
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double conjugation strategy to incorporate lipid adjuvants into multiantigenic vaccines†
Waleed M.
Hussein
a,
Tzu-Yu
Liu
a,
Pirashanthini
Maruthayanar
b,
Saori
Mukaida
a,
Peter M.
Moyle
c,
James W.
Wells
b,
Istvan
Toth
*acd and
Mariusz
Skwarczynski
*a
aSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: m.skwarczynski@uq.edu.au; i.toth@uq.edu.au
bThe University of Queensland Diamantina Institute, The University of Queensland, Translational Research Institute, Brisbane, Australia
cSchool of Pharmacy, The University of Queensland, Brisbane, QLD 4102, Australia
dInstitute for Molecular Bioscience, The University of Queensland, Brisbane, QLD 4072, Australia
First published on 4th January 2016
Abstract
Conjugation of multiple peptides by their N-termini is a promising technique to produce branched multiantigenic vaccines. We established a double conjugation strategy that combines a mercapto-acryloyl Michael addition and a copper-catalysed alkyne-azide 1,3-dipolar cycloaddition (CuAAC) reaction to synthesise self-adjuvanting branched multiantigenic vaccine candidates. These vaccine candidates aim to treat cervical cancer and include two HPV-16 derived epitopes and a novel self-adjuvanting moiety. This is the first report of mercapto-acryloyl conjugation applied to the hetero conjugation of two unprotected peptides by their N-termini followed by a CuAAC reaction to conjugate a novel synthetic lipoalkyne self-adjuvanting moiety. In vivo experiments showed that the most promising vaccine candidate completely eradicated tumours in 46% of the mice (6 out of 13 mice).
Introduction
The ability to develop safe vaccines using minimal microbial components has triggered rapid growth in research into peptide-based vaccines.1 However, the inability of peptides in isolation to stimulate the immune system is one of the key challenges in the development of peptide-based vaccines. Therefore, an adjuvant (immunostimulant) is necessary to stimulate a potent immune response against peptide epitopes.2 However, the use of adjuvants is usually associated with side effects and substantial toxicity that has limited the number of adjuvants approved for human use.3 Only alum has been approved as a general human adjuvant, while just a few others were approved for particular vaccine formulation e.g. MF59, ASO3 and ASO4.4 Unfortunately for anticancer vaccines, adjuvants that stimulate safe and effective cytotoxic T lymphocyte (CTL) responses are scarce.5 To overcome this problem, peptide vaccine research has turned its focus to the development of self-adjuvanting delivery systems. These vaccines combine peptide epitopes and immunostimulatory moieties (for example lipidic or polymeric entities) in a single covalently-linked conjugate, thereby ensuring co-delivery of the antigen to antigen presenting cells (APCs) activated by immunostimulatory moieties. This combined presentation helps to enhance vaccine potency and to avoid undesirable side effects that result from using classical adjuvants.6Every year, approximately 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 women are newly diagnosed with cervical cancer around the world, making it the second most common cancer among women. According to experimental and epidemiological studies, human papilloma virus (HPV) is the main cause of cervical cancer.7 Two high-risk genotypes, HPV types 16 (HPV-16) and 18 (HPV-18) are responsible for 70% of all cervical cancers.8
000 women are newly diagnosed with cervical cancer around the world, making it the second most common cancer among women. According to experimental and epidemiological studies, human papilloma virus (HPV) is the main cause of cervical cancer.7 Two high-risk genotypes, HPV types 16 (HPV-16) and 18 (HPV-18) are responsible for 70% of all cervical cancers.8
Prophylactic vaccines against HPV infection help to reduce the incidence of cervical cancers through the generation of neutralizing antibodies and are only effective if administered before infection with HPV.9 Hence, there is a strong demand for the development of effective therapeutic vaccines that are able to treat HPV-related cancers.10 The HPV genome encodes two types of proteins: early proteins (E1, E2, E4, E5, E6 and E7) and late proteins (L1 and L2). Expression of the E6 and E7 oncoproteins results in deregulation of the cell cycle, inactivating tumour suppressor gene products p53 and retinoblastoma protein (pRb) and leading to cancer.10
Peptide-based strategies to develop therapeutic vaccines against HPV-associated cancers have shown promising outcomes in several early stage clinical trials.10 The choice of an appropriate peptide antigen is a crucial issue in the design of synthetic peptide vaccines. Therapeutic vaccines to treat cancer must elicit cellular immunity, thus must include CTL (CD8+) epitopes.1a A CTL epitope was identified in the HPV-16 E6 protein sequence (QLLRREVYDFAFRDL; E643–57)10,11 and was previously shown to induce CTLs in vivo.11,12 Recently, our group showed that the 8Qmin peptide, a small fragment of HPV-16 E7 protein (QAEPDRAHYNIVTF; E744–57),9 that encodes CTL and T-helper cell epitopes, could reduce tumour growth and eradicate E7-expressing TC-1 tumour cells in mice through activation of CTLs13 when administered with a self-adjuvanting delivery system. Therefore, these two CTL epitopes, E643–57 and 8Qmin, were chosen as promising antigens for peptide vaccine development. We also recently demonstrated that anti-8Qmin antibodies were not produced by mice vaccinated with the 8Qmin epitope conjugated to the poly tert-butyl acrylate delivery system.14
It was reported that the orientation of antigens in a vaccine conjugate was very important for stimulating an immune response.15 Conjugation of different peptides via the C-terminus is valuable for the development of multiantigenic branched vaccines. Branched antigens tend to have increased stability to proteolysis,16 and therefore a longer circulation time in the host, providing more opportunities to be taken up by APCs. As a result, these peptides can elicit stronger in vivo immune responses than linear peptides.17 We recently reported that modification of the 8Qmin epitope from the E7 protein by replacing the C-terminal CCKCD sequence with SSKSD or SKKKK substantially diminished its immunogenicity. In contrast, deletion of the CCKCD sequence did not have any negative influence on the epitope potency.9 These results suggest that the CTL epitope is only effective if conjugated to the vaccine delivery system via its N-terminus.
The attachment of a lipidic moiety to the N-terminus of an antigenic peptide to obtain amphiphilic vaccine molecules was previously reported.6c,6d,18 However, the N-terminal conjugation of two or more different unprotected epitopes to a vaccine delivery system have not yet been described (to the best of our knowledge). Thus we established a double conjugation synthetic technique to allow the conjugation of different unprotected peptides, E643–57 and 8Qmin, via their N-termini in order to produce novel branched multiantigenic immunotherapeutics.
