
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diene hydroaminomethylation via ruthenium-catalyzed C–C bond forming transfer hydrogenation: beyond carbonylation†
Susumu
Oda
,
Jana
Franke
and
Michael J.
Krische
*
Department of Chemistry, University of Texas at Austin, Austin, Texas, 78712 USA. E-mail: mkrische@mail.utexas.edu
First published on 17th November 2015
Abstract
Under the conditions of ruthenium catalyzed transfer hydrogenation using 2-propanol as terminal reductant, 1,3-dienes engage in reductive C–C coupling with formaldimines obtained in situ from 1,3,5-tris(aryl)-hexahydro-1,3,5-triazines to form homoallylic amines. Deuterium labelling studies corroborate a mechanism involving reversible diene hydroruthenation to form an allylruthenium complex that engages in turn-over limiting imine addition. Protonolysis of the resulting amidoruthenium species releases product and delivers a ruthenium alkoxide, which upon β-hydride elimination closes the catalytic cycle. These transformations, which include enantioselective variants, represent the first examples of diene hydroaminomethylation.
Introduction
Rhodium catalyzed hydroformylation-reductive amination or “hydroaminomethylation”1 of α-olefins (Scheme 1, eqn (1)) has emerged as an important method for the synthesis of N-containing compounds, including pharmaceutical ingredients (e.g. cinacalcet,2,3a ibutilide,2,3b and fexofenadine2,3c). Following its discovery at BASF in 1949 by Reppe,4 hydroaminomethylation initially received only a modest level of attention from academic and industrial researchers.5 The systematic studies of Eilbracht in the late 1990's6 brought rhodium catalyzed hydroaminomethylation to the forefront of research, and in the last 15 years significant progress in this area was made. Notable achievements include the design of catalytic systems enabling direct use of ammonia,6c,7 the ability to control regioselectivity in reactions of terminal8a as well as internal8b alkenes via ligand control8 or use of directing groups,9 and the development of the first ruthenium catalyzed carbonylative hydroaminomethylations.10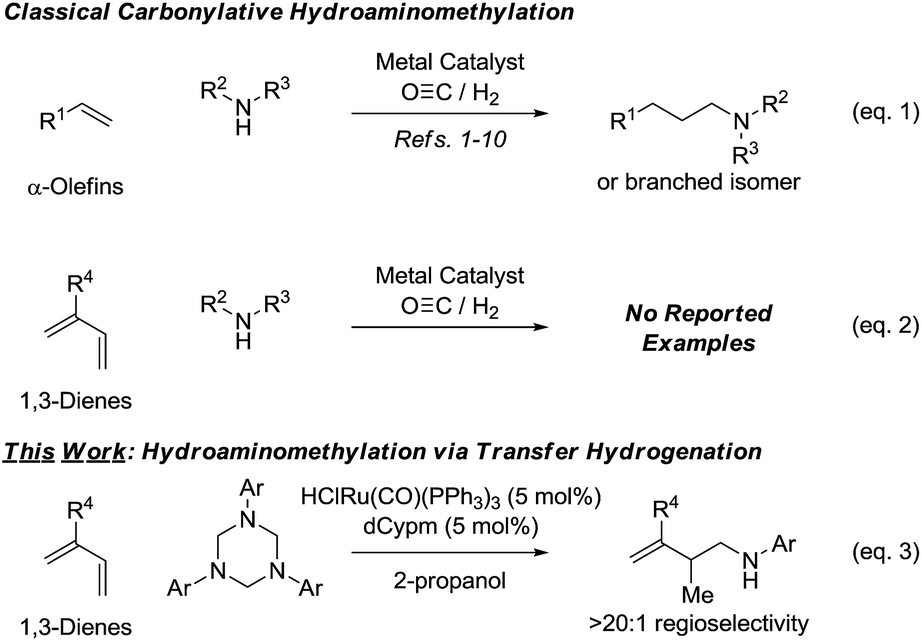 | ||
| Scheme 1 Hydroaminomethylation via carbonylation or 2-propanol mediated reductive coupling. | ||
Despite these advances, existing catalysts for hydroaminomethylation via hydroformylation-reductive amination are restricted to the use of nonconjugated alkenes, typically α-olefins. The carbonylative hydroaminomethylation of other π-unsaturated reactants, such as 1,3-dienes, has not been reported, as regioselectivity and “over-hydroformylation” to form dialdehydes are difficult to control (Scheme 1, eqn (2)).11 In connection with our exploration of hydrogenation and transfer hydrogenation in the context of reductive C–C coupling, we have found that paraformaldehyde serves as a convenient and inexpensive C1-building block for the hydrohydroxymethylation of 1,3-dienes,12 allenes13,14 and alkynes.15 Most importantly, reductive couplings of paraformaldehyde provide access to products of hydrohydroxymethylation that cannot be formed selectively under hydroformylation conditions.16
