed by flow cytometry (Fig. 4). When the cells expressed REX(A)–TERC(Qm) RNA in which the mutation prevented G-quadruplex formation, they showed a certain level of background fluorescence (Fig. 4A, green curve), probably as a result of self-assembly of the two split haves of eGFP.32 When REX(A)–TERC(Q) RNA was expressed, however, a new population of cells with much higher fluorescence intensities emerged, resulting in an additional peak at the right hand side of the histogram (Fig. 4A, red curve). Again, expression of the other two RNAs carrying REX(A)–G4T(Q) and REX(A)–PITX(Q), respectively, also showed increased fluorescence in the cells compared with those expressing the corresponding RNA mutants (Fig. 4B and C, red versus green curve). The cells expressing wild and mutated RNAs displayed little difference in their forward scatter (FSC) signal (Fig. 4D–F). The RNA expression levels were also similar between the two species (Fig. 4G–I). These two results support that the skewed distribution in eGFP fluorescence in the cells expressing G-quadruplex-bearing RNAs was the result of G-quadruplex formation, rather than possible variation in cell size or RNA level.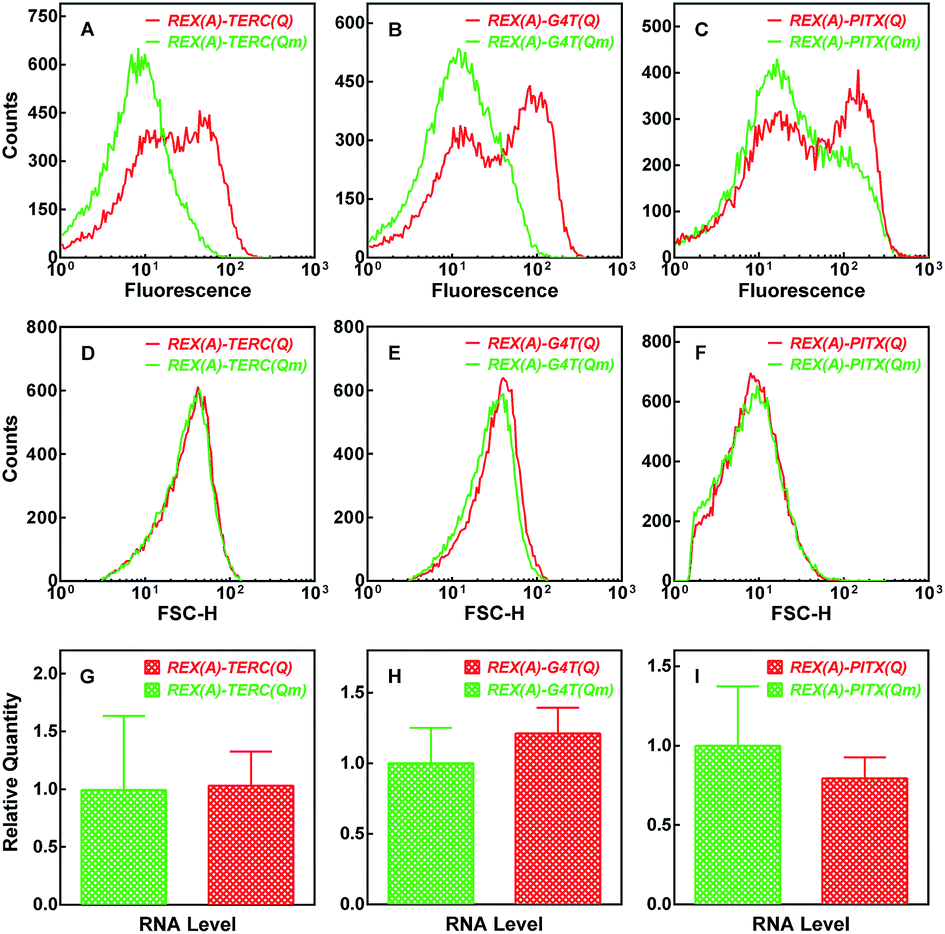 | ||
| Fig. 4 Detection of BiFC fluorescence in E. coli cells by flow cytometry. Distribution of (A–C) eGFP fluorescence and (D–F) forward scattering intensity was collected from cells expressing GN-REX(A) and RHAU(Q)-GC probe proteins plus the indicated RNA. (G–I) Relative RNA expression (mean ± SD of three experiments) assayed by qPCR. | ||
To test if the enhancement in fluorescence involved target RNA, we replaced the RNA expressing plasmid (Fig. S1A) with a new one (Fig. S4A†) by which the expression of REX(A)–TERC(Q) and REX(A)–TERC(Qm) RNA can be independently induced by anhydrotetracycline (ATc). After testing several induction intervals, we found a greater difference in fluorescence between cells expressing REX(A)–TERC(Q) and REX(A)–TERC(Qm) when they were induced at about the same time as the probe proteins. As is shown in Fig. 5A, the enhancement of fluorescence increased over time after the induction of probe proteins in the absence of ATc, which might have resulted from a background expression of target RNAs. When the RNAs were simultaneously induced with the proteins, the fluorescence enhancement was initially reduced (red versus green curve) probably because of a reduction in the protein expression due to competition for resources from the RNA expression. At a later time, however, the fluorescence enhancement became significantly greater than that in the cells without ATc treatment. This increment in fluorescence enhancement upon RNA induction clearly showed that the greater fluorescence in the REX(A)–TERC(Q)-expressing cells was an RNA- and G-quadruplex-dependent event.