Results and discussion
We designed and synthesised immunostimulatory lipoalkynes 1–3 (Scheme 1a). These lipoalkynes were based on the structure of Pam2Cys (di-palmitoyl-S-glycerol cysteine), a well-characterised self-adjuvanting moiety that is widely used in experimental vaccine design.19 As Pam2Cys is a thio-1,2-diglyceride ester of palmitic acid, the new 1,3-diglyceride lipoalkynes 1–3 were designed by replacing the two ester linkages in Pam2Cys with two ether bonds to increase the stability of the compounds against esterases. The two long hydrocarbon chains in Pam2Cys were modified by substituting two methylene groups with oxygen atoms in two different positions as in lipid 1 and 2, to investigate the effect of increasing the polarity (and subsequently the aqueous solubility) on the adjuvanting effect of the resulting molecules.20 For control purposes, lipid 3 contained the same hydrocarbon chain as Pam2Cys was synthesised. In contrast to Pam2Cys, lipids 1–3 have no chiral center and therefore exist as single isomers. They carry an alkyne moiety, thereby allowing easy conjugation of an antigen through a copper-catalysed alkyne-azide 1,3-dipolar cycloaddition (CuAAC) reaction.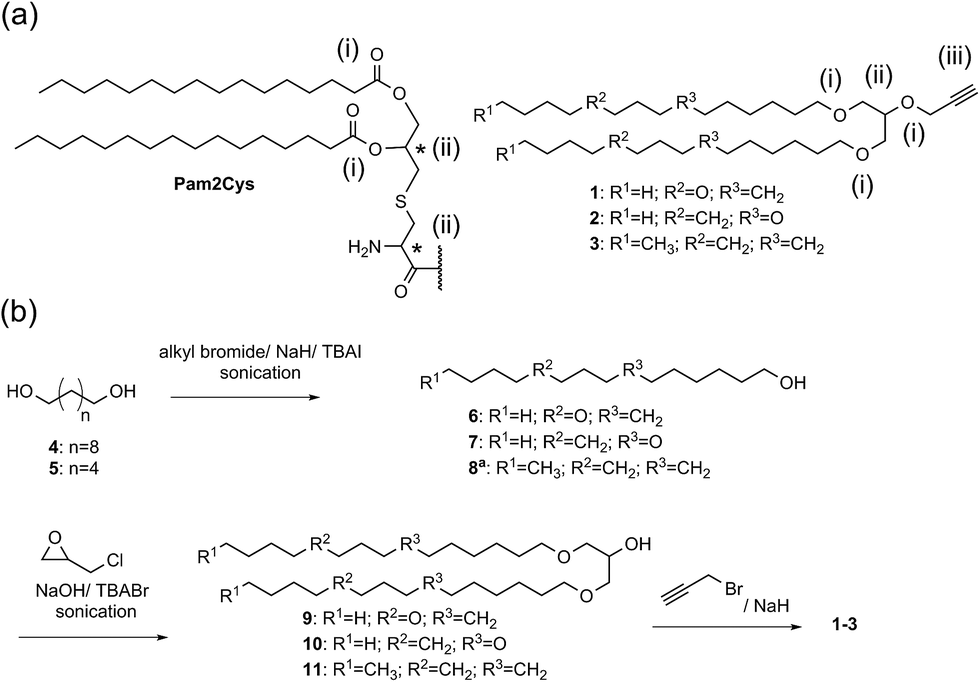 | ||
| Scheme 1 (a) Comparison between the structure of Pam2Cys and the lipoalkynes 1–3: (i) hydrolyzable ester linkages in Pam2Cys versus stable ether bonds in lipoalkynes 1–3, (ii) two chiral centers in Pam2Cys versus achiral molecule in lipoalkynes 1–3, and (iii) presence of alkyne moiety in lipoalkynes 1–3. (b) Synthesis of the lipoalkyne vaccine adjuvants 1–3. a Compound 8 is a commercially available compound. | ||
Lipoalkynes 1–2 were synthesised using three straightforward steps (Scheme 1b), while lipoalkyne 3 required only two steps to be produced. Alcohols 6–7 were prepared from diols 4–5 in 42 and 44% yields, respectively, using alkyl bromide and a phase-transfer catalyst tetrabutylammonium iodide (TBAI) in presence of DMF as a solvent under sonication conditions. The sonication of a mixture of alcohols 6–8, powdered sodium hydroxide, epichlorohydrin, and tetrabutylammonium bromide (TBABr) provided the branched alcohols 9–11. Alcohols 9–11 were treated with propargyl bromide and sodium hydride to afford the lipoalkynes 1–3 in good yields.
A double conjugation strategy was developed to produce anticancer vaccine candidates. Two peptide epitopes were combined into a multiantigenic construct via thioether conjugation using an acryloyl peptide. The reaction was examined on two model short peptides (12 and 13), where one peptide carried both mercapto and azide groups at its N-terminus (12) and the other peptide had an acryloyl moiety attached to its N-terminus (13) (Scheme 2).
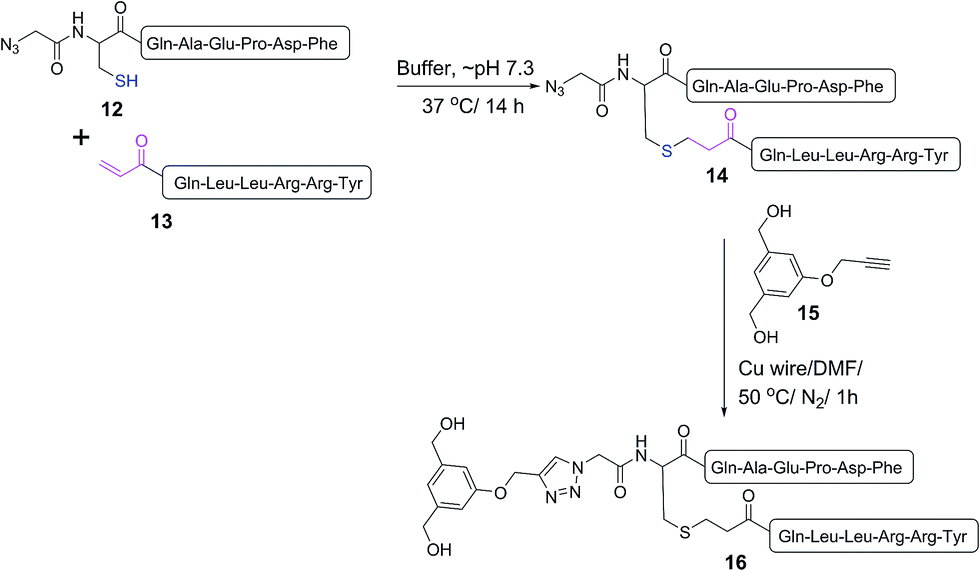 | ||
| Scheme 2 Model double conjugation through a Michael addition, between 12 and 13, followed by a CuAAC reaction, between 14 and 15. | ||
The mercapto-acryloyl conjugation conditions were optimised; ∼pH 7.3, 37 °C, 14 h in the presence of denaturants (6 M guanidine) was found to be optimal. The product 14 of this conjugation was reacted with a model alkyne 15 producing the desired conjugate 16 (Scheme 2, Fig. 1 and 2). The ability of double conjugation strategy to be performed in a one pot reaction was also demonstrated (Scheme 3 and Fig. 3).