These results supported the feasibly of corresponding hydroaminomethylations wherein π-unsaturated reactants are reductively coupled with formaldimines. In proof-of-concept studies, it was found that 1,1-disubstituted allenes engage in regioselective reductive coupling with formaldimines derived in situ through cracking of 1,3,5-tris(aryl)-hexahydro-1,3,5-triazines under the conditions of ruthenium catalyzed transfer hydrogenation employing 2-propanol as terminal reductant.17 Corresponding hydroaminomethylations of 1,3-dienes such as butadiene, isoprene and myrcene, which are important feedstock chemicals, would be even more desirable, however, competing aza-Diels–Alder cycloaddition18 and alkene isomerization19 of the homoallylic amines products rendered the outcome of such processes uncertain. Here, we report that ruthenium complexes modified by dCypm (bis(dicyclohexylphosphino)methane) catalyze the 2-propanol mediated reductive coupling of 2-substituted 1,3-dienes with 1,3,5-tris(aryl)-hexahydro-1,3,5-triazines to form products of hydroaminomethylation as single regioisomers with complete suppression of olefin isomerization in all but one case (Scheme 1, eqn (3)). These transformations represent the first examples of diene hydroaminomethylation.20,21
Results and discussion
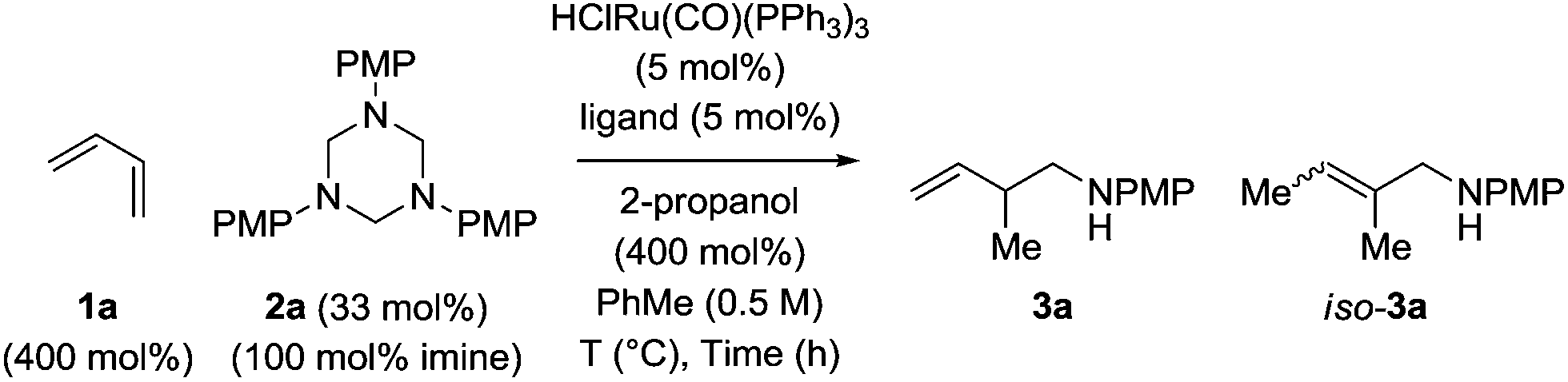
Hexahydro-1,3,5-triazine 2a is a white, crystalline solid conveniently prepared through the condensation of paraformaldehyde and para-anisidine.22 In an initial experiment, butadiene 1a was exposed to hexahydro-1,3,5-triazine 2a in the presence of 2-propanol (400 mol%) and commercial HClRu(CO)(PPh3)3 (5 mol%) in toluene solvent (0.5 M) at 120 °C. The targeted product of hydroaminomethylation 3a was obtained as a single isomer in 11% yield (Table 1, entry 1). A series of phosphine ligands were evaluated for their ability to enhance conversion. The isolated yield of 3a was not improved upon use of the monodentate phosphine ligands, for example PCy3 (Table 1, entry 2). A series of chelating bis(diphenylphosphino)-substituted ligands were screened, including dppf, dppm and dppe (Table 1, entries 3–5). The isolated yield of 3a was increased to 81% using dppe, however, substantial quantities of olefin isomerization product, allylic amine iso-3a, was formed (Table 1, entry 5).19 The chelating ligands dCypm and dCype, which incorporate bis(dicyclohexylphosphino) moieties, provided superior results, delivering the homoallylic amine 3a in 86% (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3a:iso-3a) and 71% (20:1, 3a:iso-3a) isolated yields, respectively (Table 1, entries 6 and 7). Attempts to enhance the performance of the dCypm-modified catalyst through variation of reaction temperature (Table 1, entries 8 and 9) or reaction time (Table 1, entry 10) did not avail additional improvement (Table 2).
:1, 3a:iso-3a) and 71% (20:1, 3a:iso-3a) isolated yields, respectively (Table 1, entries 6 and 7). Attempts to enhance the performance of the dCypm-modified catalyst through variation of reaction temperature (Table 1, entries 8 and 9) or reaction time (Table 1, entry 10) did not avail additional improvement (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | T (°C) | Time (h) | Yield 3a |
3a:iso-3a |
