
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly phosphorescent platinum(II) emitters: photophysics, materials and biological applications†
Kai
Li
ab,
Glenna So
Ming Tong
a,
Qingyun
Wan
a,
Gang
Cheng
ab,
Wai-Yip
Tong
a,
Wai-Hung
Ang
a,
Wai-Lun
Kwong
a and
Chi-Ming
Che
*ab
aState Key Laboratory of Synthetic Chemistry, Institute of Molecular Functional Materials, HKU-CAS Joint Laboratory on New Materials and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: cmche@hku.hk
bHKU Shenzhen Institute of Research and Innovation, Shenzhen 518053, China
First published on 7th January 2016
Abstract
In recent years a blossoming interest in the synthesis, photophysics and application of phosphorescent Pt(II) complexes, particularly on their uses in bioimaging, photocatalysis and phosphorescent organic light-emitting diodes (OLEDs), has been witnessed. The superior performance of phosphorescent Pt(II) complexes in these applications is linked to their diverse spectroscopic and photophysical properties, which can be systematically modulated by appropriate choices of auxiliary ligands. Meanwhile, an important criterion for the practical application of phosphorescent metal complexes is their stability which is crucial for biological utilization and industrial OLED applications. Taking both the luminescence properties and stability into consideration, chelating ligands having rigid scaffolds and with strong σ-donor atoms are advantageous for the construction of highly robust phosphorescent Pt(II) complexes. The square-planar coordination geometry endows Pt(II) complexes with the intriguing spectroscopic and photophysical properties associated with their intermolecular interactions in both the ground and excited states. In this article, we discuss the design and synthesis of phosphorescent Pt(II) complexes with elaboration on the effects of ligands on the structure and luminescence properties. Based on their photophysical and emission properties, we intend to shed light on the great promise of highly robust phosphorescent Pt(II) emitters in an array of applications from molecular materials to biosensors.
1 Introduction
Phosphorescent transition metal complexes are distinct from pure organic luminophores due to their characteristic long emission lifetime, large absorption–emission Stokes shift, and tuneable excited states. The efficient phosphorescence from transition metal complexes at room temperature is attributed to the heavy atom effect that induces strong spin–orbit coupling (SOC), facilitating both fast intersystem crossing (ISC) and the formally spin-forbidden triplet radiative decay.1,2 In the literature, there are numerous reports on the photophysical and photochemical properties of transition metal complexes, particularly those of Ru(II), Ir(III), and Pt(II).3–7 Platinum, being a third-row transition element, has the second largest SOC constant. In contrast to d6 Ru(II) and Ir(III) complexes that have an octahedral coordination geometry, d8 Pt(II) complexes usually adopt a square planar coordination geometry with open axial coordination sites allowing for structural distortion, inner sphere substrate binding, and intermolecular interactions, all of which can significantly alter the ground state and excited state properties.8 We and others have been working to develop new classes of phosphorescent Pt(II) complexes and explore their applications in luminescent chemosensing,9–12 photocatalysis,13–16 optical limiting,17 bioimaging,18–21 and organic light-emitting diodes (OLEDs)22 (Fig. 1).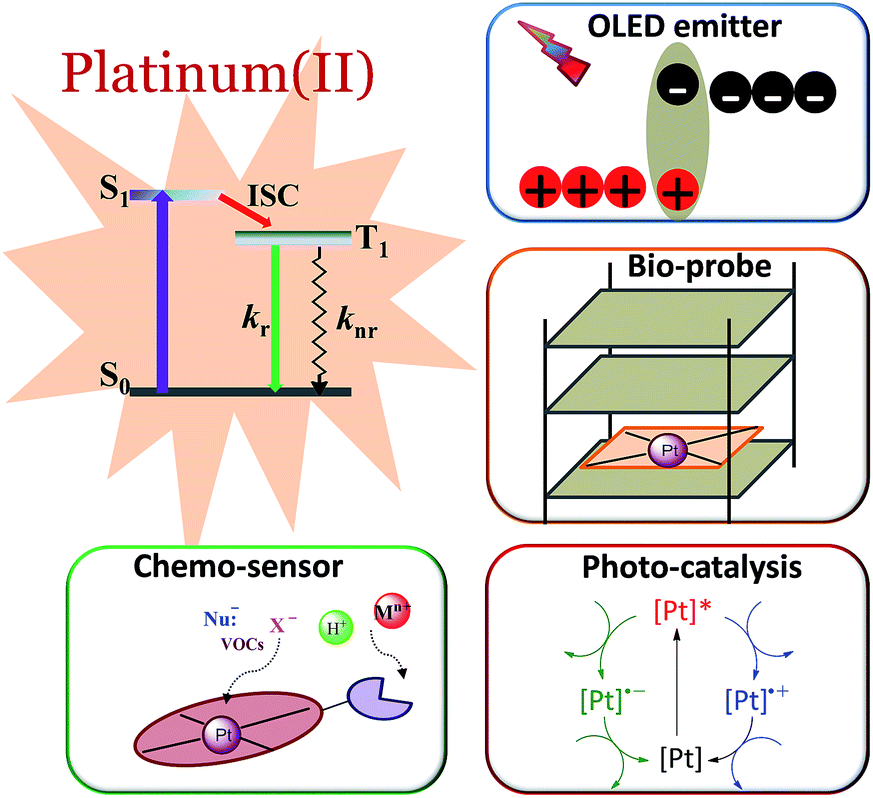 | ||
| Fig. 1 Various applications of phosphorescent Pt(II) complexes. | ||
State-of-the-art phosphorescent OLEDs based on Ir(III) and Pt(II) emitters have exhibited high efficiencies with an external quantum efficiency (EQE) > 20%. However, one of the obstacles in the commercialization of phosphorescent OLEDs is the stability of the various materials used in the device fabrication. Hence, a high robustness of phosphorescent transition metal complexes is essential for enabling their practical application. In this regard, triplet emitters should be (1) tolerant to thermal deposition during device fabrication, (2) resistant to structural rearrangement as encountered in some Ir(III)-OLED emitters during thermal treatment23–25 and (3) stable against chemical degradation that leads to device operational aging.26 As bioimaging agents, the phosphorescent metal complexes should preferably be kinetically inert against ligand-exchange reactions or biological reduction under physiological conditions.18 In photocatalysis, the metal complex acting as the photosensitizer would have to be stable against photobleaching and solvent-induced decomposition in order to afford a high product turnover.27
The robustness of a phosphorescent metal complex is a crucial issue because the presence of electron(s) in the metal–ligand anti-bonding dσ orbitals (for d6 and d8 electronic configuration), either through photoexcitation or thermal population, will decrease the strength of the M–L bond, resulting in its rupture. It is well-documented that the photosubstitution/photodissociation of [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) in solution occurs via thermally populated metal-centred (MC) d–d states.28,29 The rupture of Ir–L bonds via strongly structurally distorted d–d states has been reported to account for the degradation of Ir(III) dopants in OLEDs.26
In this perspective, we describe the factors that affect the non-radiative decay rate and stability of luminescent Pt(II) complexes, followed by a discussion of the recent advances made in phosphorescent Pt(II) complexes which display high emission efficiency and/or high stability. In particular, the latest reports on luminescent Pt(II) complexes supported by tetradentate ligands are summarized. Based on experimental findings and theoretical calculations, we try to correlate superior emission properties and robustness with structure in selected classes of phosphorescent Pt(II) complexes. In addition to being used as phosphorescent OLED dopant materials, the self-assembly and bioimaging applications of luminescent Pt(II) complexes are highlighted.