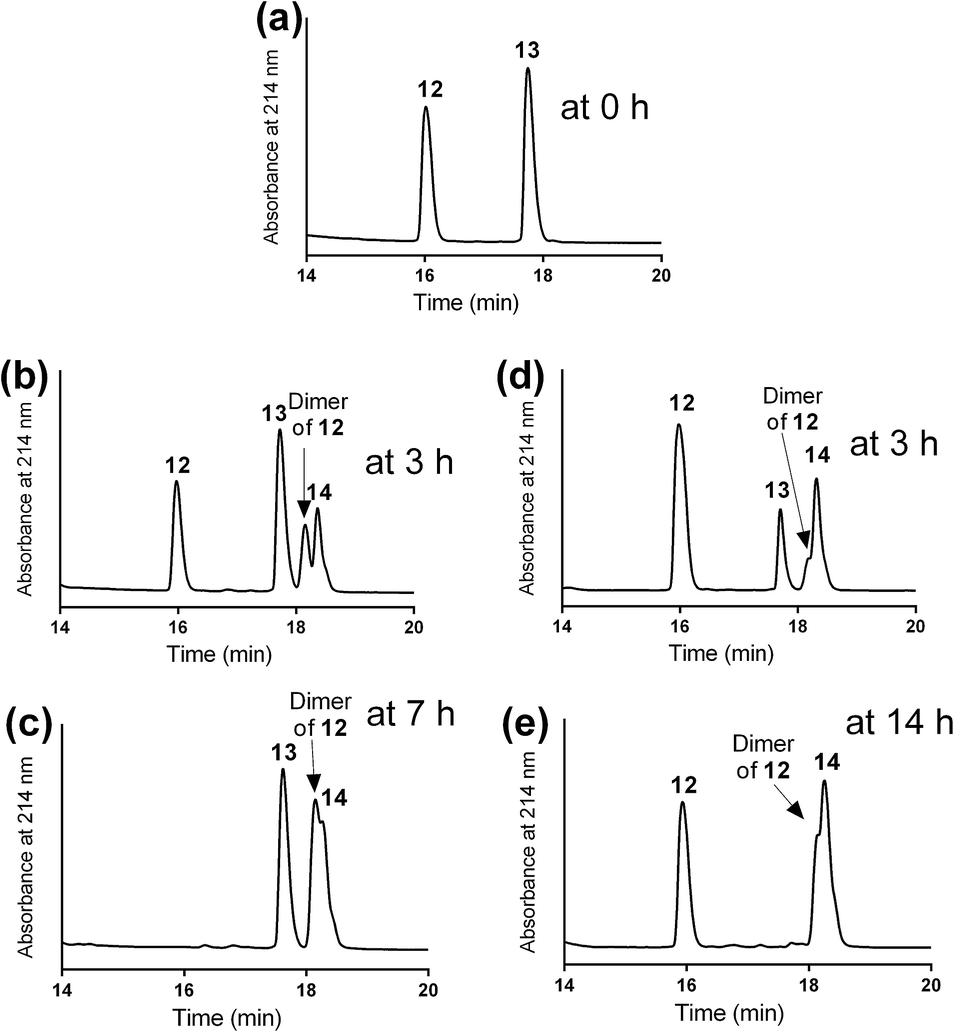 | ||
| Fig. 1 Optimising the conditions for mercapto-acryloyl conjugation between model mercapto-azide (12) that used in excess and acryloyl derivative (13) peptides (a) at 0 time; (b) at 3 h, using DMF as a solvent and two drops of DIPEA. New products started to form including the mercapto-acryloyl conjugation product (14) and the dimer of 12; (c) at 7 h in DMF/DIPEA the reaction was completed with the formation of peptide 14 and the dimer of 12 as major products with the remaining of the majority of the acryloyl peptide 13; (d) at 3 h, using guanidine buffer as a solvent (∼pH 7.3) at 37 °C, new products formed, the mercapto-acryloyl conjugation product (14) together with the dimer of 12; (e) at 14 h in a guanidine buffer, the reaction was completed by formation of peptide 14 as a major product together with the dimer of 12 and complete consumption of the acryloyl peptide 13. The reaction progress was monitored by HPLC and the products were detected by mass spectrometry. | ||
 | ||
| Fig. 2 A copper-catalysed alkyne-azide 1,3-dipolar cycloaddition (CuAAC) model reaction between model conjugation product (14) and model alkyne (15) in DMF in presence of Cu wire at 50 °C under a nitrogen atmosphere (a) at 0 time; (b) at 1 h the reaction was completed by the complete consumption of 14 and formation of the model CuAAC product 16. | ||
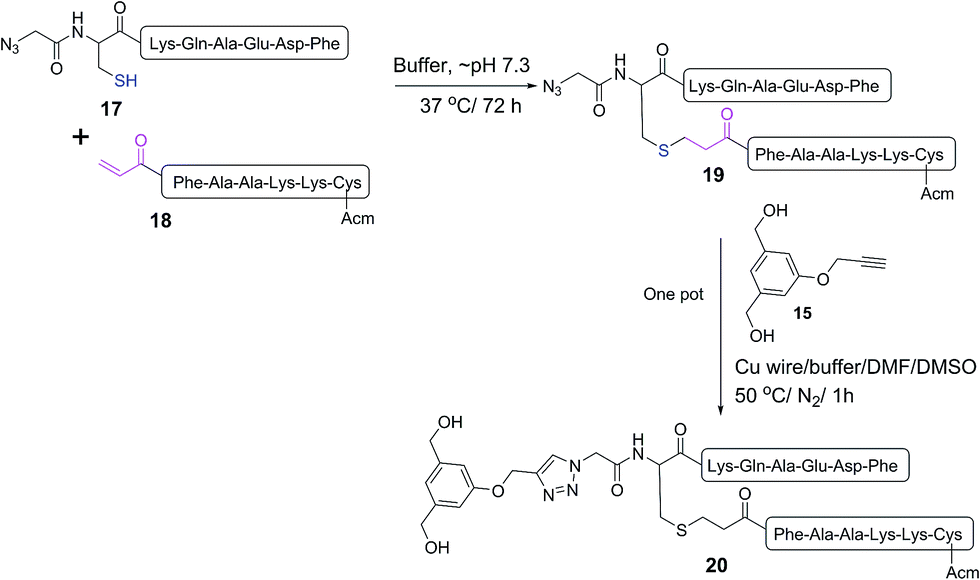 | ||
| Scheme 3 One pot reaction of model double conjugation through mercapto-acryloyl reaction, between 17 and 18 model peptides, followed by azide-alkyne reaction, between compounds 19 and 15. | ||
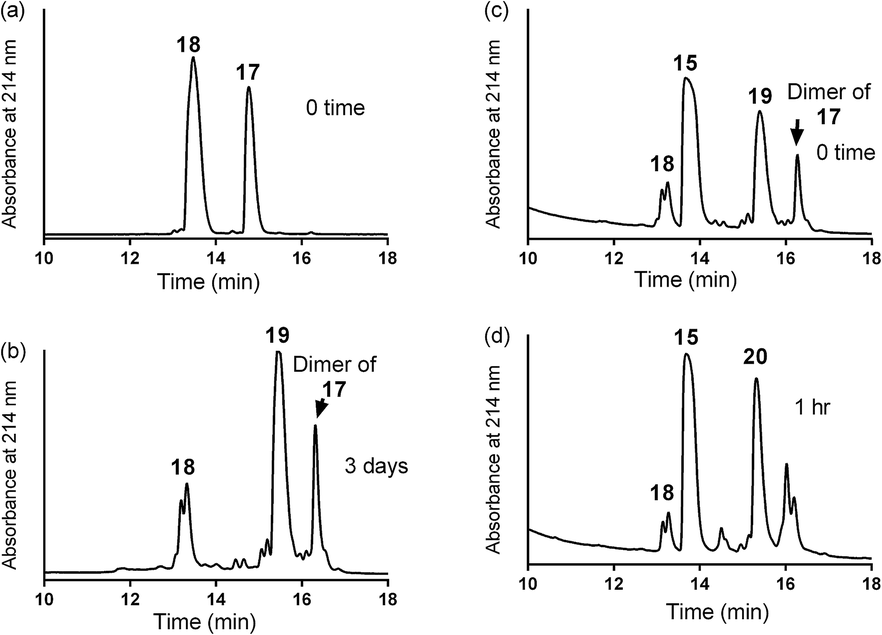 | ||
| Fig. 3 One pot double conjugation model reaction. Mercapto-acryloyl conjugation between mercapto-azide (17) and acryloyl derivative (18) at ∼pH 7.3 (guanidine buffer), 37 °C (a) at 0 time; (b) at 72 h the reaction was completed by the complete consumption of 17 and formation of the conjugation product (19) together with the disulfide dimer of 17. In a one pot reaction, a CuAAC model reaction between model conjugation product (19) and model alkyne (15) in guanidine buffer in presence of Cu wire at 50 °C under a nitrogen atmosphere (c) at 0 time; (d) at 1 h the reaction was completed by the complete consumption of 19 and formation of the model CuAAC product 20. The reaction progress was monitored by HPLC and the products were detected by Mass spec. | ||
The new vaccine candidates, lipopeptides 24–26, were synthesised using the developed conjugation method. First, the N-terminal amine moieties of 8Qmin and E643–57 were modified using stepwise SPPS. Fmoc-cysteine and azidoacetic acid were coupled to 8Qmin to produce mercapto-azide derivative 21. The second peptide (E643–57) was modified with acrylic acid to afford acryloyl derivative 22. The two modified unprotected peptides (21 and 22) were then conjugated via a Michael addition mercapto-acrylate reaction to produce azide derivative 23.