| a Yields are of material isolated by silica gel chromatography. Isomeric ratios were determined via1H NMR analysis. PMP = para-methoxyphenyl, dppf (1,1′-bis(diphenylphosphino)ferrocene), dppm (bis(diphenylphosphino)methane), dppe (1,2-bis(diphenylphosphino)ethane), dCypm (bis(dicyclohexylphosphino)methane), dCype (1,2-bis(dicyclohexylphosphino)ethane). b PCy3 (10 mol%). See ESI† for further details. | |||||
| 1 | — | 120 | 24 | 11% | >20:1 |
| 2b | PCy3 | 120 | 24 | 10% | >20:1 |
| 3 | dppf | 120 | 24 | 19% | >20:1 |
| 4 | dppm | 120 | 24 | 34% | 4:1 |
| 5 | dppe | 120 | 24 | 81% | 4:1 |
| 6 | dCypm | 120 | 24 | 86% |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 7 | dCype | 120 | 24 | 71% | 20:1 |
| 8 | dCypm | 110 | 24 | 65% | 10:1 |
| 9 | dCypm | 140 | 24 | 82% | 8:1 |
| 10 | dCypm | 120 | 12 | 74% | 10:1 |
|
|
|||
|---|---|---|---|
| Entry | 1,3-Diene | Product | Yield (3:iso-3) |
| a Yields are of material isolated by silica gel chromatography. Isomeric ratios were determined via1H NMR analysis. b PhMe (0.5 M), 120 °C. See ESI† for further experimental details. | |||
| 1b |
|

|
86% yield 10:1 (3a:iso-3a) |
| 2 |
|
|
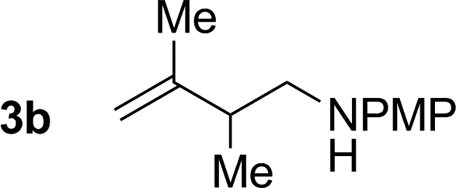
79% yield >20:1 (3b:iso-3b) |
| 3 |

|
|
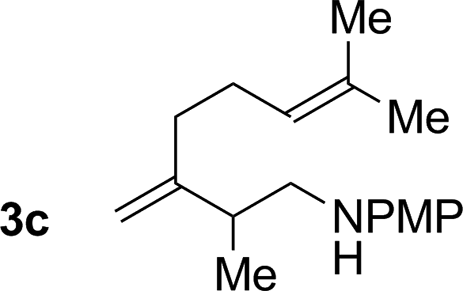
76% yield >20:1 (3c:iso-3c) |
| 4 |
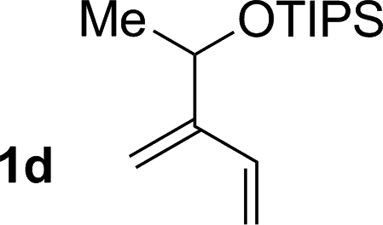
|
|
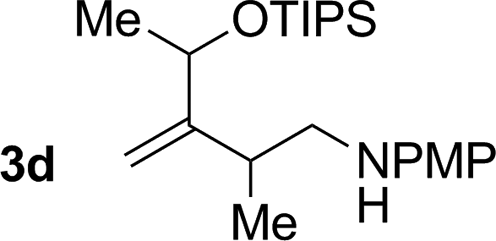
71% yield, 2:1 dr >20:1 (3d:iso-3d) |
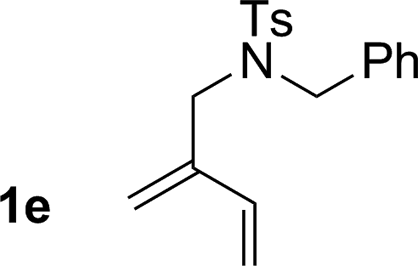
| 5 |
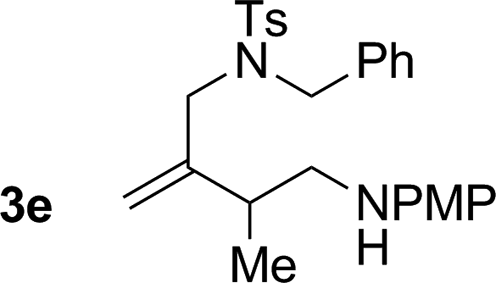
|
|
74% yield >20:1 (3e:iso-3e) |
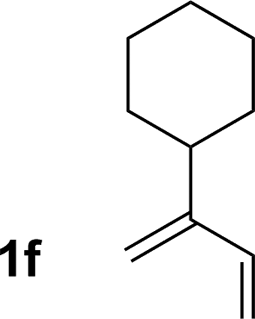
| 6 |
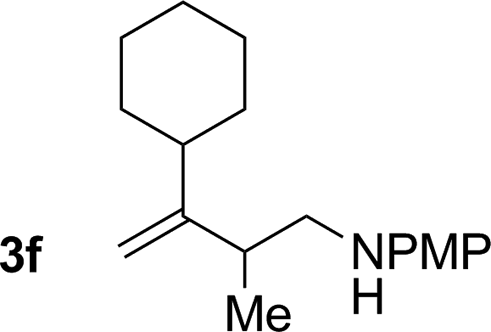
|
|
70% yield >20:1 (3f:iso-3f) |
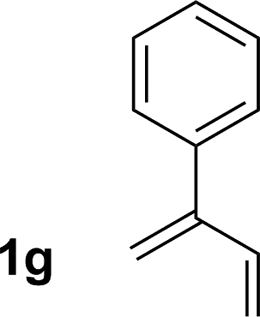
| 7 |
|

|
77% yield >20:1 (3g:iso-3g) |
| 8 |

|
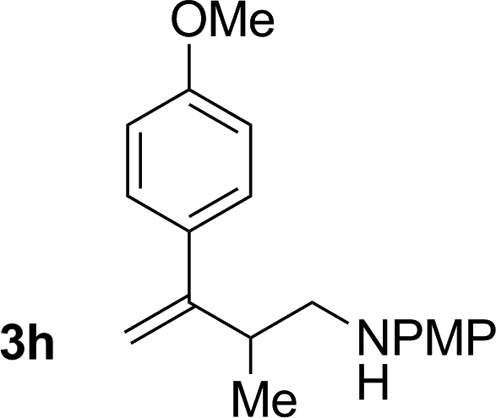
|
72% yield >20:1 (3h:iso-3h) |
| 9 |
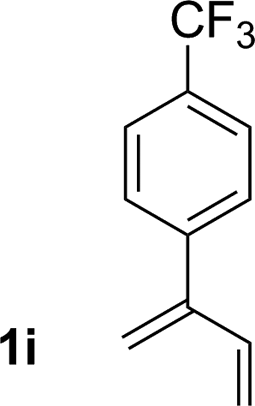
|
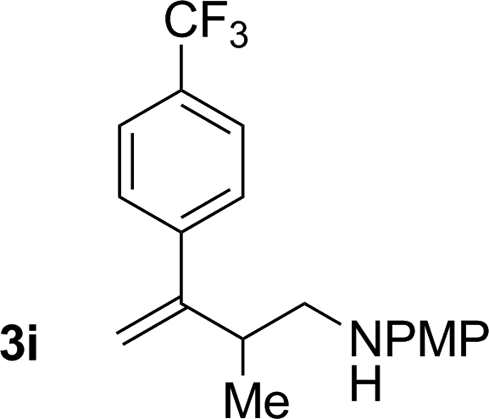
|
81% yield >20:1 (3i:iso-3i) |
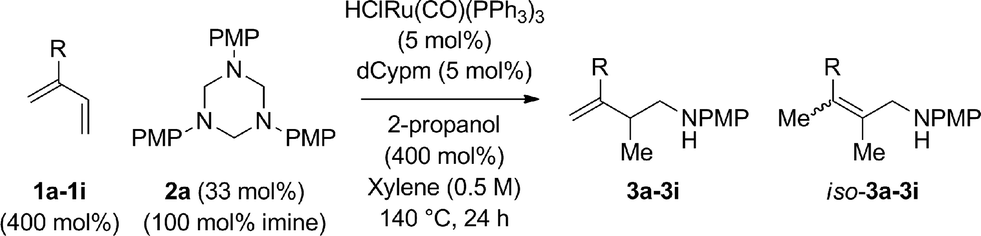
An attempt was made to apply these optimal conditions (Table, entry 6) to a series of 2-substituted 1,3-dienes 1b–1i, however, at 120 °C the desired products 3b–3i were accompanied by significant quantities of the corresponding aza-Diels–Alder [4 + 2] cycloadducts.18 2-Substituted dienes display an enhanced conformational preference for the s-cis conformer, which increases their rate of Diels–Alder cycloaddition relative to butadiene. At slightly higher temperatures (140 °C in xylene solvent), competing Diels–Alder reaction decelerates with respect to hydroaminomethylation and could be completely suppressed. Using these slightly modified conditions, 2-substituted 1,3-dienes 1b–1i were reacted with hexahydro-1,3,5-triazine 2a to furnish the products of hydroaminomethylation 3b–3i in good yield as single regioisomers, and isomeric allylic amines iso-3b–3i were not observed. Notably, a range of substituents are tolerated at the 2-position of the diene, including branched aliphatic moieties (1d, 1f), groups with allylic heteroatoms (1d, 1e) and aryl groups (1g–1i). Under the present conditions, 1-substituted dienes engage in reductive coupling, however, lower conversions and selectivities were observed. To illustrate the utility of homoallylic amines 3a–3i, the hydroaminomethylation product 3b was transformed into the trisubstituted piperidine 4bvia Prins reaction with glyoxylic acid mono-hydrate (Scheme 2, eqn (1)).23 Additionally, adduct 3i was subjected to N-allylation and ring closing metathesis to form the disubstituted piperidine 4i (Scheme 2, eqn (2)).24