2 Design principles for highly robust, strongly emissive Pt(II) complexes
2.1 Factors affecting phosphorescence quantum yield
The phosphorescence quantum yield (ϕP) of the triplet excited state is determined by the radiative (kr) and non-radiative (knr) decay rates: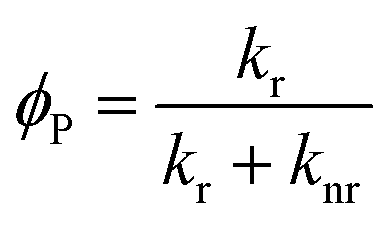
Among all the variables, suppressing the non-radiative decay rate knr is instrumental to achieving efficient phosphorescence. To this end, two main approaches have been employed: (1) decreasing the excited state structural distortion, and (2) altering the coordination environment so as to destabilize the metal-centred (MC) ligand-field excited states.
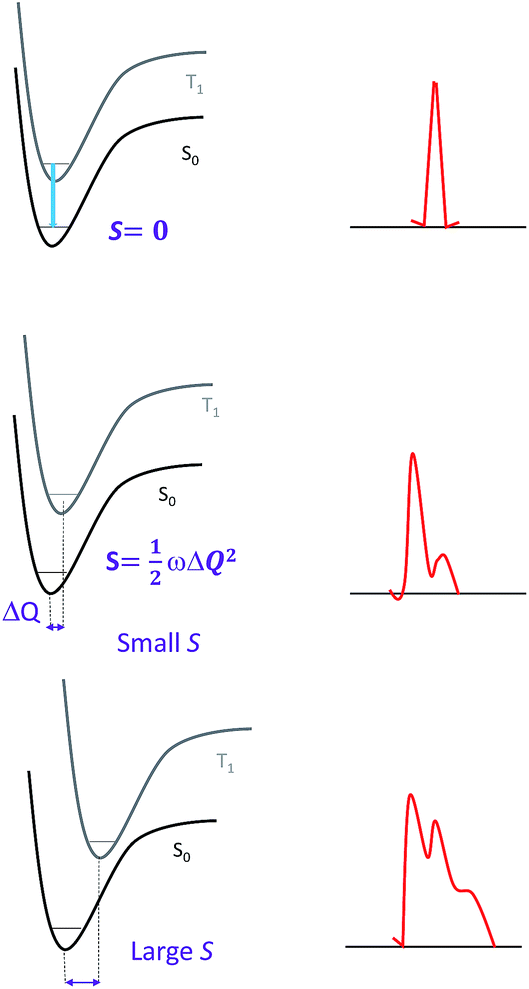 | ||
| Fig. 2 Correlations between the structural distortion of the triplet excited state with respect to the ground state and the emission spectrum: (top) sharp line emission with no structural distortion (S = 0); (middle) narrow bandwidth spectrum with a small structural distortion (i.e., S is small); and (bottom) broad bandwidth spectrum yet with the vibrational signature of a larger structural distortion (i.e., S is large); for an even larger S, only a broadband spectrum is observed. | ||
Since the overall emission bandwidth is affected by the intensity of all the vibronic progressions, a smaller S also implies a spectrum of narrow bandwidth and higher colour purity. Thus, complexes with highly rigid scaffolds are advantageous for developing luminescent materials with high emission quantum efficiencies.