A solution of mercapto-azide 21 (2 equiv.) and the acryloyl derivative 22 (1 equiv.) were gently shaken in denaturing buffer comprised of 6 M guanidine, 50 mM sodium phosphate, 20% acetonitrile, 5 mM EDTA, at ∼pH 7.3 to afford the azide derivative 23 in an excellent isolated yield of 90% (Scheme 4). The reaction was monitored by analytical HPLC and mass spectroscopy (Fig. 4). The second conjugation between the azide derivative 23 (1 equiv.) and the lipoalkynes 1–3 (1.5 equiv.) was achieved in degassed DMF under a nitrogen atmosphere using the CuAAC reaction in the presence of copper wire6b for 4 hours at 50 °C to produce the final lipopeptides 24–26 in 49–87% isolated yields (Scheme 4). The final products 24–26 were structurally well-defined, with only one stereoisomer present, and the synthesis was simple and reproducible.
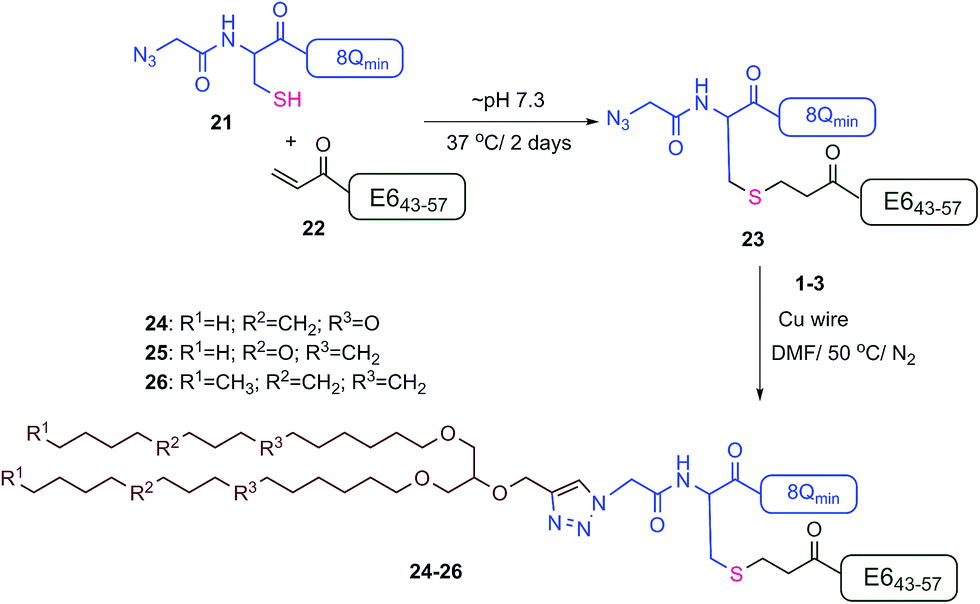 | ||
| Scheme 4 Synthesis of lipopeptides 24–26 using the double conjugation strategy. | ||
 | ||
| Fig. 4 Mercapto-acryloyl conjugation between mercapto-azide (21) and acryloyl derivative (22) at pH 7.5, 37 °C (a) at 0 time; (b) at 48 h the reaction was completed by the complete consumption of 22 and formation of the multiantigenic conjugation product (23) together with the dimer of 21. The reaction progress was monitored by HPLC and the products were detected by MS. | ||
The therapeutic effect of the multiantigenic conjugates on established HPV tumour was evaluated in a mouse model, 6–8 week old, female C57BL/6 mice. The design of compounds 1–3 was based on the structure of Pam2Cys, hence Pam2Cys conjugated to the 8Qmin/E643–57 epitopes was used as a control (29) (Scheme 6). The therapeutic importance of incorporating two epitopes in one molecular entity (24–26) was explored by synthesising compounds 27 and 28, which were comprised of lipid 1 conjugated to 8Qmin or E643–57, respectively. At day zero, mice (8/group) were implanted in the side flank with TC-1 tumour cells expressing the E6/E7 oncoproteins.21 On day 3 mice were immunised with either lipopeptides 24–26 (100 μg/100 μL sterile PBS), a physical mixture of lipid 1 conjugated with 8Qmin (27) and lipid 1 conjugated with E643–57 (28) (100 μg/100 μL sterile PBS, 1:1) (Scheme 5), 8Qmin/E643–57 epitopes conjugated with Pam2Cys (29) (100 μg/100 μL sterile PBS) as a positive control, or PBS (100 μL) as a negative control. The Kaplan–Meier survival curve (Fig. 5a) showed that all of the mice treated with PBS were euthanised due to tumour burden by day 45. In contrast, mice treated with lipopeptide 24 and 25 demonstrated 38% (3 out of 8 mice) and 25% (2 out of 8 mice) survival rates, respectively, which was significantly better than that for mice treated with the positive control (8Qmin/E643–57-Pam2Cys (29), 0% survival, 0 out of 8 mice). Among tested groups, the slowest tumour growth was observed in mice immunised with vaccine candidate 24 (Fig. 6a). The physical mixture of 27 and 28 did not slow down tumour growth significantly and only one mouse treated with the mixture survived to the end of the experiment. We proposed that the physical mixture of 27 and 28 would allow each epitope to be taken up by different APCs, thus the immune stimulating effect of T-helper epitope present in 8Qmin may not enhance the immune response against the E643–57 epitope. It was reported that the co-recognition of T-helper and CTL epitopes by the same APC was essential for the efficient stimulation of cellular immunity.22 Interestingly, the biological study showed that Pam2Cys analogue (29) and the most hydrophobic vaccine candidate 26 induced very weak antitumour responses. In tumour challenge, 0/5 and 1/5 mice survived on day 60 for 29 and 26, respectively (Fig. 5a) despite the well-proven ability of Pam2Cys to induce cellular immune responses.23 This might be explained by the formation of large aggregates (>5 μm in diameter) by conjugates 26 and 29 while compounds 24 and 25 formed particles of submicron size (0.3–0.8 μm as measured by dynamic light scattering) under aqueous conditions. This observation is in the agreement with well-known phenomena that the immune responses are highly dependent on the vaccine particle size.2 This size difference may have arisen because the presence of oxygen atoms in the hydrocarbon chain in both lipids 1 and 2 increased the solubility of the latter compounds (24 and 25).