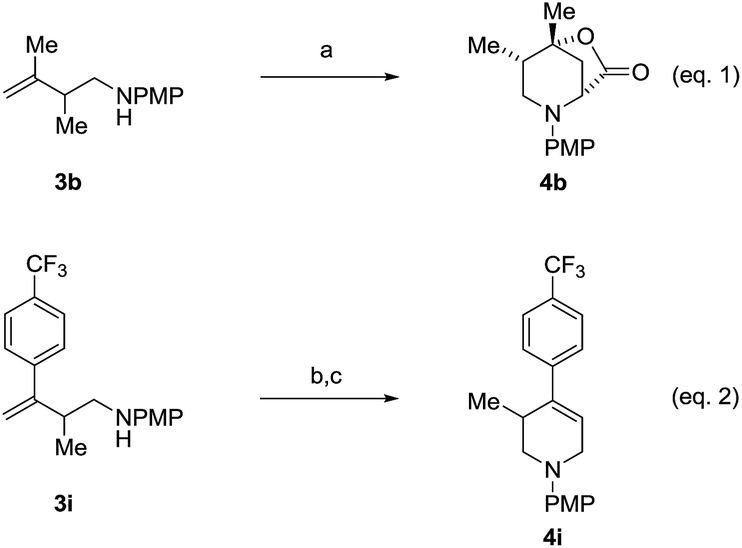 | ||
Scheme 2 Conversion of hydroaminomethylation products 3b and 3i to compounds 4b and 4i, respectively. aYields are of material isolated by silica gel chromatography. (a) (HO)2CCO2H, MeCN–H2O, 25 °C, 80% yield, 10:1 dr (b) BrCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, K2CO3, DMF, 25 °C, 75% yield. (c) Grubbs-II, DCM, 40 °C, 72% yield. See ESI† for further experimental details. CH2, K2CO3, DMF, 25 °C, 75% yield. (c) Grubbs-II, DCM, 40 °C, 72% yield. See ESI† for further experimental details. | ||
Variation of the 1,3,5-tris(aryl)-hexahydro-1,3,5-triazine was subsequently investigated in the hydroaminomethylation of butadiene 1a (Table 3). N-Aryl substituted triazines 2a–2f were subjected to optimal conditions identified for the hydroaminomethylation of butadiene using triazine 2a (Table 1, entry 6). Electron rich N-aryl triazines 2a–2c, including ortho-substituted triazine 2c, undergo hydroaminomethylation efficiently to afford the branched homoallylic amines 3a–5a with complete regiocontrol. In each case, small quantities of the allylic amines iso-3a–5a were observed as side-products. Electron neutral triazine 2d and electron deficient triazines 2e and 2f were converted to the respective homoallylic amines 6a, 7a and 8a in good yield, although increased quantities of the allylic amine side-products were observed. As illustrated in the formation of 8a, nitrogen bearing heterocycles are tolerated. Attempted use of N-alkyl, N-acyl and N-sulfonyl triazines failed in the coupling with dienes under these initially developed conditions. It should be noted that the 3a–8a:iso-3a–8a ratio does not change as a function of conversion or reaction time, suggesting olefin isomerization is kinetically controlled, perhaps occurring by way of the homoallylic amidoruthenium intermediate (vida supra).
| a Yields are of material isolated by silica gel chromatography. Isomeric ratios were determined via1H NMR analysis. See ESI† for further experimental details. |
|---|
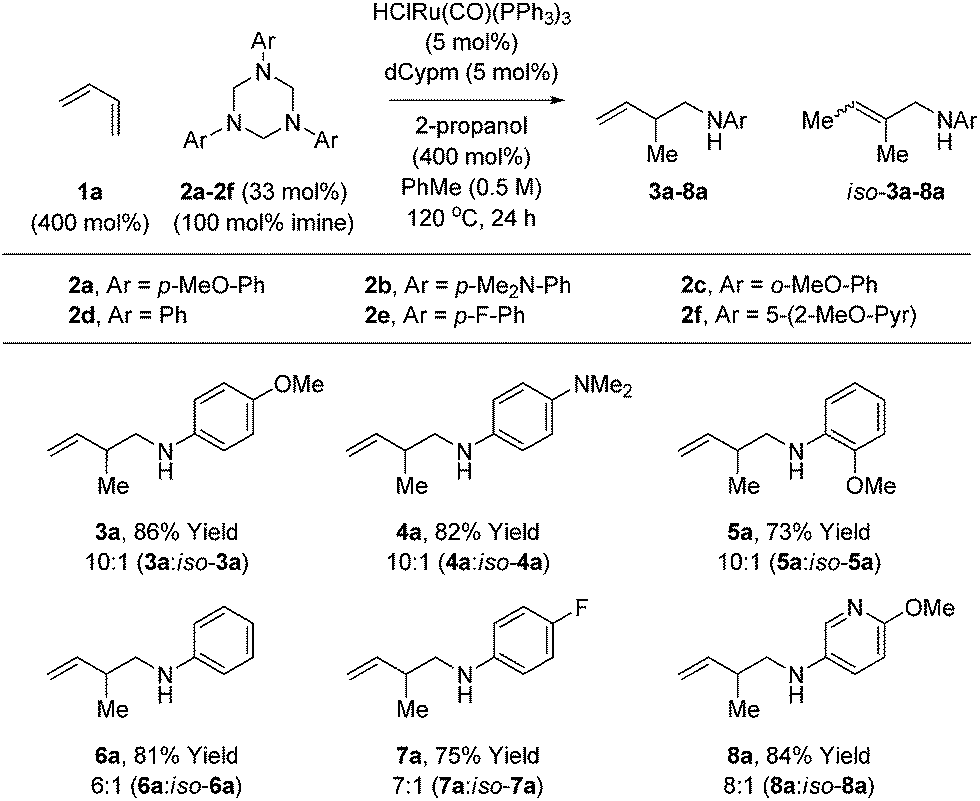
|
Having established favourable conditions for diene hydroaminomethylation, enantioselective variants were investigated in reactions of butadiene 1a. A survey of triazines 2a–2f revealed that triazine 2c derived from ortho-anisidine provided the highest levels of enantiomeric enrichment. Among various chiral phosphine ligands, (R)-MeO-furyl-BIPHEP provided the highest levels of enantiocontrol. In the presence of this chiral ligand, the reaction of 1,3-butadiene 1a with N-ortho-methoxyphenyl triazine 2c delivered the homoallylic amine 5a in 49% yield as a 94:6 ratio of enantiomers in the absence of allylic amine side-product iso-5a (Scheme 3). Application of these initially developed conditions for enantioselective hydroaminomethylation to isoprene resulted in the formation of homoallylic amine 5b in 54% yield as a 93:7 ratio of enantiomers (Scheme 3). The absolute stereochemical assignment of 5a was determined by single crystal X-ray diffraction analysis of the corresponding 4-nitrobenzenesulfonamide.