Spectroscopic and computational studies have revealed that the non-radiative decay rate of luminescent pincer-type cyclometalated Pt(II) complexes can be profoundly affected by the ligands coordinated to Pt(II). As depicted in Fig. 3, there is usually a significant structural distortion from a coplanar ground state geometry to a bent conformation in the T1 excited state for the cyclometalated Pt(II) complexes that are non- or weakly emissive.30–32 Computational studies revealed that the quasi-degeneracy of the HOMO and H−1 orbitals and steric strain between the cyclometalated and ancillary ligands contribute to this type of excited state structural distortion that leads to very fast non-radiative decay.31,32 It has been pointed out that, in the design of efficient phosphorescent Pt(II) emitters, the relative dispositions of coordinating donor atoms to the Pt(II) ion and the site of π-conjugation should be taken into consideration.31,32
 | ||
| Fig. 3 Optimized geometries of the ground state S0 (left) and the lowest triplet excited state T1 (right) of [Pt(C^N^N)Cl] 4 (adapted with permission from ref. 31. Copyright 2009, Wiley-VCH Verlag GmbH & Co. KGaA). | ||
2.2 Factors affecting the stability of phosphorescent metal complexes
Degradation of phosphorescent transition metal complexes usually occurs via M–L bond rupture. The tactics to reduce the propensity of M–L bond dissociation lie in (1) strengthening M–L bonds using strong σ-donor ligands and (2) using multidentate ligands. In general, transition metal complexes supported by multidentate ligands are more stable than those with monodentate ligands having comparable donor strength (termed the chelate effect); this is the result of the entropy effect in chelation reactions.33 In addition, the stability of transition metal complexes is influenced by the chelate ring size: 6-membered rings confer higher stability than 5-membered ones.332.3 Multidentate ligands for high efficiency and stability
Considering the structural factors that affect emission efficiency, colour purity, and robustness, rigid multidentate ligand scaffolds containing strong σ-donor atoms are beneficial for the development of highly robust phosphorescent Pt(II) complexes (Scheme 1). This design principle is reminiscent of the Ru(II) polypyridyl complexes supported by “cage” or “hemicage” ligands, which have been shown to display superior emission properties and high photostability against light-induced substitution and decomposition reactions.28,29,34,35 In the literature, there are few examples of the “one metal ion + one ligand” approach to develop luminescent d6 Ir(III) complexes.36 This may be due to the formidable challenges in synthesizing chemically inert, rigid hexadentate ligand scaffolds. In this regard, Pt(II) complexes are advantageous over d6 octahedral metal complexes because tetradentate ligand scaffolds are relatively easy to construct and modify (Scheme 1). Indeed, the recently developed Pt(II) complexes supported by tetradentate ligands have shown impressive emission efficiencies (ϕP ∼ 1) and exceptional thermal stability (Td > 400 °C).37 | ||
| Scheme 1 Illustration of the “one metal ion + one ligand” approach. | ||
3 Advances in luminescent Pt(II) complexes
In this section, we describe the structures and emission properties of various types of Pt(II) complexes which display high emission efficiency and/or high stability. Their pertinent photophysical data are summarized in Tables 1 and 2. Additionally, the performances of OLEDs doped with selected examples of these Pt(II) emitters are discussed.| Coordination mode | Complex | Medium | λ max/nm | ϕ | τ/μs | k r/104 s−1 | k nr/104 s−1 | T d/°C | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a The lifetime τ0 at infinite dilution determined from the linear variation of the observed emission decay rate constant, kobs, as a function of the concentration of the complex. b Value too low to be reported. c Value not available from the literature. | |||||||||
| [Pt(C^N)2] | 1 | THF | 582 | 0.24 | 5.5 | 4.4 | 13.8 | —c | 40 |
| [Pt(C^N)(L^X)] | 2 | 2-MeTHF | 485 | 0.22 | 4.5 | 4.9 | 17.3 | —c | 41 |
| 3 | CH2Cl2 | 595 | 0.47 | 2.6 | 18 | 21 | —c | 43 | |
| [Pt(C^N^N)X] | 4 | CH2Cl2 | 565 | 0.025 | 0.51 | 4.9 | 191 | —c | 44 |
| 5 | CH2Cl2 | 533, 569 (sh) | 0.68 | 7 | 9.7 | 4.6 | 532 | 46 | |
| 6 | CH2Cl2 | 521, 556 (sh) | 0.99 | 5.5 | 18 | —b | —c | 32 | |
| 7 | CH2Cl2 | 588, 632, 687 (sh) | 0.25 | 10.1 | 2.5 | 7.5 | —c | 32 | |
| 8 | CH2Cl2 | 495, 531 | 0.56 | 9.2 | 6.1 | 4.8 | —c | 50 | |
| [Pt(N^C^N)X] | 9 | CH2Cl2 | 491, 524, 562 | 0.6 | 7.2a | 8.3 | 5.6 | —c | 51 |
| 10 | CH2Cl2 | 518, 548 | 0.37 | 5.0a | 7.4 | 12.6 | —c | 54 | |
| 11 | CH2Cl2 | 496 | 0.6 | 8.6a | 7.0 | 4.7 | — | 55 | |
| [Pt(C^N^C)L] | 12 | CH2Cl2 | 589 | 0.26 | 15.8 | 1.6 | 4.7 | 343 | 65 |
| 13 | 2-MeTHF | 511 | 0.82 | 42 | 2.0 | 0.43 | —c | 30 | |
3.1 Luminescent Pt(II) complexes containing bidentate ligands
Homoleptic bis-cyclometalated Pt(II) complexes were reported as early as 1984 by von Zelewsky et al.38–40 This type of complex is known for its photochemical reactivity towards halogenated hydrocarbons. For example, the strongly orange light-emitting Pt(thpy)2 (1) (ϕ = 0.24 in THF) underwent an oxidative addition reaction with halogen-containing molecules, affording the corresponding Pt(IV) complexes which show blue-shifted phosphorescence.40 In view of the harsh conditions required for their preparation and their intriguing photolability, the phosphorescent bis-cyclometalated Pt(II) complexes have been sparsely used as robust emitters. Alternatively, a number of heteroleptic cyclometalated Pt(II) complexes with the formula [Pt(C^N)(acac)] (C^N denotes C-deprotonated 2-phenylpyridyl ligands and their derivatives; acac denotes deprotonated acetylacetone) have been reported and show intense phosphorescence in solution at room temperature.41,42 For example, 2 is strongly emissive with λmax = 485 nm (ϕ = 0.22) in 2-methyltetrahydrofuran (2-MeTHF).41 It was reported that modification of the substitution on the cyclometalating C^N ligand could tune the emission energy to cover the visible spectral region.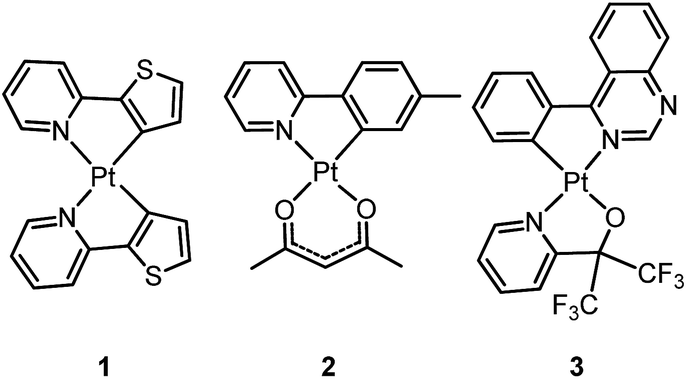
In the literature, heteroleptic cyclometalated Pt(II) and Ir(III) complexes, i.e., [Pt(C^N)(L^X)] and [Ir(C^N)2(L^X)], have usually been used in the design of high performance triplet emitters.5,7 These complexes could be obtained from chloro-bridged [Pt(C^N)(μ-Cl)]2 or [Ir(C^N)2(μ-Cl)]2 dinuclear intermediates. Hence, modification of the cyclometalated and ancillary L^X moieties could be performed to systematically tune the physical and photophysical properties of these metal emitters. However, unlike the extensive reports on [Ir(C^N)2(L^X)] with L^X covering diverse mono-anionic bidentate ligands,5,7 only deprotonated acetylacetone (acac) and its derivatives are utilized as L^X ligands in the development of luminescent Pt(II) complexes of the type [Pt(C^N)(L^X)].42 At this juncture, it should be noted that complex 3, bearing 2-pyridyl hexafluoro-propoxide as the ancillary ligand, was reported to exhibit a high emission quantum yield of 0.47 (λmax = 595 nm) in CH2Cl2.43 This finding highlights the promising prospect that phosphorescent [Pt(C^N)(L^X)] complexes have and indicates that they deserve further attention.