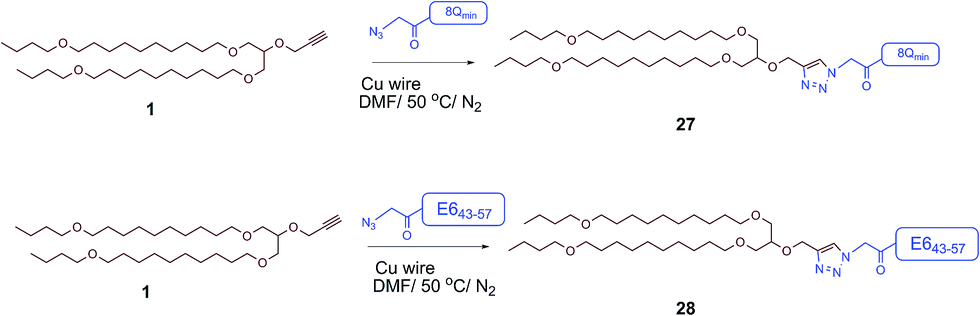 | ||
| Scheme 5 Synthesis of 27 (lipid 1 conjugated with 8Qmin) and 28 (lipid 1 conjugated with E643–57). | ||
 | ||
| Scheme 6 Synthesis of 8Qmin/E643–57-Pam2Cys (29). | ||
 | ||
| Fig. 5 Tumor challenge experiments. C57BL/6 (8/group) were inoculated subcutaneously in the right flank with 1 × 105 TC-1 cells per mouse (day 0) and immunised with: (a) lipopeptides 24–26; a physical mixture of lipid 1 conjugated with 8Qmin (27) and lipid 1 conjugated with E643–57 (28); 8Qmin/E643–57 epitopes conjugated with Pam2Cys (29) as a positive control; or PBS as a negative control on day 3 (8/group) or (b) lipopeptide 24; a mixture of 8Qmin–E643–57 (1:1) + ISA51 as a positive control; or PBS as a negative control on day 3 (13/group). Survival rate was monitored over 60 days post implantation and plotted as a Kaplan–Meier survival curve. Mice were euthanised when tumor volume reached 1 cm3 or started bleeding. The survival rate of each group was compared to the negative control (PBS) and was analysed using the log-rank (Mantel–Cox) test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). | ||
 | ||
| Fig. 6 Tumor challenge experiments. (a) C57BL/6 (8 per group) were inoculated subcutaneously in the right flank with 1 × 105 TC-1 tumour cells per mouse (day 0) and immunised with lipopeptide 24, a mixture of lipid 1 conjugated with 8Qmin (27) and lipid 1 conjugated with E643–57 (28) (1:1), 8Qmin/E643–57-Pam2Cys (29) as a positive control, or PBS as a negative control on day 3. (b) C57BL/6 mice (13 per group) were inoculated subcutaneously in the right flank with 1 × 105 TC-1 tumour cells per mouse (day 0) and immunised with lipopeptide 24, a mixture of 8Qmin + E643–57 (1:1) + ISA51 as a positive control, or PBS as a negative control on day 3. Mean tumour volume (cm3) in different groups of mice shown up to day 24 after tumour implantation (when the first mouse was euthanised). (c) Tumour volume (cm3) in individual TC-1 tumour bearing mice (C57BL/6 mice, 13 per group) treated with lipopeptide 24 or (d) 8Qmin + E643–57 + ISA51 shown over 60 days post implantation. Mean tumour volume (cm3) in different groups of mice (C57BL/6 mice, 13 per group) shown up to day 24 after tumour implantation (when the first mouse was euthanised). | ||
Compound 29 (which bore a Pam2Cys moiety) induced unexpectedly weak antitumour responses, therefore two additional independent experiments (with 5 + 8 mice per group) were performed to further investigate the antitumour potency of the lead vaccine candidate. Incomplete Freund's adjuvant (Montanide ISA51) was chosen as an adjuvant in an emulsion with 8Qmin and E643–57 epitopes for formulation of the positive control.
Female C57BL/6 (6–8 weeks old) mice (5 + 8/group) were immunized with either lipopeptide 24 (100 μg/100 μL sterile PBS), 30 μg of a mixture of 8Qmin and E643–57 emulsified in a total volume of 100 μL of incomplete Freund's adjuvant (Montanide ISA51)/PBS (1:1, v/v) as a positive control. The Kaplan–Meier survival curve (Fig. 5b) showed that 46% (6 out of 13 mice) of mice treated with lipopeptide 24 survived over 60 days. Vaccination with lipopeptide 24 induced significantly better survival in tumour-bearing mice than treatment with the PBS as a negative control (p = 0.0002) (Fig. 5b). As shown in Fig. 6b, tumour-bearing mice treated with lipopeptide 24 showed slow tumour growth. It is particularly noteworthy that 46% of mice (6 out of 13 mice) treated with lipopeptide 24 were tumour free after 60 days (Fig. 6c). The therapeutic efficacy of 24 was similar to that of positive control (a mixture of 8Qmin + E643–57 + ISA51) (Fig. 5b and 6b–d). We also demonstrated that both E744–57 and E643–57 peptides were active in lipopeptide 24 by assessing recall IFN-γ production by CD8 T-cells from immunised mice in response to MHC class I-restricted E7 (RAHYNIVTF) or E6 (YDFAFRDL) peptide restimulation by ELISPOT (Fig. 7).
 | ||
| Fig. 7 Assessment of CD8+ T-cell response to vaccination. Mice were immunised with lipopeptide 24, a mixture of E643–57 and 8Qmin (E6/E7), an “irrelevant” lipopeptide (KQAEDKVKASREAKKQVEKALEQLEDKVK – conjugated with lipid 1), or PBS subcutaneously in both flanks. Ten days later, spleens were harvested and IFN-γ production in response to (a) short E6 (YDFAFRDL) or (b) short E7 (RAHYNIVTF) peptides was determined by ELISPOT (n = 9 mice/group). The data were pooled from two independent experiments and analysed using the unpaired t test (*p < 0.05; ***p < 0.001). | ||
Conclusions
We established a synthetic double conjugation pathway to develop multiantigenic lipopeptide conjugates as self-adjuvanting therapeutic vaccine candidates to treat HPV-related cancers. The method involved a Michael addition mercapto-acryloyl reaction between two unprotected peptides followed by an azide-alkyne click reaction to give the final lipopeptide products. Three novel lipidic self-adjuvanting moieties were synthesised, conjugated with the multiantigenic branched peptide and were found to stimulate significantly better survival in an in vivo murine HPV model than mice treated with the Pam2Cys analogue 29, without an external adjuvant and after only a single immunization.Our double conjugation strategy provided an overall yield 50–80%; can be applied to a wide variety of synthetic applications; was simple to perform; and can be applied on unprotected peptides. This strategy could also be used to rapidly produce libraries of different vaccine constructs from a small selection of starting components (e.g. a variety of epitopes and adjuvants). It is anticipated that this technique will be widely used in the chemical synthesis of branched multiantigenic peptides and self-adjuvanting vaccines.