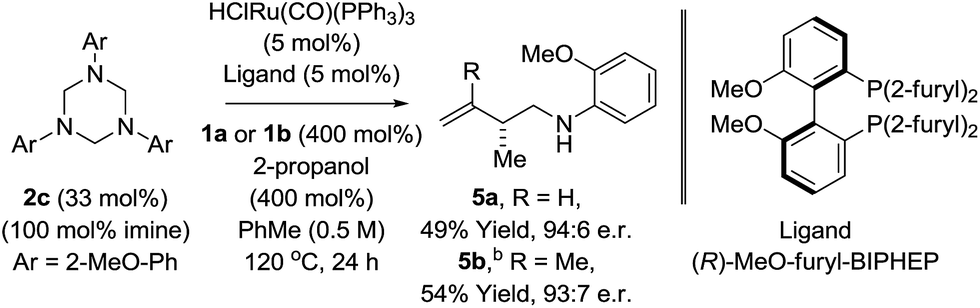 | ||
| Scheme 3 Enantioselective ruthenium catalyzed hydroaminomethylation of butadiene 1a and isoprene 1b. aYields are of material isolated by silica gel chromatography. Enantiomeric ratios were determined by chiral stationary phase HPLC analysis. bXylene (0.5 M), 140 °C. See ESI† for further experimental details. | ||
Mechanistic studies
To illuminate key features of the catalytic mechanism, deuterium labelling studies of the ruthenium catalyzed hydroaminomethylation of isoprene 1b were performed (Scheme 4). Hydroaminomethylation of isoprene 1b using the deuterated triazine deuterio-2a provided deuterio-3b with complete retention of deuterium at the methylene carbon bearing nitrogen (>95% 2H). Deuterium was not detected at any other position. This experiment suggests deuterio-3b is inert with respect to amine dehydrogenation under these conditions. In a second experiment, isoprene 1b was subjected to hydroaminomethylation using triazine 2a in the presence of d8-2-propanol. As anticipated, the product deuterio-3b incorporates significant quantities of deuterium at the methyl group (65% 2H). However, deuterium also is incorporated at the terminal vinylic positions (21% 2H) and, to a lesser extent, the allylic position (10% 2H). These data corroborate a scenario wherein rapid, reversible and non-regioselective diene hydrometalation occurs in advance of turn-over limiting imine addition. Reversible hydrometalation accounts for incomplete deuterium incorporation. Adventitious water also may diminish the extent of deuterium incorporation.25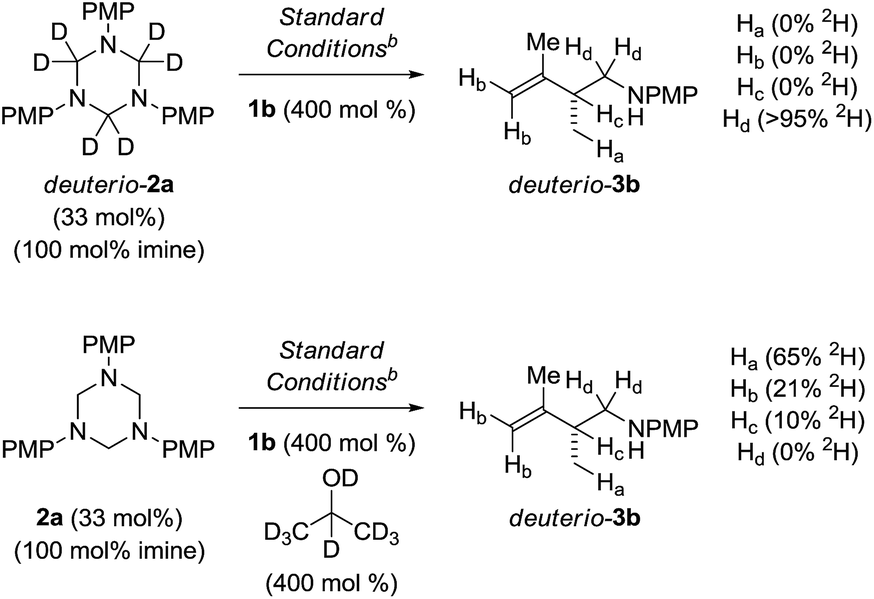 | ||
| Scheme 4 Deuterium labelling studies of the ruthenium catalyzed hydroaminomethylation of isoprene 1b. aYields are of material isolated by silica gel chromatography. bXylene (0.5 M), 140 °C. See ESI† for further experimental details. | ||
Guided by these data, a mechanism for ruthenium catalyzed diene hydroaminomethylation via transfer hydrogenation was proposed (Scheme 5). Diene hydroruthenation delivers a nucleophilic allylruthenium complex. The stoichiometric reaction of HXRu(CO)(PPh3)3 (X = Cl, Br) with dienes (or allenes) to form π-allylruthenium complexes has been reported.26 In the case of isoprene 1b,26acis-stereochemistry between the methyl groups of the resulting π-allyl are observed. Notably, HClRu(CO)(PPh3)3 hydrometalates 1,1-dimethylallene to initially form a 1,1-dimethyl substituted π-allylruthenium complex that rearranges to the cis-1,2-dimethyl substituted π-allylruthenium complex,26c suggesting cis-stereochemistry represents a thermodynamic rather than kinetic preference. Intervention of a single geometrical isomer at the stage of the σ-allylruthenium intermediate and ensuing transition state for imine addition appears consistent with the relatively high levels of enantioselectivity observed in the asymmetric hydroaminomethylation of isoprene (Scheme 3). Protonolytic cleavage of the amidoruthenium complex derived upon imine addition mediated by isopropanol releases the product of hydroaminomethylation 3b and regenerates the ruthenium hydride to close the catalytic cycle.