3.2 Luminescent Pt(II) complexes containing a terdentate C-deprotonated ligand
In 1999, Che and co-workers described luminescent pincer-type Pt(II) complexes supported by C-deprotonated 6-aryl-2,2′-bipyridine ligands.44 Compared to [Pt(trpy)Cl]+ (trpy = 2,2′:6′,2′′-terpyridine), which is non-emissive under ambient conditions, complex 4 is emissive in CH2Cl2 (ϕ = 0.025) at room temperature, with the lowest triplet excited state having a 3MLCT character. It was suggested that the covalent Pt–C bond is the root cause for the enhanced emission properties of this complex in solution. Modification of either the C^N^N moiety or ancillary ligand has led to a number of luminescent Pt(II) complexes with improved emission quantum yields and enhanced stability.45,46 For example, by extending π-conjugation of the C-deprotonated (C^N^N) moiety, strongly luminescent 5 (λmax = 533 nm and ϕ = 0.68 in CH2Cl2) was prepared.46 Computational studies revealed that the extended π-conjugation of the cyclometalated ligand in 5 removes the quasi-degeneracy of the HOMO and H−1 orbitals, and hence the structural distortion between the T1 excited state and ground state is smaller when compared to that of 4. This accounts for the low knr value of 5 in comparison to 4 (knr = 1.9 × 106 s−1 for 4 and 4.6 × 104 s−1 for 5).31 It is of note that, because of the rigid π-conjugated terdentate cyclometalated ligand, 5 has a high thermal stability with a decomposition temperature (Td) of 532 °C in N2. Using 5 as a light-emitting material, a high-performance yellowish-green OLED was fabricated with a maximum brightness of 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2. Che and co-workers also reported an efficient white OLED (WOLED) by combining emissions from a blue fluorescent material and a green-yellow emitting analogue of 5 in which the tBu groups were changed into CF3.47 The device having dual emitting layers exhibited a balanced white-light with a CIE of (0.30, 0.32). The maximum EQE reached 11.8%. Very recently, Che and co-workers reported that 6 (λmax = 521 nm) displays a phosphorescence efficiency of almost one in room temperature solution.32 Computational studies revealed that 6 shows the smallest structural distortion from S0 to T1 along the high-frequency normal modes of the complexes studied therein. The excited state geometry distortion was found to be highly dependent on the site of π-conjugation and nature of the additional ancillary ligand. A yellowish-green OLED doped with 6 showed a maximum EQE of 22.8%, which is among the highest values ever reported for OLEDs with Pt(II) dopants. With a thiophene-containing π-conjugated ligand, complex 7 has an emission with λmax = 588 nm in CH2Cl2.32 An efficient red-emitting OLED fabricated using 7 as the dopant showed a maximum EQE of 22.1%, which is comparable to the best values of red-emitting OLEDs based on Ir(III) complexes.48 The pincer-type cyclometalated Pt(II) complexes have been employed as photosensitizers or photocatalysts for solar energy conversions. Fu and co-workers showed that [Pt(C^N^NPhMe)Cl] (HC^N^NPhMe = 4-(p-tolyl)-6-phenyl-2,2′-bipyridine) was an effective photosensitizer for hydrogen evolution from water and afforded a much higher turnover number than its terpyridyl analogue under the same experimental conditions.49 Complex 6 has been examined by Che as a photocatalyst for visible light-induced reductive C–C bond formation reactions (Scheme 2).32 With diisopropylethylamine (iPr2NEt) as a sacrificial electron donor in CH3CN, and after 4–8 hours of irradiation using a blue LED, a series of alkyl bromides underwent intramolecular C–C bond formation with conversions and yields of up to 99% and 78%, respectively. This complex also catalysed the light-induced intermolecular C–C bond formation from benzyl chloride.
000 cd m−2. Che and co-workers also reported an efficient white OLED (WOLED) by combining emissions from a blue fluorescent material and a green-yellow emitting analogue of 5 in which the tBu groups were changed into CF3.47 The device having dual emitting layers exhibited a balanced white-light with a CIE of (0.30, 0.32). The maximum EQE reached 11.8%. Very recently, Che and co-workers reported that 6 (λmax = 521 nm) displays a phosphorescence efficiency of almost one in room temperature solution.32 Computational studies revealed that 6 shows the smallest structural distortion from S0 to T1 along the high-frequency normal modes of the complexes studied therein. The excited state geometry distortion was found to be highly dependent on the site of π-conjugation and nature of the additional ancillary ligand. A yellowish-green OLED doped with 6 showed a maximum EQE of 22.8%, which is among the highest values ever reported for OLEDs with Pt(II) dopants. With a thiophene-containing π-conjugated ligand, complex 7 has an emission with λmax = 588 nm in CH2Cl2.32 An efficient red-emitting OLED