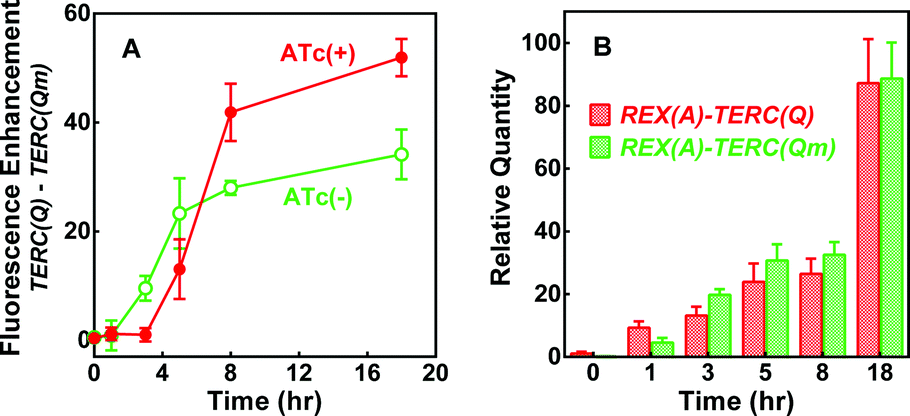 | ||
| Fig. 5 Dependence of BiFC fluorescence enhancement on target RNA expression in E. coli. (A) Time course of the difference in mean fluorescence between cells expressing RNA REX(A)–TERC(Q) and REX(A)–TERC(Qm) in the presence and absence of RNA induction by ATc. (B) RNA level in cells before and after induction with ATc. Data are given in mean ± SD of three experiments. IPTG was added at 0 h with or without ATc. Representative distribution of eGFP fluorescence of corresponding cells is given in Fig. S4B.† | ||
To verify the structural specificity of BiFC fluorescence towards the G-quadruplex, we replaced the G-quadruplex forming motif with a set of four sequences, in which the number of G3 tracts increased from 1 to 4 (ESI, Fig. S6,†1G3-4G3(Q)). The fluorescence of the corresponding cell populations showed little change with a sequential addition of a G3 tract till their number reached 3 when these changes in the sequence did not bring a possibility of G-quadruplex formation (Fig. 6A, colored curves). However, a further addition of a single G3 tract resulted in an increase in fluorescence once the sequence became capable of forming a G-quadruplex (Fig. 6A, black curve). Since the forward scatter (Fig. 6B) and RNA expression (Fig. 6C) was similar among the plasmids, this result further demonstrated that the recognition of RHAU(Q)-GC was specific to the G-quadruplex.
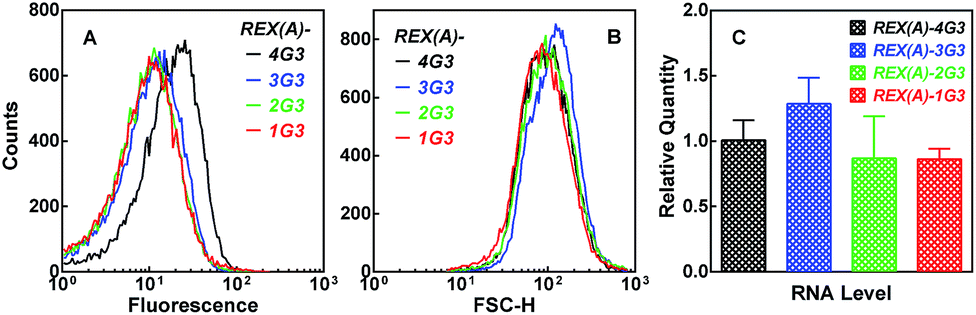 | ||
| Fig. 6 Dependence of BiFC fluorescence on the number of G3 tracts in the RNA target expressed in E. coli. Distribution of (A) eGFP fluorescence or (B) forward scattering intensity or (C) relative RNA expression (mean ± SD of three experiments) was assayed and processed as in Fig. 4. | ||
The complementation efficiency of the two eGFP halves is dependent on the distance between the two partners. To further confirm the target specificity of the fluorescence signal, we added an additional 20 nt sequence between the aptamer REX(A) and G-quadruplex TERC(Q) and G4T(Q), respectively, in the RNAs (ESI, Fig. S7†). This modification would either push the aptamer and G-quadruplexes further apart or make the linkage between them more rigid by folding into a double-stranded stem, which is expected to reduce the complementation and, as a result, BiFC fluorescence. Indeed, for the two RNAs, the fluorescence was significantly reduced (Fig. 7, blue curve) compared to that of cells expressing the corresponding RNAs without the spacer (Fig. 7, red curve).