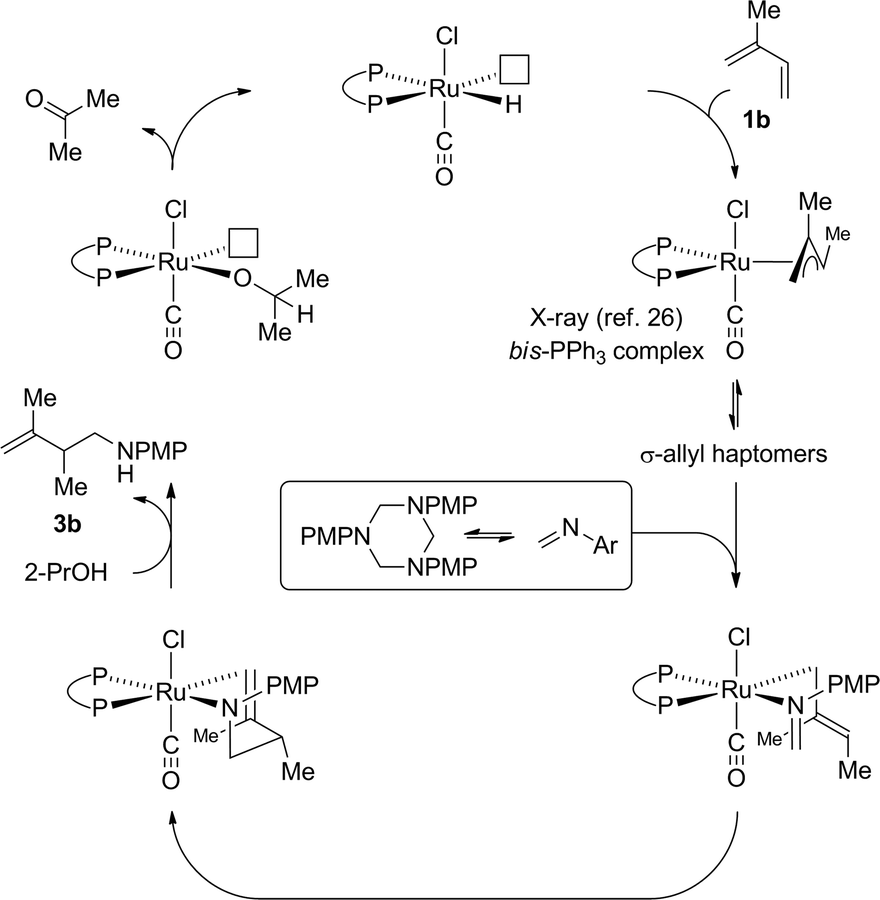 | ||
| Scheme 5 General mechanism for ruthenium catalyzed diene hydroaminomethylation via transfer hydrogenation. | ||
Conclusion
In summary, using the concepts of redox-triggered CX (X = O, N) addition,27 we report the first examples of diene hydroaminomethylation, including asymmetric variants. Specifically, ruthenium catalyzed transfer hydrogenation of 1,3-dienes in the presence of tris(aryl)-hexahydro-1,3,5-triazines results in diene–formaldimine reductive coupling to deliver homoallylic amines in good yield with complete levels of regioselectivity. While further optimization is required to enhance performance, these processes define an alternative to classical carbonylative hydroaminomethylation via hydroformylation-reductive amination, which is presently limited to reactions of non-conjugated olefins. More broadly, these studies contribute to an ever-growing body of catalytic C–C bond formations that merge the characteristics of carbonyl and imine addition with transfer hydrogenation.27
Acknowledgements
The Robert A. Welch Foundation (F-0038), the NSF (CHE-1265504) and the University of Texas Center for Green Chemistry and Catalysis are acknowledged for partial support of this research. The Deutsche Forschungsgemeinschaft (DFG) postdoctoral fellowship program (FR 3555/1-1) is acknowledged for partial support (J.F.).Notes and references
- For selected reviews on hydroaminomethylation, see: (a) P. Eilbracht, L. Bärfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rishe, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329 CrossRef CAS PubMed; (b) B. Breit and W. Seiche, Synthesis, 2001, 1 CrossRef CAS; (c) P. Eilbracht and A. M. Schmidt, Top. Organomet. Chem., 2006, 18, 65 CrossRef CAS; (d) D. Crozet, M. Urrutigoity and P. Kalck, ChemCatChem, 2011, 3, 1102 CrossRef CAS; (e) A. Behr and A. J. Vorholt, Top. Organomet. Chem., 2012, 39, 103 CrossRef CAS; (f) S. Raoufmoghaddam, Org. Biomol. Chem., 2014, 12, 7179 RSC; (g) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041 CrossRef CAS PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS PubMed.
- (a) O. Thiel, C. Bernard, R. Larsen, M. J. Martinelli and M. T. Raza, (Amgen Inc.) WO Patent 2009002427, 2008transChem. Abstr., 2007, 71, 12990 Search PubMed; (b) J. R. Briggs, J. Klosin and G. T. Whiteker, Org. Lett., 2005, 7, 4795 CrossRef CAS PubMed; (c) G. Whiteker, Top. Catal., 2010, 53, 1025 CrossRef CAS.
- Reppe's pioneering studies employ near stoichiometric loadings of Fe(CO)5: (a) W. Reppe, Experientia, 1949, 5, 93 CrossRef CAS; (b) W. Reppe and H. Vetter, Liebigs Ann. Chem., 1953, 582, 133 CrossRef CAS.