fabricated using 7 as the dopant showed a maximum EQE of 22.1%, which is comparable to the best values of red-emitting OLEDs based on Ir(III) complexes.48 The pincer-type cyclometalated Pt(II) complexes have been employed as photosensitizers or photocatalysts for solar energy conversions. Fu and co-workers showed that [Pt(C^N^NPhMe)Cl] (HC^N^NPhMe = 4-(p-tolyl)-6-phenyl-2,2′-bipyridine) was an effective photosensitizer for hydrogen evolution from water and afforded a much higher turnover number than its terpyridyl analogue under the same experimental conditions.49 Complex 6 has been examined by Che as a photocatalyst for visible light-induced reductive C–C bond formation reactions (Scheme 2).32 With diisopropylethylamine (iPr2NEt) as a sacrificial electron donor in CH3CN, and after 4–8 hours of irradiation using a blue LED, a series of alkyl bromides underwent intramolecular C–C bond formation with conversions and yields of up to 99% and 78%, respectively. This complex also catalysed the light-induced intermolecular C–C bond formation from benzyl chloride.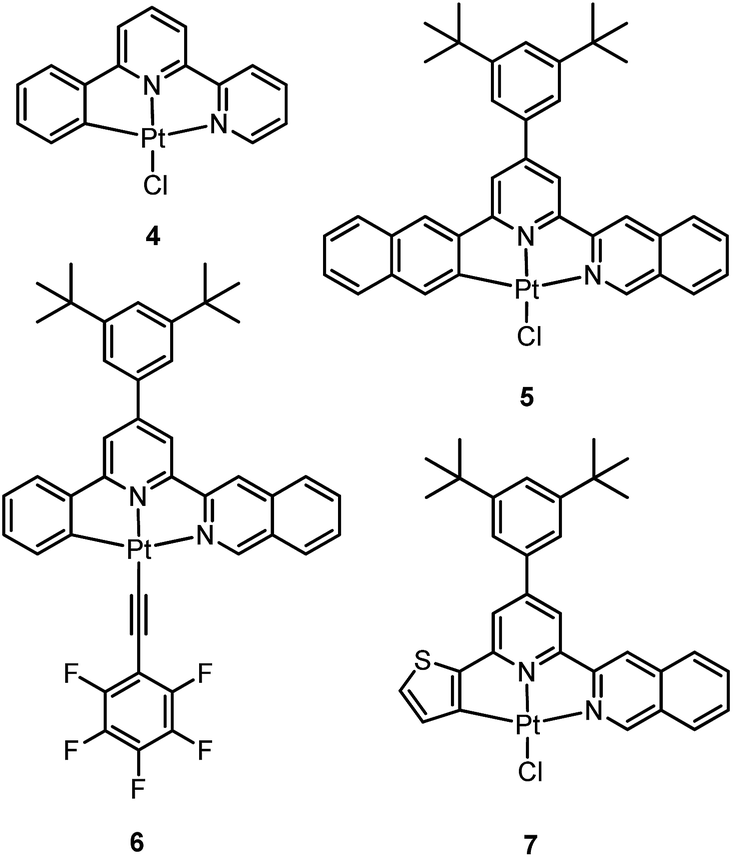
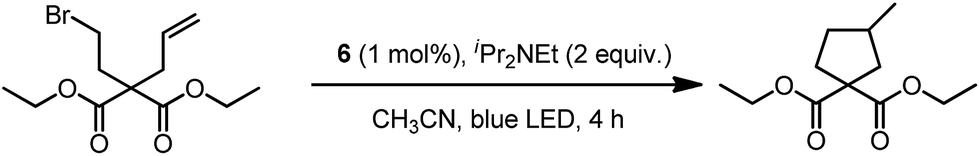 | ||
| Scheme 2 | ||
Huo and co-workers reported the structural modification of a terdentate C-deprotonated cyclometalated ligand resulting in the change of its [Pt(C^N^N)] motif from having a fused 5,5-membered metallacycle (as in 4–7) to having a 5,6-membered one (as in 8).50 Complex 8 exhibits substantially improved phosphorescence (ϕ = 0.56 and τ = 9.2 μs in CH2Cl2) compared to its fused 5,5 analogue, [Pt(C^N^N)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)] (ϕ = 0.04 and τ = 0.4 μs in CH2Cl2) (Chart S1, ESI†).45 The substantial difference in the knr values of both complexes (knr = 4.8 × 104 s−1 for 8vs. 2.4 × 106 s−1 for [Pt(C^N^N)(CCPh)]) reveals that the phosphorescence efficiency is strongly affected by the non-radiative decay rate constant. On the one hand, the slower non-radiative decay rate of 8 is probably due to the stronger donor strength of 8,50 while on the other hand, computational studies have shown that the emitting triplet excited state of [Pt(C^N^N)(CCPh)] is a 3[π(CCPh) → π*(C^N^N)] ligand-to-ligand charge transfer (LLCT) mixed with 3[dπ(Pt) → π*(C^N^N)] (MLCT), and that of 8 is 3π–π*(C^N) mixed with 3[dπ(Pt) → π*(C^N)] (Fig. S1, ESI†). These calculations are consistent with the experimental finding that [Pt(C^N^N)(CCPh)] and 8 display structureless and vibronic-structured emission profiles, respectively. As the emissive excited state of [Pt(C^N^N)(CCPh)] involves distortion of the CC bond while that of 8 does not, the presence of an additional effective high-frequency accepting mode, ωCC, would lend the former complex a faster non-radiative decay rate (see details in the ESI†). This difference in the nature of the T1 excited state is attributed to the presence of an additional amine bridge in 8 that causes the C^N moiety and pyridyl ring to be non-planar to each other, while the C-deprotonated C^N^N cyclometalated ligand has a relatively planar geometry in the case of [Pt(C^N^N)(CCPh)] (Fig. S2, ESI†). Moreover, as stated by the energy-gap law, the lower emission energy of [Pt(C^N^N)(CCPh)] may also contribute to the observed faster non-radiative decay rate.
CPh)] (ϕ = 0.04 and τ = 0.4 μs in CH2Cl2) (Chart S1, ESI†).45 The substantial difference in the knr values of both complexes (knr = 4.8 × 104 s−1 for 8vs. 2.4 × 106 s−1 for [Pt(C^N^N)(CCPh)]) reveals that the phosphorescence efficiency is strongly affected by the non-radiative decay rate constant. On the one hand, the slower non-radiative decay rate of 8 is probably due to the stronger donor strength of 8,50 while on the other hand, computational studies have shown that the emitting triplet excited state of [Pt(C^N^N)(CCPh)] is a 3[π(CCPh) → π*(C^N^N)] ligand-to-ligand charge transfer (LLCT) mixed with 3[dπ(Pt) → π*(C^N^N)] (MLCT), and that of 8 is 3π–π*(C^N) mixed with 3[dπ(Pt) → π*(C^N)] (Fig. S1, ESI†). These calculations are consistent with the experimental finding that [Pt(C^N^N)(CCPh)] and 8 display structureless and vibronic-structured emission profiles, respectively. As the emissive excited state of [Pt(C^N^N)(CCPh)] involves distortion of the CC bond while that of 8 does not, the presence of an additional effective high-frequency accepting mode, ωCC, would lend the former complex a faster non-radiative decay rate (see details in the ESI†). This difference in the nature of the T1 excited state is attributed to the presence of an additional amine bridge in 8 that causes the C^N moiety and pyridyl ring to be non-planar to each other, while the C-deprotonated C^N^N cyclometalated ligand has a relatively planar geometry in the case of [Pt(C^N^N)(CCPh)] (Fig. S2, ESI†). Moreover, as stated by the energy-gap law, the lower emission energy of [Pt(C^N^N)(CCPh)] may also contribute to the observed faster non-radiative decay rate.