Experimental section
Materials
1,3-Di(hydroxymethyl)-5-(prop-2-ynyloxy)benzene was prepared as reported.24 Protected L-amino acids were purchased from Novabiochem (Läufelfingen, Switzerland) and Mimotopes (Melbourne, Australia). pMBHA resin was purchased from Peptides International (Kentucky, USA). Rink amide MBHA resin, N,N′-dimethylformamide (DMF), dichloromethane (DCM), methanol, N,N′-diisopropylethylamine (DIPEA), piperidine and trifluoroacetic acid (TFA) were obtained from Merck (Darmstadt, Germany). Copper wire was purchased from Aldrich (Castle Hill, Australia). (Dimethylamino)-N,N-dimethyl(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yloxy)-methanim-inium hexafluorophosphate (HATU) was purchased from Mimotopes (Melbourne, Australia). HPLC grade acetonitrile was obtained from Labscan (Bangkok, Thailand). All other reagents were obtained at the highest available purity from Sigma-Aldrich (Castle Hill, NSW, Australia). Anhydrous hydrofluoric acid (HF) was supplied by BOC gases (Sydney, NSW, Australia). A Kel-F HF apparatus (Peptide Institute, Osaka, Japan) was used for HF cleavage. ESI-MS was performed using a Perkin-Elmer-Sciex API3000 instrument with Analyst 1.4 software (Applied Biosystems/MDS Sciex, Toronto, Canada). High-resolution electrospray ionization mass spectra measurements were obtained on a Bruker micrOTOF mass spectrometer by direct infusion in MeCN at 3 μL min−1 using sodium formate clusters as an internal calibrant. Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker Avance 300, 500 or 600 MHz spectrometer (Bruker Biospin, Germany). Chemical shifts are reported in parts per million (ppm) on a δ scale, relative to the solvent peak (CDCl3δH 7.24, δC 77.0). Coupling constants (J) are reported in hertz (Hz) and peak multiplicities described as singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), multiplet (m), or broad (br). Analytical RP-HPLC was performed using Shimadzu (Kyoto, Japan) instrumentation (DGU-20A5, LC-20AB, SIL-20ACHT, SPD-M10AVP) with a 1 mL min−1 flow rate and detection at 214 nm and/or evaporative light scattering detector (ELSD). Separation was achieved using a 0–100% linear gradient of solvent B over 40 min with Method A (0.1% TFA/H2O as solvent A and 90% MeCN/0.1% TFA/H2O as solvent B) and/or Method B (0.1% TFA/H2O as solvent A and 90% MeOH/0.1% TFA/H2O as solvent B) on either a Vydac analytical C4 column (214TP54; 5 μm, 4.6 mm × 250 mm) or a Vydac analytical C18 column (218TP54; 5 μm, 4.6 mm × 250 mm). Preparative RP-HPLC was performed on Shimadzu (Kyoto, Japan) instrumentation (either LC-20AT, SIL-10A, CBM-20A, SPD-20AV, FRC-10A or LC-20AP × 2, CBM-20A, SPD-20A, FRC-10A) in linear gradient mode using a 5–20 mL min−1 flow rate, with detection at 230 nm. Separations were performed with solvent A and solvent B on a Vydac preparative C18 column (218TP1022; 10 μm, 22 mm × 250 mm), Vydac semi-preparative C18 column (218TP510; 5 μm, 10 mm × 250 mm) or Vydac semi-preparative C4 column (214TP510; 5 μm, 10 mm × 250 mm). Flash chromatography was performed on Merck Kieselgel 60 as described by Still.25 Particle size was measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano Series with DTS software. Sizes were analysed using a non-invasive backscatter system. Multiplicate measurements were performed at 25 °C with scattering angle of 173° using disposable cuvettes and the number-average hydrodynamic particle diameters are reported.General procedure of manual stepwise SPPS on rink amide MBHA resin – Fmoc-chemistry
Peptides were synthesised by manual stepwise SPPS on rink amide MBHA resin (substitution ratio: 0.60 mmol g−1, 0.2 mmol scale, 0.33 g) using HATU/DIPEA Fmoc-chemistry. Amino acid activation was achieved by dissolving Fmoc-amino acid (0.84 mmol, 4.2 equiv.), in 0.5 M HATU/DMF solution (1.6 mL, 0.8 mmol, 4.0 equiv.) followed by the addition of DIPEA (146 μL, 0.84 mmol, 4.2 equiv.). Coupling cycle consisted of Fmoc deprotection with 20% of piperidine in DMF (twice, 10 and 20 min), a 1 min DMF flow-wash, followed by coupling with 4.2 equiv. of preactivated Fmoc-amino acids (2 × 1 h). Upon completion of synthesis, the resin was washed with DMF, DCM, and MeOH, then dried (vacuum desiccator). The cleavage of model mercapto-azide was carried out by stirring the resin in the solution of TFA (99%)/triisopropylsilane/water (95:2.5:2.5) for 4 h. The cleaved peptide was precipitated, filtered, and washed with ice-cold Et2O. After lyophilization, the crude peptide was obtained as an amorphous powder.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO-QLLRREVYDFAFRDL) was synthesised following the general manual stepwise SPPS HATU/DIPEA Fmoc-chemistry procedure. The coupling of acrylic acid (4.2 equiv.) was achieved using HATU (4 equiv.)/DIPEA (4.2 equiv.) at room temperature (2 × 1 h). The crude product was purified by a preparative RP-HPLC on C-18 column with a 35–55% solvent B gradient over 20 min. HPLC analysis (C-18 column, Method A): tR = 22.7 min, purity > 95%. Yield: 33%. ESI-MS: m/z 998.2 (calc 998.1) [M + 2H]2+; 665.8 (calc 665.8) [M + 3H]3+; MW 1994.3.
CHCO-QLLRREVYDFAFRDL) was synthesised following the general manual stepwise SPPS HATU/DIPEA Fmoc-chemistry procedure. The coupling of acrylic acid (4.2 equiv.) was achieved using HATU (4 equiv.)/DIPEA (4.2 equiv.) at room temperature (2 × 1 h). The crude product was purified by a preparative RP-HPLC on C-18 column with a 35–55% solvent B gradient over 20 min. HPLC analysis (C-18 column, Method A): tR = 22.7 min, purity > 95%. Yield: 33%. ESI-MS: m/z 998.2 (calc 998.1) [M + 2H]2+; 665.8 (calc 665.8) [M + 3H]3+; MW 1994.3.
Preparation of guanidine buffer
6 M guanidine, 50 mM sodium phosphate, 20% acetonitrile, 5 mM EDTA, ∼pH 7.3.ESI-MS: m/z 1466.2 (calc 1466.1) [M + 3H]3+; 1100.0 (calc 1099.8) [M + 4H]4+; 880.2 (calc 880.0) [M + 5H]5+; MW 4395.1.
ESI-MS: m/z 1466.2 (calc 1466.1) [M + 3H]3+; 1100.0 (calc 1099.8) [M + 4H]4+; 880.0 (calc 880.0) [M + 5H]5+; MW 4395.1.
Model compounds
CHCO-Gln-Leu-Leu-Arg-Arg-Tyr-NH2) was synthesised following the general manual stepwise SPPS HATU/DIPEA Fmoc-chemistry procedure. The coupling of acrylic acid (4.2 equiv.) was achieved using HATU (4 equiv.)/DIPEA (4.2 equiv.) at room temperature (2 × 1 h). The crude product was purified by a preparative RP-HPLC on C18 column with a 20–40% solvent B gradient over 20 min. HPLC analysis (C18 column, Method A): tR = 17.7 min, purity > 95%. Yield: 40%. ESI-MS: m/z 901.9 (calc 902.1) [M + H]+; 451.4 (calc 451.5) [M + 2H]2+; MW 901.1.