- (a) H.de V. Finch and R. E. Meeker, (Shell Oil Company) US Pat., 3234283, 1966transChem. Abstr., 1965, 62, 14500b Search PubMed; (b) G. Biale, (Union Oil Company) US Pat., 3513200, 1970transChem. Abstr., 1970, 73, 34776a Search PubMed; (c) A. F. M. Iqbal, Helv. Chim. Acta, 1971, 54, 1440 CrossRef CAS; (d) T. Imai, (Uop Inc.) US. Pat., 4220764, 1978transChem. Abstr., 1980, 93, 239429d Search PubMed; (e) R. M. Laine, J. Org. Chem., 1980, 45, 3370 CrossRef CAS; (f) F. Jachimowicz, (W. R. Grace and Co.) Belgian Patent 887630, 1980transChem. Abstr., 1981, 95, 152491k Search PubMed; (g) F. Jachimowicz and J. W. Raksis, J. Org. Chem., 1982, 47, 445 CrossRef CAS; (h) K. Murata, A. Matsuda and T. Masuda, J. Mol. Catal., 1984, 23, 121 CrossRef CAS; (i) F. Jachimowicz and P. Manson, (W. R. Grace and Co.) Canadian Patent 123 1199, 1984transChem. Abstr., 1988, 109, 38485u Search PubMed; (j) E. E. MacEntire and J. F. Knifion, (Texaco Development Corp.) EP 240193, 1987transChem. Abstr., 1989, 110, 134785h Search PubMed; (k) M. D. Jones, J. Organomet. Chem., 1989, 366, 403 CrossRef CAS; (l) E. Drent and A. J. M. Breed, (Shell Int. Res. M.) EP 457386, 1992transChem. Abstr., 1992, 116, 83212h Search PubMed; (m) S. Törös, I. Gémes-Pésci, B. Heil, S. Mahó and Z. Tuba, J. Chem. Soc., Chem. Commun., 1992, 858 RSC; (n) T. Baig and P. Kalck, J. Chem. Soc., Chem. Commun., 1992, 1373 RSC; (o) T. Baig, J. Molinier and P. Kalck, J. Organomet. Chem., 1993, 455, 219 CrossRef CAS; (p) G. Diekhaus, D. Kampmann, C. Kniep, T. Müller, J. Walter and J. Weber, (Hoechst AG) DE 4334809, 1993transChem. Abstr., 1995, 122, 314160g Search PubMed; (q) J. J. Brunet, D. Neibecker, F. Agbossou and R. S. Srivastava, J. Mol. Catal., 1994, 87, 223 CrossRef CAS; (r) M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpainter, J. Mol. Catal. A: Chem., 1995, 104, 17 CrossRef CAS.
- (a) T. Rische and P. Eilbracht, Synthesis, 1997, 1331 CrossRef CAS; (b) C. L. Kranemann and P. Eilbracht, Synthesis, 1998, 71 CrossRef CAS; (c) T. Rische, B. Kitsos-Rzychon and P. Eilbracht, Tetrahedron, 1998, 54, 2723 CrossRef CAS; (d) L. Bärfacker, C. Hollmann and P. Eilbracht, Tetrahedron, 1998, 54, 4493 CrossRef; (e) T. Rische and P. Eilbracht, Tetrahedron, 1998, 54, 8441 CrossRef CAS; (f) L. Bärfacker, D. El Tom and P. Eilbracht, Tetrahedron Lett., 1999, 40, 4031 CrossRef; (g) C. L. Kranemann, B. Costisella and P. Eilbracht, Tetrahedron Lett., 1999, 40, 7773 CrossRef CAS; (h) T. Rische, L. Bärfacker and P. Eilbracht, Eur. J. Org. Chem., 1999, 653 CrossRef CAS; (i) P. Eilbracht, C. L. Kranemann and L. Bärfacker, Eur. J. Org. Chem., 1999, 1907 CrossRef CAS; (j) T. Rische and P. Eilbracht, Tetrahedron, 1999, 55, 1915 CrossRef CAS; (k) T. Rische and P. Eilbracht, Tetrahedron, 1999, 55, 3917 CrossRef CAS; (l) C. L. Kranemann, B. E. Kitsos-Rzychon and P. Eilbracht, Tetrahedron, 1999, 55, 4721 CrossRef CAS; (m) L. Bärfacker, T. Rische and P. Eilbracht, Tetrahedron, 1999, 55, 7177 CrossRef; (n) T. Rische and P. Eilbracht, Tetrahedron, 1999, 55, 7841 CrossRef CAS; (o) T. Rische, K.-S. Müller and P. Eilbracht, Tetrahedron, 1999, 55, 9801 CrossRef CAS; (p) C. L. Kranemann and P. Eilbracht, Eur. J. Org. Chem., 2000, 2367 CrossRef CAS; (q) A. Behr, M. Fiene, C. Buss and P. Eilbracht, Eur. J. Lipid Sci. Technol., 2000, 102, 467 CrossRef CAS.
- (a) B. Zimmermann, J. Herwig and M. Beller, Angew. Chem., Int. Ed., 1999, 38, 2372 CrossRef CAS; (b) B. Zimmermann, J. Herwig and M. Beller, Chem. Ind., 2001, 82, 521 CAS; (c) H. Klein, R. Jackstell, M. Kant, A. Martin and M. Beller, Chem. Eng. Technol., 2007, 30, 721 CrossRef CAS.
- (a) M. Ahmed, A. M. Seayad, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2003, 125, 10311 CrossRef CAS PubMed; (b) M. Ahmed, R. P. J. Bronger, R. Jackstell, P. C. J. Kamer, P. W. N. M. van Leeuwen and M. Beller, Chem.–Eur. J., 2006, 12, 8979 CrossRef CAS PubMed.
- B. Breit, Tetrahedron Lett., 1998, 39, 5163 CrossRef CAS.
- L. Wu, I. Fleischer, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2013, 135, 3989 CrossRef CAS PubMed.
- For selected examples of 1,3-diene hydroformylation, see: (a) W. H. Clement and M. Orchin, Ind. Eng. Chem. Prod. Res. Dev., 1965, 4, 283 CrossRef CAS; (b) B. Fell and H. Bahrmann, J. Mol. Catal., 1977, 2, 211 CrossRef CAS; (c) H. Bahrmann and B. Fell, J. Mol. Catal., 1980, 8, 329 CrossRef CAS; (d) C. Botteghi, M. Branca and A. Saba, J. Organomet. Chem., 1980, 184, C17–C19 CrossRef CAS; (e) P. W. N. M. van Leeuwen and C. F. Roobeek, J. Mol. Catal., 1985, 31, 345 CrossRef CAS; (f) J. C. Chalchat, R. Ph. Garry, E. Lecomte and A. Michet, Flavour Fragrance J., 1991, 6, 179 CrossRef CAS; (g) S. Bertozzi, N. Campigli, G. Vitulli, R. Lazzaroni and P. Salvadori, J. Organomet. Chem., 1995, 487, 41 CrossRef CAS; (h) T. Horiuchi, T. Ohta, K. Nozaki and H. Takaya, Chem. Commun., 1996, 155 RSC; (i) T. Horiuchi, T. Ohta, E. Shirakawa, K. Nozaki and H. Takaya, Tetrahedron, 1997, 53, 7795 CrossRef CAS; (j) P. Liu and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 10772 CrossRef CAS PubMed; (k) H. J. V. Barros, B. E. Hanson, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2004, 278, 57 CrossRef CAS; (l) H. J. V. Barros, J. G. da Silva, C. C. Guimarães, E. N. dos Santos and E. V. Gusevskaya, Organometallics, 2008, 27, 4523 CrossRef CAS; (m) A. L. Watkins and C. R. Landis, Org. Lett., 2011, 13, 164 CrossRef CAS PubMed; (n) T. E. Smith, S. J. Fink, Z. G. Levine, K. A. McClelland, A. A. Zackheim and M. E. Daub, Org. Lett., 2012, 14, 1452 CrossRef CAS PubMed.