Williams and co-workers described Pt(II) complexes (9 and its derivatives) supported by terdentate cyclometalated ligands derived from C-deprotonated 1,3-bis(2-pyridyl)benzene (N^C^N) ligands.51 A high emission efficiency of 0.6 was recorded for 9 in CH2Cl2 and the emitting triplet excited state has been characterized to be metal-perturbed 3IL (IL = intraligand) in nature. Compared to [Pt(C^N^N)Cl] (4), shorter Pt–C bonds were observed for 9 and its derivatives.51,52 This may result in 9 having a higher-lying d–d excited state. The finding of the emission lifetime to be insensitive to temperature rules out the possibility of a non-radiative decay pathway occurring via a d–d state.53 The [Pt(N^C^N)Cl] complexes show a strong tendency to undergo excimer formation at elevated concentrations in solution, and aggregation and/or excimer formation in the solid state. Taking advantage of this feature, modification of either the cyclometalating moiety or variation of the fourth auxiliary ligand can tune the energy of frontier molecular orbitals and/or perturb the extent of intermolecular interactions, leading to a shift in the emission to the near-infrared (NIR) region. For instance, attaching electron-withdrawing –CF3 groups stabilizes the pyridyl-localized LUMO of 10 and hence its excimeric emission occurs at a low energy of 756 nm in CH2Cl2.54 Coordination of SCN− to Pt(II), as in the case of 11, does not affect the excited state energy in comparison to its chloride counterpart, but alters the solid-state packing, resulting in a shorter Pt⋯Pt contact of 3.3 Å. As a consequence, an OLED based on a neat-film of 11 shows EL at 855 nm.55 In addition to the promising NIR emission due to aggregation or excimer formation, cyclometalated pincer-type Pt(II) complexes are also promising candidate materials for single-dopant WOLEDs through a combination of both their monomeric blue-to-green emission and excimeric red emission (vide infra).56–58
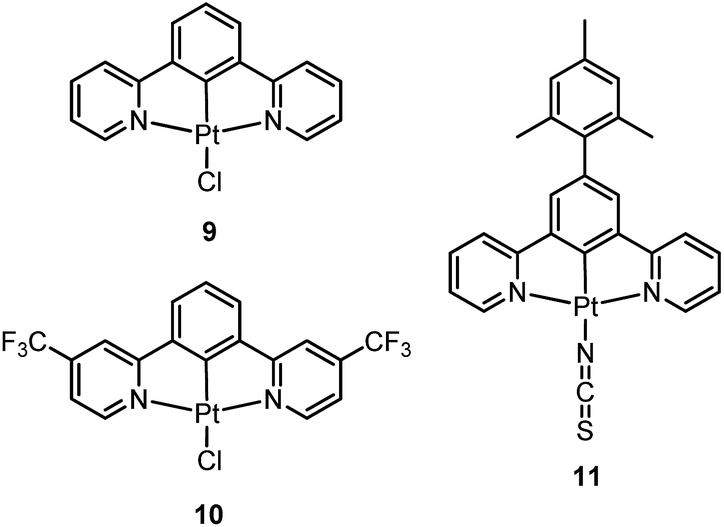
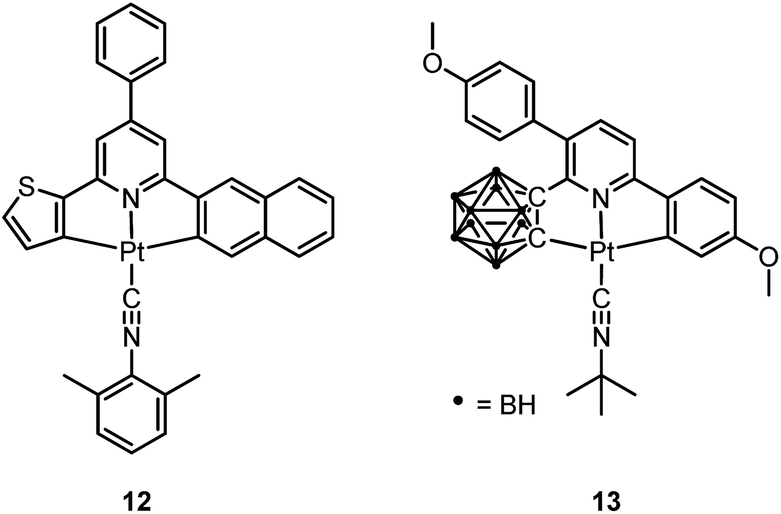
Pt(II) complexes supported by doubly C-deprotonated 2,6-diphenylpyridine (C^N^C) ligands constitute another class of cyclometalated Pt(II) complexes which possess rich photophysical properties.59–64 Despite the presence of two covalent Pt–C bonds, most of the [Pt(C^N^C)L] type complexes are non-emissive or weakly emissive in solution at room temperature. Computational studies revealed that these complexes undergo significant structural distortions upon shifting from S0 to T1 state.31 In 2012, Che and co-workers reported the first examples of organoplatinum(II) complexes bearing functionalized (C^N^C) ligands that are emissive in solution at room temperature (e.g., ϕ = 0.26 for 12).65 The emission switch-on property is achieved by extending the π-conjugation of the (C^N^C) ligand, thereby shifting the lowest electronic excited states to be predominantly 3IL in nature. Because of the more rigid ligand framework, the excited state structural distortion of the new emitters is greatly reduced. High thermal stability (Td > 300 °C) renders 12 a suitable dopant for OLED applications; a red OLED with CIE coordinates of (0.65, 0.35) was fabricated and a maximum EQE of 12.6% was obtained. Very recently, Yersin et al. developed a brightly luminescent Pt(II) complex 13 (ϕ = 0.82 in 2-MeTHF) which is supported by a bulky and rigid carboranyl-phenylpyridine ligand.30 Photophysical studies revealed an exceptionally small non-radiative decay rate constant that is in the order of 103 s−1. DFT calculations showed that no bent conformation, which is commonly encountered in other non-emissive [Pt(C^N^C)X] (X = auxiliary ligand) complexes, can be observed in the T1 excited state of 13, thereby significantly decreasing the non-radiative decay rate.