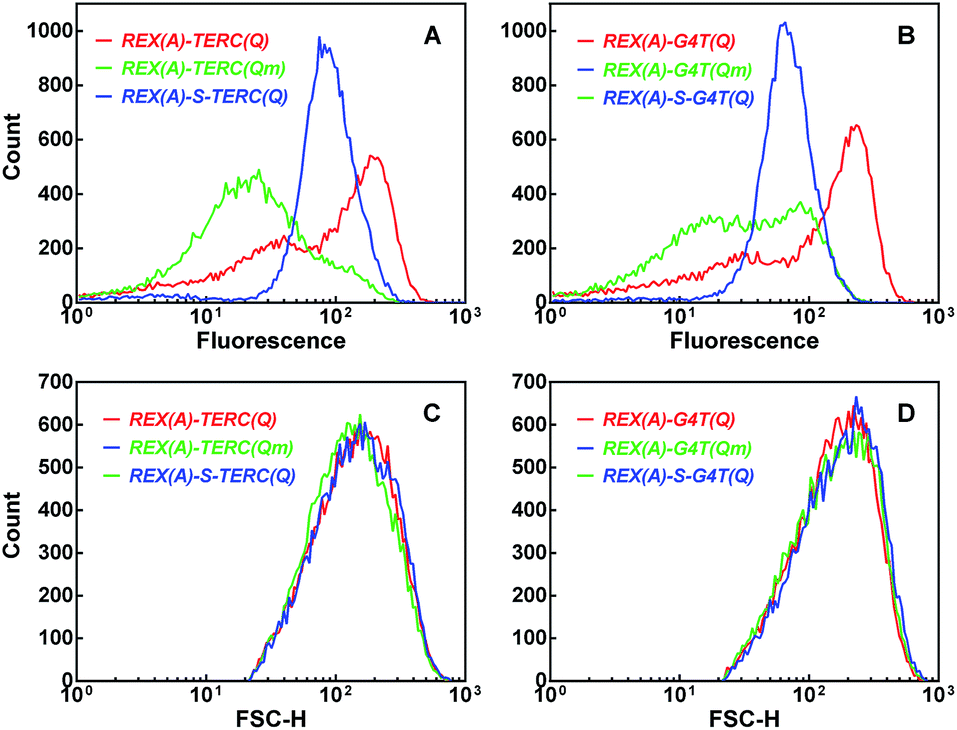 | ||
| Fig. 7 Dependence of BiFC fluorescence on the distance between the aptamer and G-quadruplex targets. Distribution of (A and B) eGFP fluorescence or (C and D) forward scattering intensity was collected by flow cytometry and processed as in Fig. 4. Cells expressed GN-REX(A) and RHAU(Q)-GC plus the indicated RNA without (red, and green curves) or with a 20 bp stem spacer (blue curve) between the aptamer and G-quadruplex motif. | ||
Immuno-detection of probe protein-G-quadruplex interaction in living E. coli
We verified the interaction between RHAU(Q)-GC and RNA G-quadruplexes in cells by a pull-down experiment. The target RNA was pulled down by an immobilized DNA oligonucleotide that was complementary to the 22 nt region at the 5′ end of the RNAs. The protein associated with the RNA was then detected by a monoclonal antibody against eGFP. While information regarding the amino acid sequence recognized by the antibody is not available from the manufacturer, the antibody appeared to recognize both fragments of eGFP (Fig. 8). When IPTG was not added to induce the expression of the probe proteins and target RNA or when the RNA-expressing plasmid was omitted, only a weak background was detected (Fig. 8A, lanes 7–10, top gel). When REX(A)–TERC(Q) was expressed, the two protein bands showed dramatically enhanced intensity at similar magnitudes (lane 11). Mutation at the G-quadruplex moiety significantly reduced the intensity of the RHAU(Q)-GC band as expected (lane 12). In the same lane, GN-REX(A) displayed a smaller decrease in intensity, which might be resulting from the loss of RHAU(Q)-GC bound to RNA. A fraction of the GN-REX(A) proteins on the RNA might be simply associated with RHAU(Q)-GC by self-assembly without itself binding the REX(A) aptamer such that they were lost when the binding of RHAU(Q)-GC on RNA was reduced. Therefore the reduction in the signal for both proteins signified that the binding of RHAU(Q)-GC to RNA was G-quadruplex specific. As expected, hydrolysis of RNA with RNase A (lanes 13–14) significantly reduced the intensity of the two probe proteins, indicating that the proteins were pulled down by the target RNA via a protein–RNA interaction. Similar results were also obtained with REX(A)–G4T(Q) RNA and its mutant (Fig. 8B, lanes 7–14, top gel). The results for both RNAs therefore confirmed the interaction between the probe proteins and their target RNAs in the cells.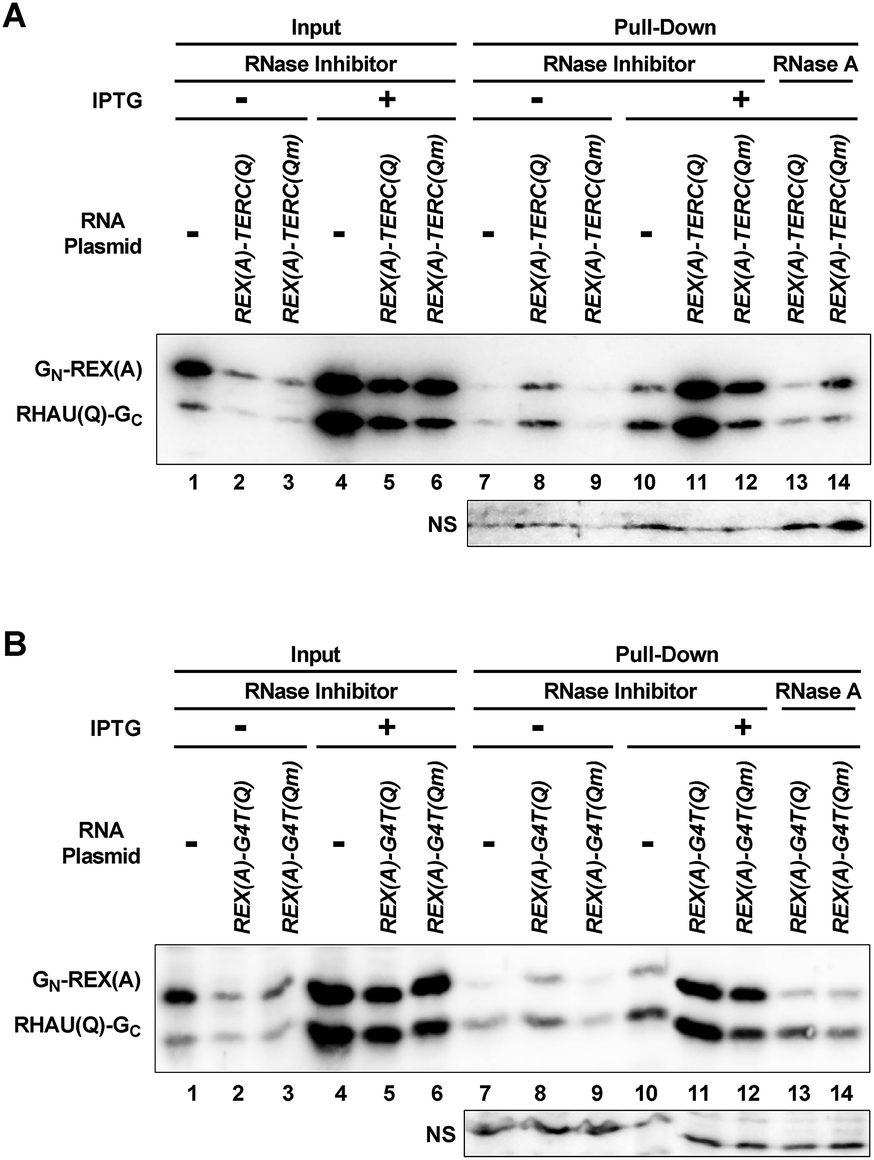 | ||
| Fig. 8 Pull-down of G-quadruplex-recognizing proteins by RNA from E. coli. Cells were lysed in the presence of RNase inhibitor or RNase A. Probe proteins associated with the indicated target RNAs were pulled down with an immobilized DNA oligomer complementary to the 5′ end of the RNA and detected by an antibody against eGFP. “NS” indicates a non-specifically stained band that can serve as a loading control. | ||
Discussion