- For catalytic reductive coupling of 1,3-dienes with paraformaldehyde, see: (a) T. Smejkal, H. Han, B. Breit and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 10366 CrossRef CAS PubMed; (b) A. Köpfer, B. Sam, B. Breit and M. J. Krische, Chem. Sci., 2013, 4, 1876 RSC.
- For catalytic reductive coupling of allenes to paraformaldehyde, see: (a) M.-Y. Ngai, E. Skucas and M. J. Krische, Org. Lett., 2008, 10, 2705 CrossRef CAS PubMed; (b) B. Sam, T. P. Montgomery and M. J. Krische, Org. Lett., 2013, 15, 3790 CrossRef CAS PubMed.
- For catalytic redox-neutral coupling of allenes with methanol, see: J. Moran, A. Preetz, R. A. Mesch and M. J. Krische, Nat. Chem., 2011, 3, 287 CrossRef CAS PubMed.
- C. C. Bausch, R. L. Patman, B. Breit and M. J. Krische, Angew. Chem., Int. Ed., 2011, 50, 5687 CrossRef CAS PubMed.
- For a review on the use of paraformaldehyde and methanol as C1-feedstocks in metal catalyzed C–C coupling, see: B. Sam, B. Breit and M. J. Krische, Angew. Chem., Int. Ed., 2015, 53, 3267 CrossRef PubMed.
- S. Oda, B. Sam and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 8525 CrossRef CAS PubMed.
- For selected examples of the thermal Diels–Alder [4 + 2] cycloaddition of 1,3-dienes with formaldimines, see: (a) S. D. Larsen and P. A. Grieco, J. Am. Chem. Soc., 1985, 107, 1768 CrossRef CAS; (b) L. Bhat, A. G. Steinig, R. Appelbe and A. De Meijere, Eur. J. Org. Chem., 2001, 1673 CrossRef CAS.
- For selected examples of ruthenium catalyzed alkene isomerization, see: (a) C. Cadot, P. I. Dalko and J. Cossy, Tetrahedron Lett., 2002, 43, 1839 CrossRef CAS; (b) T. J. Donohoe, T. J. C. O'Riordan and C. P. Rosa, Angew. Chem., Int. Ed., 2009, 48, 1014 CrossRef CAS PubMed; (c) C. R. Larsen and D. B. Grotjahn, J. Am. Chem. Soc., 2012, 134, 10357 CrossRef CAS PubMed; (d) J. R. Clark, J. R. Griffiths and S. T. Diver, J. Am. Chem. Soc., 2013, 135, 3327 CrossRef CAS PubMed.
- For nickel catalyzed diene–imine reductive couplings and related multi-component processes, see: (a) M. Kimura, A. Miyachi, K. Kojima, S. Tanaka and Y. Tamaru, J. Am. Chem. Soc., 2004, 126, 14360 CrossRef CAS PubMed; (b) K. Kojima, M. Kimura and Y. Tamaru, Chem. Commun., 2005, 4717 RSC; (c) M. Kimura, K. Kojima, Y. Tatsuyama and Y. Tamaru, J. Am. Chem. Soc., 2006, 128, 6332 CrossRef CAS PubMed; (d) M. Kimura, Y. Tatsuyama, K. Kojima and Y. Tamaru, Org. Lett., 2007, 9, 1871 CrossRef CAS PubMed.
- For the ruthenium catalyzed C–C coupling of 1,3-dienes with carbonyl partners or imines, see: (a) F. Shibahara, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6338 CrossRef CAS PubMed; (b) J. R. Zbieg, E. Yamaguchi, E. L. McInturff and M. J. Krische, Science, 2012, 336, 324 CrossRef CAS PubMed; (c) S. Zhu, X. Lu, Y. Luo, W. Zhang, H. Jiang, M. Yan and W. Zeng, Org. Lett., 2013, 15, 1440 CrossRef CAS PubMed; (d) T.-Y. Chen, R. Tsutsumi, T. P. Montgomery, I. Volchkov and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 1798 CrossRef CAS PubMed.
- (a) C. A. Bischoff and F. Reinfeld, Chem. Ber., 1903, 36, 41 CrossRef CAS; (b) A. G. Giumanini, G. Verardo, E. Zangrando and L. Lassiani, J. Prakt. Chem., 1987, 329, 1087 CrossRef CAS; (c) A. G. Giumanini, N. Toniutti, G. Verardo and M. Merli, Eur. J. Org. Chem., 1999, 141 CrossRef CAS.
- D. J. Bennet and N. M. Hamilton, Tetrahedron Lett., 2000, 41, 7961 CrossRef.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef CAS PubMed.
- S. K. S. Tse, P. Xue, Z. Lin and G. Jia, Adv. Synth. Catal., 2010, 352, 1512 CrossRef CAS.
- For the stoichiometric reaction of HXRu(CO)(PPh3)3 (X = Cl, Br) with dienes or allenes to furnish π-allylruthenium complexes, see: (a) K. Hiraki, N. Ochi, Y. Sasada, H. Hayashida, Y. Fuchita and S. Yamanaka, J. Chem. Soc., Dalton Trans., 1985, 873 RSC; (b) A. F. Hill, C. T. Ho and J. D. E. T. Wilton-Ely, Chem. Commun., 1997, 2207 RSC; (c) P. Xue, S. Bi, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Organometallics, 2004, 23, 4735 CrossRef CAS.
- J. M. Ketcham, I. Shin, T. P. Montgomery and M. J. Krische, Angew. Chem., Int. Ed., 2014, 53, 9142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data for new compounds, including scanned images of 1H and 13C NMR spectra. Single crystal X-ray diffraction data for a derivative of 5a. CCDC 1430833. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03854e |
| This journal is © The Royal Society of Chemistry 2016 |