3.3 Luminescent Pt(II) complexes supported by a tetradentate ligand
Pt(II) porphyrin complexes are well-known to be robust phosphorescent emitters as a result of the rigid macrocyclic porphyrin ligand scaffold. With an extended π-conjugated porphyrin ligand, these complexes display red or NIR 3IL emission with very long lifetimes.66–69 In recent years, there has been a surge of interest to develop luminescent Pt(II) complexes supported by non-porphyrin tetradentate ligands. By judiciously designing the tetradentate ligand, the emission energy can be tuned throughout the entire visible spectral region. This type of Pt(II) complex usually has high thermal stability and the emissive excited states show a relatively slow non-radiative decay rate.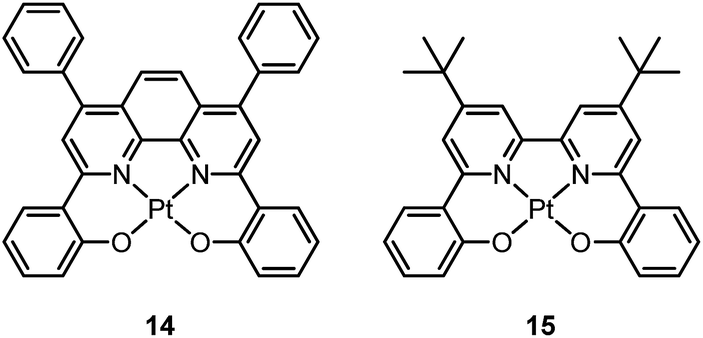
In 2003, Che and co-workers reported the first series of phosphorescent Pt(II) complexes supported by tetradentate dianionic bis(phenoxy)diimine (N2O2) ligands (14 and 15) that have fused 6,5,6-membered rings.70 The Pt(II) ions in the crystal structures of both 14 and 15 adopt a planar coordination geometry and the two Pt(N2O2) frameworks are highly planar (Fig. 4). Complexes 14 and 15 are stable up to 440 and 530 °C, respectively, in N2. Notably, they also show very high thermal stability in air with significant weight loss at temperatures above approximately 380 °C. Complexes 14 and 15 display structureless emissions in CH2Cl2 at room temperature with λmax = 586 nm (ϕ = 0.6; τ = 5.3 μs) and 595 nm (ϕ = 0.1; τ = 1.9 μs), respectively. Their potentials as phosphorescent OLED emitters were examined. The maximum brightness (9330 cd cm−2) and power efficiency (1.44 lm W−1) were recorded for a device using 15 as the dopant. Despite the much higher emission quantum yield of 14 in CH2Cl2, the device doped with 14 exhibited a low performance that was ascribed to intermolecular quenching (including self-quenching). In contrast, the bulky tert-butyl groups in 15 are believed to reduce the intermolecular interactions.
 | ||
| Fig. 4 Perspective view (50% thermal probability) (left) and packing arrangement along the ab plane (right) of 15 (adapted with permission from ref. 70. Copyright 2003, Wiley-VCH Verlag GmbH & Co. KGaA). | ||
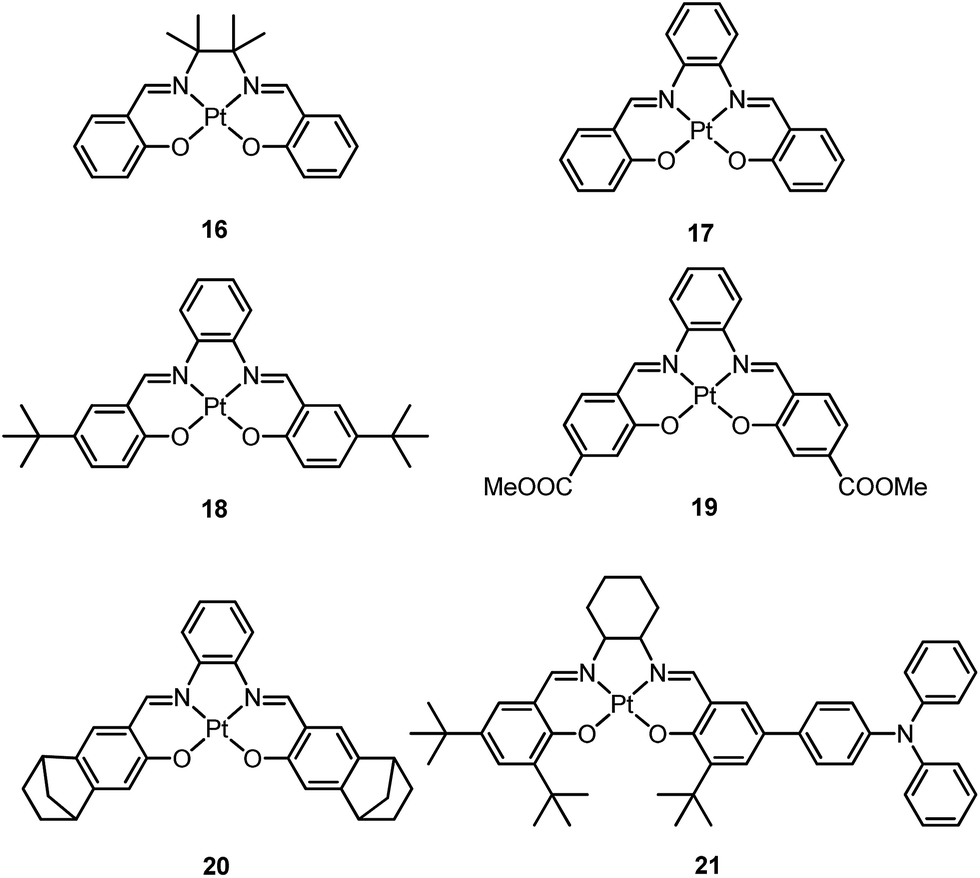
In 2004, Che and co-workers reported another tetradentate N2O2 ligand system, i.e., Schiff base, that can be used for the preparation of phosphorescent Pt(II) emitters.71 Detailed photophysical studies of a series of Pt(II) Schiff base complexes were later presented in 2010.72 These types of complex are attractive in the context of practical applications because of the straightforward preparation of the Schiff base ligands and the simplicity of scaling up for mass production. It is also well-known that Schiff bases readily form stable transition metal complexes. Not surprisingly, all the Pt(II) Schiff base complexes are stable in the solid state, and no decomposition or ligand dissociation was observed in EtOH, 2-propanol, DMSO, or CH3CN under ambient conditions for one month. The Pt(II) Schiff base complexes display excellent thermal stability with Td of up to 495 °C in N2.72 Complex 16 shows an emission with λmax = 546 nm in benzene. This complex exhibits the highest reported emission quantum yield (ϕ = 0.27) among this class of Pt(II) emitters. Structural diversity of the Schiff base ligand allows facile tailoring of both the physical and photophysical properties of this class of complexes. Replacing the ethylene linker with a phenyl ring resulted in a red-shifted emission as in the case of 17 (λmax = 628 nm and ϕ = 0.26 in benzene). Zero-field splitting (ZFS) can be used to estimate the extent of MLCT perturbation and SOC efficiency of the emitting triplet state as well as to assess the suitability