In summary, we successfully detected G-quadruplex formation in living bacterial cells with sequence and structure specificity using plasmids expressing RNA and target-recognizing fusion probe proteins. Our results provide evidence that a G-quadruplex can form in living cells under a truly natural condition. By the in vitro EMSA assays, we proved that the probe proteins can specifically recognize and simultaneously bind to their intended targets on RNA (Fig. 2). As a result, bimolecular complementation took place as expected between the two proteins, giving rise to fluorescence to signal the presence of a G-quadruplex structure (Fig. 3). The binding was suppressed in a solution of Li+ that does not stabilize a G-quadruplex (Fig. S5†). In addition, mutation in the G-quadruplex forming motifs to prevent G-quadruplex formation abolished the binding of the G-quadruplex probe protein to RNA (Fig. 2 and 3). Our in vivo assays further showed that these properties led to a successful application of the BiFC system in living cells (Fig. 4–6, and S4†). The correlation between the increase in fluorescence and capability of the sequences to form a G-quadruplex confirmed the structure specificity (Fig. 4, 5 and S4†). The dependence of the fluorescence emission of eGFP on the distance between the two probe proteins supported the complementation interaction between the two split halves of eGFP (Fig. 7).The BiFC events mediated by RHAU indicate not only a formation of a G-quadruplex, but also an interaction between the target G-quadruplex and RHAU in cells. RHAU is able to bind both intra- and intermolecular G-quadruplexes.30 There were two or three G-tracts in our RNA mutants REX(A)–TERC(Qm), REX(A)–G4T(Qm), REX(A)–PITX(Qm), REX(A)–3G3, and REX(A)–2G3. They are capable of forming intermolecular G-quadruplexes and, therefore, might also lead to BiFC. Because these RNAs were used as controls, the fluorescence enhancement associated with the wild RNAs was an indication of intramolecular G-quadruplex formation. A recent study shows that RHAU binds parallel quadruplexes, but not non-parallel ones,33 suggesting that RHAU only binds the folded form. This property eases the concern on a possibility that the protein may fold a sequence into a G-quadruplex.
The bimolecular complementation strategy in BiFC and the choice of plasmid-generated RNA transcript offer several advantages. Firstly, the detection can be conducted in living cells in a natural cellular environment. Secondly, negative controls can be arranged in BiFC to strictly evaluate the structural specificity. When G-quadruplexes in endogenous nucleic acid molecules are to be examined, it is technically difficult to have a negative control. Thirdly, the construction of a plasmid made it possible to study any G-quadruplex-forming RNA sequence by simply inserting a corresponding DNA between the promoter and terminator (Fig. S1†). Lastly, BiFC is normally irreversible.32 This is a desired property that should enhance the detection sensitivity because BiFC is expected to accumulate even when an interacting G-quadruplex dissociates or unfolds.