CHCO-Phe-Ala-Ala-Lys-Lys-Cys(Acm)-NH2) was synthesised following the general manual stepwise SPPS HATU/DIPEA Fmoc-chemistry procedure. The coupling of acrylic acid (4.2 equiv.) was achieved using HATU (4 equiv.)/DIPEA (4.2 equiv.) at room temperature (2 × 1 h). The crude product was purified by a preparative RP-HPLC on C18 column with a 20–40% solvent B gradient over 20 min. HPLC analysis (C18 column, Method A): tR = 14.1 min and (C4 column, Method A): tR = 8.4 min, purity > 95%. Yield: 27%. ESI-MS: m/z 791.5 (calc 791.4) [M + H]+; MW 791.
:DMSO (1:1) (0.5 mL) and copper wire (60 mg) was added to the reaction mixture. The air in the reaction mixture was removed by nitrogen bubbling. The reaction mixture was stirred at 50 °C under nitrogen. The progress of the reaction was monitored by analytical HPLC (C-18 column) until 19 was completely consumed after 1 h and gave the final product 20.
ESI-MS of compound 19: m/z 1713.2 (calc 1714.0) [M + H]+; 857.1 (calc 857.5) [M + 2H]2+; 571.7 (calc 572.0) [M + 3H]3+; MW 1713.
ESI-MS of compound 20: m/z 953.7 (calc 953.6) [M + 2H]2+; 636.2 (calc 636.1) [M + 3H]3+; MW 1905.
Particle size measurement
The final constructs of lipopeptides 24–26 and 29 were dissolved in sterile PBS using a vortex until a homogenous solution was obtained. The particle sizes were measured by dynamic light scattering. The measurements were repeated at least five times. All compounds formed particles with rather high polydispersity. Compound 24 and 25 formed submicron particles (24: 450–750 nm, PDI = 0.25–0.55 and 25: 350–550 nm, PDI is 0.20–0.40). Compounds 26 and 29 formed large aggregates (visible to the naked eye) with sizes above the upper detection level of the instrument (>5 μm).Biological assay
:1); 8Qmin/E643–57-Pam2Cys (29) (100 μg/100 μL sterile PBS) as a positive control; or 100 μL PBS as a negative control. Each mouse received a single immunization only. The size of the tumour was measured by palpation and calipers every two days and reported as the average tumour size across the group of five mice or as tumour size in individual mice.30 Tumour volume was calculated using the formula V (cm3) = 3.14 × [largest diameter × (perpendicular diameter)2]/6.30b The mice were euthanised when tumour reached 1 cm3 or started bleeding to avoid unnecessary suffering.
The second tumor challenge study was executed with 24 and antigens administered with commercial adjuvant, in the similar manner as described above. Two independent experiments were performed with total 13 C57BL/6 mice per group (5 + 8 mice per group) and the list of the immunization compounds was: lipopeptide 24; 30 μg of a mixture of 8Qmin and E643–57 emulsified in a total volume of 100 μL of Montanide ISA51 (Seppic, France)/PBS (1:1, v/v) as a positive control; and PBS as a negative control.
IFN-gamma ELISPOT assays
Splenocytes were harvested from the spleens of naïve and immunised mice and depleted of red-blood cells using Red Blood Cell Lysing Buffer (0.155 M ammonium chloride in 0.01 M Tris–HCl buffer, Sigma). Splenocytes were then resuspended in RPMI (Sigma) supplemented with 20% FCS (Sigma), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin, and 50 μM β-mercaptoethanol. The cells were plated at 5 × 105 cells per well in triplicate in ELISPOT plates (Millipore Biotec) which had been previously coated with 5 μg mL−1 IFN-γ capture mAb (clone 14-7313-85 eBioscience) in PBS at 4 °C overnight and then blocked with RPMI/20% FCS at room temperature for 3 hours. E7 (RAHYNIVTF) and E6 (YDFAFRDL) peptides (10 μg mL−1) were added alongside 10 ng mL−1 rhIL-2 (R&D systems) to a final volume of 200 μL per well. Plates were incubated for 18 hours at 37 °C, washed, and a biotinylated IFN-γ detection Ab (2 μg mL−1; clone R4-6A2; eBioscience) in 1% BSA added at room temperature for 3 hours. After washing, a streptavidin–HRP complex (DakoCytomation) in 1% BSA was added for 1 h at room temperature, plates were washed again, and bound cytokine was visualised with 3-amino-9-ethylcarbozole (Calbiochem). Spots were counted with an ELISPOT reader (Autoimmun Diagnostika). An “irrelevant” lipopeptide (KQAEDKVKASREAKKQVEKALEQLEDKVK – conjugated with lipid 1) was used as a control in this experiment. Group A streptococcal B-cell epitope (J14) was selected as the irrelevant peptide.31Acknowledgements
This work was supported by the National Health and Medical Research Council of Australia (NHMRC 1006454). We thank Dr Zeinab Khalil from the Institute for Molecular Bioscience, The University of Queensland for her assistance in acquiring the high-resolution mass spectroscopy data. We thank Thalia Guerin for her critical review of this manuscript.References
- (a) A. W. Purcell, J. McCluskey and J. Rossjohn, Nat. Rev. Drug Discovery, 2007, 6, 404–414 CrossRef CAS PubMed; (b) J. Pol, N. Bloy, A. Buque, A. Eggermont, I. Cremer, C. Sautes-Fridman, J. Galon, E. Tartour, L. Zitvogel, G. Kroemer and L. Galluzzi, OncoImmunology, 2015, 4, e974411-1–e974411-12 Search PubMed.
- M. Skwarczynski and I. Toth, Nanomedicine, 2014, 9, 2657–2669 CrossRef CAS PubMed.
- S. G. Reed, S. Bertholet, R. N. Coler and M. Friede, Trends Immunol., 2009, 30, 23–32 CrossRef CAS PubMed.
- R. Rappuoli, M. Pizza, G. Del Giudice and E. de Gregorio, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12288–12293 CrossRef CAS PubMed.
- (a) F. Azmi, A. A. A. Fuaad, M. Skwarczynski and I. Toth, Hum. Vaccines Immunother., 2014, 10, 778–796 CrossRef CAS; (b) R. J. Hopkins, N. F. Daczkowski, P. E. Kaptur, D. Muse, E. Sheldon, C. LaForce, S. Sari, T. L. Rudge and E. Bernton, Vaccine, 2013, 31, 3051–3058 CrossRef CAS PubMed.
- (a) P. M. Moyle, C. Olive, M.-F. Ho, M. F. Good and I. Toth, J. Med. Chem., 2006, 49, 6364–6370 CrossRef CAS PubMed; (b) M. Skwarczynski, M. Zaman, C. N. Urbani, I. C. Lin, Z. Jia, M. R. Batzloff, M. F. Good, M. J. Monteiro and I. Toth, Angew. Chem., Int. Ed., 2010, 49, 5742–5745 CrossRef CAS PubMed; (c) M. Black, A. Trent, Y. Kostenko, J. S. Lee, C. Olive and M. Tirrell, Adv. Mater., 2012, 24, 3845–3849 CrossRef CAS PubMed; (d) T. H. Wright, A. E. S. Brooks, A. J. Didsbury, G. M. Williams, P. W. R. Harris, P. R. Dunbar and M. A. Brimble, Angew. Chem., Int. Ed., 2013, 52, 10616–10619 CrossRef CAS PubMed; (e) F. Boato, R. M. Thomas, A. Ghasparian, A. Freund-Renard, K. Moehle and J. A. Robinson, Angew. Chem., Int. Ed., 2007, 46, 9015–9018 CrossRef CAS PubMed.