Since the introduction of an aptamer at one side of the G-quadruplex-forming motif, the native RNA sequence is altered. This may influence but is unlikely to prevent the formation of G-quadruplexes because G-quadruplexes are stable structures. On the other hand, the formation of an aptamer reduces its interaction with the rest of the RNA and such interaction may be further minimized when the aptamer is bound by a probe protein. RNA–protein interaction constantly takes place in cells. The disturbance brought by the aptamer and its interaction with a probe protein may not be different from those by other RNA–protein interactions naturally occurring in cells. Therefore, once G-quadruplexes are detected, it is most likely that G-quadruplexes would also form in the endogenous native sequence. For future development of the method, the aptamer may be removed if DNA G-quadruplexes are detected using ZFN, TALEN, and CRISPR/Cas techniques in which recognition of virtually any sequence is possible.34
Our data demonstrated that certain factors affect the detection of G-quadruplexes. The detection relies on a proper G-quadruplex-binding protein. Similar to antibodies and chemical ligands, a protein may recognize better some types of G-quadruplexes than others. This might be why the BiFC fluorescence varied when different G-quadruplex targets were used (Fig. 3). For the same reason, when a G-quadruplex-forming sequence has more than four G-tracts and therefore has a chance to adopt different forms of G-quadruplexes, some may be better captured than others depending on the selectivity of the probe protein. Therefore, testing different binding proteins may be required when a positive signal is not seen with a G-quadruplex forming sequence. When a protein is able to bind a G-quadruplex target, the distance between the G-quadruplex and aptamer is critical for a given fusion probe protein pair (Fig. 7). The optimal distance may also vary with different probe protein pairs depending on their size and spatial orientation when bound to their targets. Our method also inherits an issue from BiFC in that self-assembly can occur between the two halves of eGFP,32 resulting in background fluorescence especially when the two proteins are expressed at a high level. Given this, the timing of RNA and probe protein expression becomes important. If the concentration of RNA outweighs that of the probe proteins, the two halves of the protein tend to bind to different RNA molecules, reducing their chance to meet with each other. On the other hand, if the RNA expression lags behind that of the proteins, the level of proteins may become too high leading to high fluorescence background as a result of self-assembly. It seems that inducing both RNA and protein expression at the same time is a practical option to produce optimal results and simplify the expression system. While a thorough exploration of these technical issues is not within the scope of this work, our study demonstrates that the BiFC-based technique should benefit studies towards a specific G-quadruplex in a sequence of interest in a natural cellular environment, thus, providing a complementation to the existing methods.
Experimental section
RNA- and protein-expressing plasmids
An IPTG-inducible RNA-expressing plasmid was constructed based on the pET28b (Novagen) backbone (Fig. S1A†) to which an anti-REX RNA aptamer, REX(A), was inserted between Xba I and Xho I sites and a G-quadruplex forming sequence, GQ, or a mutant was inserted between Xho I and Bpu1102 I sites downstream of the T7 promoter.An ATc-inducible RNA-expressing plasmid was constructed based on the pETDuet-1 (Novagen) backbone (Fig. S4A†) to which 2 × tetO, a REX(A)–TERC(Q) or REX(A)–TERC(Qm) sequence, and a T7 terminator was inserted between the Xba I and Hind III site downstream of the first T7 promoter. A TetR protein coding sequence was amplified from the pLenti6/TR vector (Invitrogen) and inserted between the Bgl II and Kpn I site downstream of the second T7 promoter of the modified plasmid. Then the LacI promoter and part of the LacI coding sequence were cut out at the Sph I and Mlu I site. Finally, the second T7 promoter was replaced by a lacI promoter by insertion at the Not I and Bgl II site.
The protein-expressing plasmid for BiFC was constructed based on pACYCDuet1 (Novagen) to express two fusion proteins simultaneously (Fig. S1B†).35 The aptamer-binding protein, GN-REX(A), is a REX protein fused to the C-terminus of a N-terminal fragment (aa 1–158) of eGFP. The G-quadruplex-binding protein, RHAU(Q)-GC, consisted of a RHAU protein fragment (aa 53–105) fused to the N-terminus of a C-terminal fragment (aa 159–238) of eGFP. The sequence of the two components in each of the fusion proteins was separated by the same flexible 10-mer oligopeptide. For protein purification, the coding sequences of GN-REX(A) and RHAU(Q)-GC were amplified from the pACYCDuet1 plasmid by PCR and inserted into the pET28b plasmid between the Nde I and Xho I sites downstream of a T7 promoter.
RNA preparation
To prepare RNA for EMSA and in vitro BiFC assays, the RNA-expressing plasmid was linearized by cutting at the Bpu1102 I site of pET28b. The resulting DNA (50 ng μl−1) was transcribed at 37 °C for 3 h in 80 mM HEPES-LiOH buffer (pH 7.5), 10 mM MgCl2, 50 mM LiCl, 10 mM DTT, 1 mM spermidine, 4 U μl−1 T7 RNA polymerase (Fermentas, USA), 2 mM rNTPs, 1 U μl−1 RiboLock RNase Inhibitor (Fermentas, USA), and 0.025 U μl−1 pyrophosphatase, inorganic (Fermentas, USA). The samples were then treated at 37 °C for 30 min with DNase I at a final concentration of 0.04 U μl−1 to remove DNA, followed by an addition of 20 mM EDTA final concentration to stop the reaction. RNA transcript was purified using the RNAclean Kit (TIANGEN, China) and stored at −70 °C in RNase-free water containing 1 U μl−1 of RiboLock RNase Inhibitor (Fermentas, USA). The preparation of RNA of the quadruplex core or the corresponding mutant sequence for circular dichroism (CD) spectroscopy was carried out in the same way except that synthesized DNA was used as a template at a final concentration of 0.1 μM.Protein expression and purification
The protein-expressing plasmid was transformed into the BL21 (DE3) E. coli strain (TransGen Biotech) and the expressed proteins in an inclusion body were purified as described36 with modification. Briefly, transformed E. coli cells were grown at 37 °C to OD600 ≈ 0.5, then protein expression was induced by adding 0.5 mM IPTG and lowering the temperature to 25 °C. After an overnight growth, cells were collected by centrifugation and resuspended in 50 mM Tris–HCl (pH 8.5), 25% sucrose, 1 mM EDTA, and 10 mM DTT. They were then disrupted by freezing at −70 °C followed by sonication with a Vibra-Cell (Sonics, USA) in an ice-water bath for 30 min in cycles of 3 s on and 7 s off. After centrifugation, the pellet was resuspended in washing buffer (50 mM Tris–HCl, pH 8.5, 0.5% TritonX-100, 100 mM NaCl, 1 mM DTT, and 1 mM NaEDTA) by a brief sonication. The sample was centrifuged and the pellet was solubilized in a buffer of 25 mM MES (pH 8.5), 8 M urea, 10 mM NaEDTA, 0.1 mM DTT. The solution was added slowly drop by drop to a refolding buffer (50 mM Tris–HCl, pH 8.5, 500 mM NaCl, 1 mM DTT) to a 1:150 fold dilution and kept at 4 °C overnight. The refolded His-tagged protein was purified with the His GraviTrap column (GE Healthcare) and concentrated by ultrafiltration with the Ultra-4 Centrifugal Filter Units (Amicon, Millipore). Purified protein (Fig. S3†) was stored in 20 mM Tris–HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, and 50% (v/v) glycerol at −70 °C until use.
Circular dichroism (CD) spectroscopy
0.5 μM RNA obtained by in vitro transcription was denatured at 95 °C for 5 min in 10 mM Tris–HCl (pH 7.4), 50 mM KCl, 1 mM EDTA, and quickly cooled on ice. CD spectra were measured at 25 °C on a Chirascan-plus CD spectrometer (Applied Photophysics, United Kingdom) with a 10 mm pathlength quartz cuvette.Electrophoretic mobility shift assay (EMSA)
An EMSA was carried out as described.37 RNA (0.4 μM) was mixed with a synthetic 3′ 6-FAM labeled DNA oligomer (0.8 μM) complementary to a 22 nt (ref. 32) region at the 5′ end of the RNA in 10 mM Tris–HCl (pH 7.4), 50 mM KCl, and 1 mM EDTA. The mixture was heated to 95 °C and slowly cooled down to 25 °C. The RNA (0.04 μM) was then incubated with GN-REX(A) or RHAU(Q)-GC or both at the indicated concentration in a buffer of 10 mM Tris–HCl, 100 mM KCl, 1 mM EDTA, 1 mM DTT, 5%(v/w) glycerol, 0.4 U μl−1 RiboLock RNase inhibitor (Fermentas, USA), and 110 ng μl−1 yeast tRNA. RNA–protein complexes were resolved on a 8% non-denaturing polyacrylamide gel (37.5:1 acrylamide:bis) at 4 °C for 90 min in 1× TBE buffer containing 75 mM KCl.
Spectroscopy of real-time BiFC in vitro
In vitro real-time monitoring of BiFC by eGFP fluorescence was performed in a 96-Well Black Solid Plate (Costar, Corning). First, 1 μM RNA in 10 mM Tris–HCl (PH 7.4), 50 mM KCl, and 1 mM EDTA was heated to 95 °C and then slowly cooled down to 25 °C. Then 10 μl RNA was added to the 96-Well plate containing 100 μl of 20 mM Tris–HCl (pH 7.4), 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.4 U μl−1 RiboLock RNase Inhibitor (Fermentas, USA), and 0.2 μM fusion protein each or both. Fluorescence was recorded at 507 nm with excitation at 480 nm at a 2 min interval in a Synergy 4 Hybrid Microplate Reader (BioTek, USA) at 25 °C.Flow cytometry of BiFC in cells
For simultaneous expression of RNA and probe proteins, RNA and protein expressing plasmids (Fig. S1†) were transformed into the BL21 (DE3) (TransGen Biotech) E. coli strain. Cells were cultured at 37 °C in 5 ml of LB medium on a shaking incubator overnight. The culture was then diluted 100 times in fresh LB media and maintained at 37 °C for about 2–3 h until OD600 ≈ 0.5. Then 0.1 mM IPTG was added to induce the expression of RNA and probe proteins overnight at 25 °C. The cells were collected by centrifugation at 4000 × g for 2 min and washed twice with PBS by filtration on a 0.22 μm filter. They were then diluted in PBS to an OD600 ≈ 0.05. Fluorescence was measured on a FACSCalibur flow cytometer (Becton-Dickinson, USA) using the FL1 channel (excitation at 488 nm, emission filter at 530/30 nm).To monitor BiFC with independent induction of target RNA, the RNA (Fig. S4A) and probe protein (Fig. S1B†) expressing plasmids were transformed into BL21 (DE3). A colony was selected to start the culturing in a LB medium with 100 mg ml−1 of ampicillin and 34 mg ml−1 of chloramphenicol at 37 °C overnight. The cultures were then diluted 100 times into fresh LB media and maintained at 37 °C for about 3 h until OD600 ≈ 0.4. Then 0.1 mM IPTG was added together with or without 2 μg ml−1 anhydrotetracycline (ATc) to induce the expression of probe proteins/RNA or protein only. Cells were collected at different time points and analyzed by flow cytometry as aforementioned. This RNA expressing plasmid was only used in the experiments for Fig. S4.†
Quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from the E. coli cells using the RNAprep Pure Cell/Bacteria Kit (TIANGEN, China). RNA was then polyadenylated by E. coli poly(A) polymerase (M0276S, NEB) in a 50 μl volume containing 50 mM Tris–HCl (pH 7.9), 250 mM NaCl, 10 mM MgCl2, 1.5 μg RNA, 2.5 U poly(A) polymerase, and 1 mM ATP. cDNA was generated from the purified tailed RNA using M-MLV Reverse Transcriptase (M368B, Promega). Briefly, 1 μg of tailed RNA and 0.05 nmol Oligo(dT)18 in a total volume of 15 μl in RNase free water were heated at 70 °C for 5 min and then cooled immediately on ice for 5 min. They were then mixed in a total volume of 50 μl 50 mM Tris–HCl (pH 8.3) buffer with a 4 U μl−1 M-MLV RT (H−) point mutant, 