- H.-J. Cho, Y.-K. Oh and Y. B. Kim, Expert Opin. Ther. Pat., 2011, 21, 295–309 CrossRef CAS PubMed.
- O. Peralta-Zaragoza, V. H. Bermudez-Morales, C. Perez-Plasencia, J. Salazar-Leon, C. Gomez-Ceron and V. Madrid-Marina, OncoTargets Ther., 2012, 5, 315–328 CrossRef CAS PubMed.
- T.-Y. Liu, W. M. Hussein, Z. Jia, Z. M. Ziora, N. A. J. McMillan, M. J. Monteiro, I. Toth and M. Skwarczynski, Biomacromolecules, 2013, 14, 2798–2806 CrossRef CAS PubMed.
- T.-Y. Liu, W. M. Hussein, I. Toth and M. Skwarczynski, Curr. Top. Med. Chem., 2012, 12, 1581–1592 CrossRef CAS PubMed.
- A. K. Sarkar, G. Tortolero-Luna, P. N. Nehete, R. B. Arlinghaus, M. F. Mitchell and K. J. Sastry, Viral Immunol., 1995, 8, 165–174 CrossRef CAS PubMed.
- P. R. Manuri, B. Nehete, P. N. Nehete, R. Reisenauer, S. Wardell, A. N. Courtney, R. Gambhira, D. Lomada, A. K. Chopra and K. J. Sastry, Vaccine, 2007, 25, 3302–3310 CrossRef CAS PubMed.
- T. Y. Liu, A. K. Giddam, W. M. Hussein, Z. F. Jia, N. A. J. McMillan, M. J. Monteiro, I. Toth and M. Skwarczynski, Curr. Drug Delivery, 2015, 12, 3–8 CrossRef CAS.
- T. Y. Liu, W. M. Hussein, A. K. Giddam, Z. Jia, J. M. Reiman, M. Zaman, N. A. McMillan, M. F. Good, M. J. Monteiro, I. Toth and M. Skwarczynski, J. Med. Chem., 2015, 58, 888–896 CrossRef CAS PubMed.
- (a) A. B. M. Abdel-Aal, M. R. Batzloff, Y. Fujita, N. Barozzi, A. Faria, P. Simerska, P. M. Moyle, M. F. Good and I. Toth, J. Med. Chem., 2008, 51, 167–172 CrossRef CAS PubMed; (b) W. G. Zeng, S. Ghosh, M. Macris, J. Pagnon and D. C. Jackson, Vaccine, 2001, 19, 3843–3852 CrossRef CAS PubMed.
- L. Bracci, C. Falciani, B. Lelli, L. Lozzi, Y. Runci, A. Pini, M. G. de Montis, A. Tagliamonte and P. Neri, J. Biol. Chem., 2003, 278, 46590–46595 CrossRef CAS PubMed.
- X.-D. Tang, G.-Z. Wang, J. Guo, M.-H. Lue, C. Li, N. Li, Y.-L. Chao, C.-Z. Li, Y.-Y. Wu, C.-J. Hu, D.-C. Fang and S.-M. Yang, Mol. Cancer Ther., 2012, 11, 1183–1192 CrossRef CAS PubMed.
- (a) H. P. Liu, K. D. Moynihan, Y. R. Zheng, G. L. Szeto, A. V. Li, B. Huang, D. S. van Egeren, C. Park and D. J. Irvine, Nature, 2014, 507, 519–522 CrossRef CAS PubMed; (b) M. Skwarczynski, A. A. H. A. Fuaad, L. Rustanti, Z. M. Ziora, M. Aqil, M. R. Batzloff, M. F. Good and I. Toth, Drug Delivery Lett., 2011, 1, 2–8 CAS.
- W.-G. Zeng, E. Eriksson, B. Chua, L. Grollo and D. C. Jackson, Amino Acids, 2010, 39, 471–480 CrossRef CAS PubMed.
- W. Zeng, S. Ghosh, Y. F. Lau, L. E. Brown and D. C. Jackson, J. Immunol., 2002, 169, 4905–4912 CrossRef.
- J. Tang, R. Yin, Y. Tian, Z. Huang, J. Shi, X. Fu, L. Wang, Y. Wu, F. Hao and B. Ni, Vaccine, 2012, 30, 1071–1082 CrossRef CAS PubMed.
- G. Behrens, M. Li, C. M. Smith, G. T. Belz, J. Mintern, F. R. Carbone and W. R. Heath, Immunol. Cell Biol., 2004, 82, 84–90 CrossRef CAS PubMed.
- (a) J. C. A. Cobbin, W. Zeng, D. C. Jackson and L. E. Brown, PLoS One, 2014, 9, e115356 Search PubMed; (b) G. Deliyannis, K. Kedzierska, Y. F. Lau, W. G. Zeng, S. J. Turner, D. C. Jackson and L. E. Brown, Eur. J. Immunol., 2006, 36, 770–778 CrossRef CAS PubMed; (c) D. C. Jackson, Y. F. Lau, T. Le, A. Suhrbier, G. Deliyannis, C. Cheers, C. Smith, W. G. Zeng and L. E. Brown, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15440–15445 CrossRef CAS PubMed; (d) Y. F. Lau, G. Deliyannis, W. Zeng, A. Mansell, D. C. Jackson and L. E. Brown, Int. Immunol., 2006, 18, 1801–1813 CrossRef CAS PubMed.
- C. N. Urbani, D. E. Lonsdale, C. A. Bell, M. R. Whittaker and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1533–1547 CrossRef CAS.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- W. M. Hussein, B. P. Ross, M. J. Landsberg, D. Levy, B. Hankamer and R. P. McGeary, J. Org. Chem., 2009, 74, 1473–1479 CrossRef CAS PubMed.
- J. M. Dyke, A. P. Groves, A. Morris, J. S. Ogden, A. A. Dias, A. M. S. Oliveira, M. L. Costa, M. T. Barros, M. H. Cabral and A. M. C. Moutinho, J. Am. Chem. Soc., 1997, 119, 6883–6887 CrossRef CAS.
- (a) B. Y. Chua, W. Zeng and D. C. Jackson, Methods Mol. Biol., 2008, 494, 247–261 CrossRef CAS PubMed; (b) J. W. Metzger, K. H. Wiesmuller and G. Jung, Int. J. Pept. Protein Res., 1991, 38, 545–554 CrossRef CAS PubMed.
- K.-Y. Lin, F. G. Guarnieri, K. F. Staveley-O'Carroll, H. I. Levitsky, J. T. August and D. M. Pardoll, Cancer Res., 1996, 56, 21–26 CAS.
- (a) C.-F. Hung, W.-F. Cheng, C.-Y. Chai, K.-F. Hsu, L. He, M. Ling and T. C. Wu, J. Immunol., 2001, 166, 5733–5740 CrossRef CAS PubMed; (b) Q. Zeng, S. Peng, A. Monie, M. Yang, X. Pang, C.-F. Hung and T. C. Wu, Hum. Gene Ther., 2011, 22, 809–819 CrossRef CAS PubMed.
- W. A. Hayman, E. R. Brandt, W. A. Relf, J. Cooper, A. Saul and M. F. Good, Int. Immunol., 1997, 9, 1723–1733 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structures, spectroscopic data with 1H and C13 NMR spectra, HPLC profiles and MS spectra. See DOI: 10.1039/c5sc03859f |
| This journal is © The Royal Society of Chemistry 2016 |