0.25 mM dNTP each, 75 mM LiCl, 3 mM MgCl2 and 10 mM DTT. After a reaction with 3 cycles of 45 °C for 5 min, 48 °C for 5 min, 52 °C for 5 min, and then followed by 52 °C for 30 min, a qPCR reaction was carried out in 20 μl mixture containing 10 μl GoTaq qPCR Master Mix (A6001, Promega), 1 μl 1/5 diluted cDNA, 0.2 μM forward primer and 0.2 μM reverse primer on the real-time cycler qTower (Analytik Jena AG, Jena, Germany) with the following setting: 95 °C for 2 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 60 s. The forward and reverse primer sequences for the target RNA were 5′-TTAGGCGACGGTACGCAAGT-3′ and 5′-TCGAGAAATAGGCCGG-CG-3′, respectively. For IPTG-induced RNA expression (Fig. S1A†), an internal control was amplified from KanR using a primer pair of 5′-GCAATCAGGTGCGACAATCT-3′ (forward) and 5′-TCATTGGCAACGCTACCTTT-3′ (reverse). For ATc-induced RNA expression (Fig. S4A†), an internal control was amplified from CmR using a primer pair of 5′-CACATTCTTGCCCGCCTGAT-3′ (forward) and 5′-TCACCGTCTTTCATTGCCATAC-3′ (reverse).Immunoblotting of RNA–protein interaction
Cells from 4 ml of culture were pelleted and resuspended in 300 μl ice-cold lysis buffer containing 50 mM Tris–HCl, pH 7.4, 150 mM KCl, 1 mM EDTA, 1 mM DTT, 0.1 mM AEBSF, 1 U μl−1 RiboLock RNase Inhibitor (Fermentas, USA) or 10 μg ml−1 RNase A (Fermentas, USA), followed by sonication for 15 min with 5 s on and 5 s off cycles in an ice-water bath (Bioruptor-Plus, Diagenode). The lysate was centrifuged at 16000 × g for 20 min and the supernatant was collected and incubated at room temperature under constant rotation for 1 h with 20 μl of streptavidin-agarose beads (S1638, Sigma) that were pre-loaded with a biotinylated DNA oligomer complementary to the 22 nt region at the 5′ end of the target RNA. After centrifugation at 2000 × g for 1 min, resin-bound RNA-protein complexes were washed three times with a 10 resin bed volume of washing buffer (50 mM Tris–HCl, pH 7.4, 150 mM KCl, 1 mM EDTA, 1 mM DTT, and 0.1 mM AEBSF). The RNA–protein complexes were then released by digestion with RNase H (Fermentas, USA) in 20 mM Tris–HCl (pH 7.8), 40 mM KCl, 10 mM MgCl2, and 1 mM DTT. After centrifugation at 12000 × g for 1 min, the supernatant was collected and resolved on a 15% Tris–glycine PAGE gel. The proteins were transferred to a Protran BA 85 nitrocellulose membrane (Whatman–Schlicher and Schuell) using a semi-dry electroblotting system (Pyxis Protein Transfer/Staining System). The membrane was blocked by 5% fat-free milk in TBST buffer (50 mM Tris–HCl, PH 7.6, 150 mM NaCl, 0.1% Tween 20) for 1 h at room temperature and washed three times by TBST buffer each for 5 min at room temperature. The membrane was then incubated overnight at 4 °C with anti-GFP antibody (E385, abcam) at a 1:2000 dilution in TBST buffer containing 5% fat-free milk. After washing three times with TBST buffer, the membrane was incubated with 1:10000 diluted HRP Goat anti-Rabbit IgG Antibody (LP1001a, abcam) for 1 h at room temperature. After another three times of washing, eGFP was detected using the eECL Western blotting kit (CW0049, CWbiotech) according to the manufacturer's instruction.
Acknowledgements
This work was supported by the Ministry of Science and Technology of China, grant # 2013CB530802 and 2012CB720601 and the National Science Foundation of China, grant # 21432008 and 31470783.Notes and references
- L. A. Cahoon and H. S. Seifert, Science, 2009, 325, 764–767 CrossRef CAS PubMed.
- R. Rodriguez, K. M. Miller, J. V. Forment, C. R. Bradshaw, M. Nikan, S. Britton, T. Oelschlaegel, B. Xhemalce, S. Balasubramanian and S. P. Jackson, Nat. Chem. Biol., 2012, 8, 301–310 CrossRef CAS PubMed.
- L. T. Gray, A. C. Vallur, J. Eddy and N. Maizels, Nat. Chem. Biol., 2014, 10, 313–318 CrossRef CAS PubMed.
- G. H. Nguyen, W. Tang, A. I. Robles, R. P. Beyer, L. T. Gray, J. A. Welsh, A. J. Schetter, K. Kumamoto, X. W. Wang, I. D. Hickson, N. Maizels, R. J. Monnat Jr and C. C. Harris, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9905–9910 CrossRef CAS PubMed.
- N. Maizels, EMBO Rep., 2015, 16, 910–922 CrossRef CAS PubMed.
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2005, 33, 2908–2916 CrossRef CAS PubMed.
- V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, Nat. Biotechnol., 2015, 33, 877–881 CrossRef PubMed.
- X. M. Li, K. W. Zheng, J. Y. Zhang, H. H. Liu, Y. D. He, B. F. Yuan, Y. H. Hao and Z. Tan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14581–14586 CrossRef CAS PubMed.
- K. W. Zheng, S. Xiao, J. Q. Liu, J. Y. Zhang, Y. H. Hao and Z. Tan, Nucleic Acids Res., 2013, 41, 5533–5541 CrossRef CAS PubMed.
- R. Y. Wu, K. W. Zheng, J. Y. Zhang, Y. H. Hao and Z. Tan, Angew. Chem., Int. Ed. Engl., 2015, 54, 2447–2451 CrossRef CAS PubMed.
- K. W. Zheng, R. Y. Wu, Y. D. He, S. Xiao, J. Y. Zhang, J. Q. Liu, Y. H. Hao and Z. Tan, Nucleic Acids Res., 2014, 42, 10832–10844 CrossRef PubMed.
- S. Xiao, J. Y. Zhang, J. Wu, R. Y. Wu, Y. Xia, K. W. Zheng, Y. H. Hao, X. Zhou and Z. Tan, Angew. Chem., Int. Ed. Engl., 2014, 53, 13110–13114 CrossRef CAS PubMed.
- Y. Xu, Y. Suzuki, K. Ito and M. Komiyama, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14579–14584 CrossRef CAS PubMed.
- G. W. Collie, S. M. Haider, S. Neidle and G. N. Parkinson, Nucleic Acids Res., 2010, 38, 5569–5580 CrossRef CAS PubMed.
- A. Randall and J. D. Griffith, J. Biol. Chem., 2009, 284, 13980–13986 CrossRef CAS PubMed.
- H. Martadinata and A. T. Phan, J. Am. Chem. Soc., 2009, 131, 2570–2578 CrossRef CAS PubMed.
- Y. Xu, K. Kaminaga and M. Komiyama, J. Am. Chem. Soc., 2008, 130, 11179–11184 CrossRef CAS PubMed.
- P. H. Wanrooij, J. P. Uhler, T. Simonsson, M. Falkenberg and C. M. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16072–16077 CrossRef CAS PubMed.
- A. Bugaut and S. Balasubramanian, Nucleic Acids Res., 2012, 40, 4727–4741 CrossRef CAS PubMed.
- S. Millevoi, H. Moine and S. Vagner, Wiley Interdiscip. Rev.: RNA, 2012, 3, 495–507 CrossRef CAS PubMed.
- C. Schaffitzel, I. Berger, J. Postberg, J. Hanes, H. J. Lipps and A. Pluckthun, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8572–8577 CrossRef CAS PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- G. Biffi, M. Di Antonio, D. Tannahill and S. Balasubramanian, Nat. Chem., 2014, 6, 75–80 CrossRef CAS PubMed.
- Y. P. Xing, C. Liu, X. H. Zhou and H. C. Shi, Sci. Rep., 2015, 5, 8125 CrossRef CAS PubMed.
- A. Laguerre, K. Hukezalie, P. Winckler, F. Katranji, G. Chanteloup, M. Pirrotta, J. M. Perrier-Cornet, J. M. Wong and D. Monchaud, J. Am. Chem. Soc., 2015, 137, 8521–8525 CrossRef CAS PubMed.
- T. K. Kerppola, Nat. Protoc., 2006, 1, 1278–1286 CrossRef PubMed.
- S. Baskerville, M. Zapp and A. D. Ellington, J. Virol., 1999, 73, 4962–4971 CAS.
- S. Lattmann, B. Giri, J. P. Vaughn, S. A. Akman and Y. Nagamine, Nucleic Acids Res., 2010, 38, 6219–6233 CrossRef CAS PubMed.
- S. Lattmann, M. B. Stadler, J. P. Vaughn, S. A. Akman and Y. Nagamine, Nucleic Acids Res., 2011, 39, 9390–9404 CrossRef CAS PubMed.
- E. P. Booy, R. Howard, O. Marushchak, E. O. Ariyo, M. Meier, S. K. Novakowski, S. R. Deo, E. Dzananovic, J. Stetefeld and S. A. McKenna, Nucleic Acids Res., 2014, 42, 3346–3361 CrossRef CAS PubMed.
- N. V. Hud, F. W. Smith, F. A. Anet and J. Feigon, Biochemistry, 1996, 35, 15383–15390 CrossRef CAS PubMed.
- Y. J. Shyu and C. D. Hu, Trends Biotechnol., 2008, 26, 622–630 CrossRef CAS PubMed.
- B. Heddi, V. V. Cheong, H. Martadinata and A. T. Phan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9608–9613 CrossRef CAS PubMed.
- T. Gaj, C. A. Gersbach and C. F. Barbas 3rd, Trends Biotechnol., 2013, 31, 397–405 CrossRef CAS PubMed.
- M. Valencia-Burton, R. M. McCullough, C. R. Cantor and N. E. Broude, Nat. Methods, 2007, 4, 421–427 CAS.
- V. V. Demidov and N. E. Broude, Nat. Protoc., 2006, 1, 714–719 CrossRef CAS PubMed.
- G. J. Smith, T. R. Sosnick, N. F. Scherer and T. Pan, RNA, 2005, 11, 234–239 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7. See DOI: 10.1039/c5sc03946k |
| This journal is © The Royal Society of Chemistry 2016 |