of an emitter for OLED fabrication.1,2 The value of ZFS of selected complexes was determined by measuring their temperature-dependent lifetimes in the range of 1.5–130 K. As shown in Fig. 5, the total ZFS for 16 was estimated to be 20 cm−1, belonging to the intermediate range in a library of triplet emitters examined by Yersin and co-workers.72 The emitting triplet state was assigned to have ILCT character with significant MLCT perturbation. DFT/TDDFT studies also supported this assignment. Che and co-workers have extended the theoretical studies to the substituent effects on the luminescence efficiency of the Pt(II) Schiff base complexes.73 Replacing the ethylene bridge (16) of the Schiff base ligand with a π-conjugated phenylene bridge (17) not only widens the 3MLCT–3d–d energy gap due to a lowered ligand π* orbital (relative to the dx2−y2 orbital) in 17 (Fig. 6), but also results in a smaller Huang–Rhys factor (SM = 1.05 and 0.89 for 16 and 17 respectively). As such, though the emission energy of 17 is smaller than that of 16 and the former should have a faster non-radiative decay rate than the latter based on the energy gap law, both complexes have similar knr values in CH3CN at room temperature (knr = 2.31 × 105 s−1 for 16 and 2.24 × 105 s−1 for 17)72 due to the smaller structural distortion in 17. It was also demonstrated in the same work that the nature of the substituents (17–19) can modify the SOC and frequency shifts of the low-frequency modes between the T1 and S0 states. Inclusion of both factors in addition to the structural distortion allowed quantitative computation of the phosphorescence efficiency. An OLED doped with 16 at an optimized doping concentration of 4.0 wt% exhibited a maximum EQE, current efficiency, power efficiency, and brightness of 11%, 31 cd A−1, 14 lm W−1, and 23000 cd m−2, respectively.71 Moreover, this device gave stable EL spectra over a wide range of operating voltages from 3 to 16 V. Using the same device configuration as that for 16, a stable red-OLED using 17 as the dopant was achieved with a maximum EQE and power efficiency of 9.4% and 4.9 lm W−1, respectively. Notably, this device showed a lifetime of more than 20000 h at 100 cd m−2 and the performance was further improved to an impressive 77000 h at 500 cd m−2 by modifying the device configuration.72 However, due to the severe self-quenching effect (kq = 6.9 × 108 M−1 s−1), the optimal device can only be fabricated with a low doping level of 17 (<1.5 wt%). Recently, Che and co-workers prepared a series of derivatives of 17 bearing bulky groups, an example of which is 20.74 Complex 20, having norbornene moieties in the ligand scaffold, displays an emission quantum yield of 0.20 with λmax = 624 nm in CH2Cl2 and a significantly reduced emission self-quenching rate constant (kq) of 1 × 107 M−1 s−1. As a result, an efficient red OLED could be fabricated at a higher doping concentration of 4 wt% and displayed a delayed efficiency roll-off. Wong et al. also reported an asymmetric Pt(II) Schiff base complex 21 with bulky tert-butyl and triphenylamino groups introduced at different sites to prevent aggregation or excimer formation.75 Using 21 as the emitter, an efficient yellow OLED (λmax = 564 nm) was fabricated at a high doping concentration (8.0 wt% in TCTA). The EQE, current efficiency, power efficiency, and maximum brightness were 8.3%, 23 cd A−1, 17 lm W−1, and 11106 cd m−2, respectively.
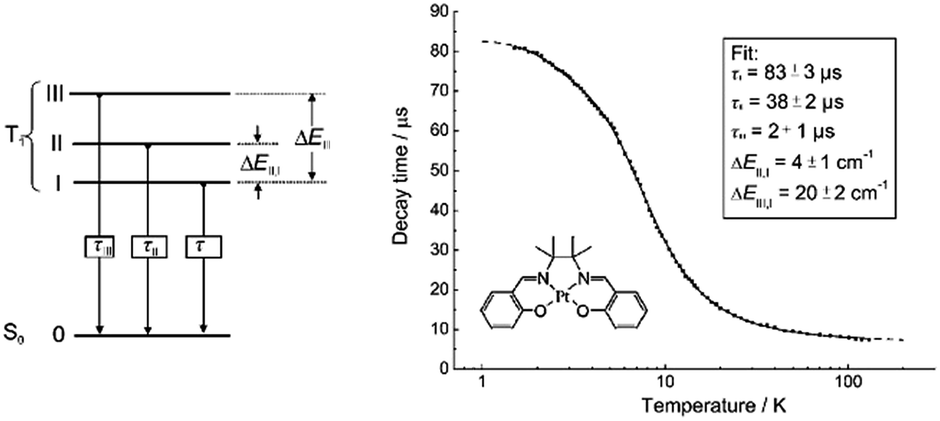 | ||
| Fig. 5 Schematic energy-state diagram for the substates of the emitting T1 state of a transition metal complex (left). Plot of emission decay times of 16 in THF (∼10−5 M) versus temperature and the fit to the equation describing the temperature dependence of the measured decay time in a model of three substates (right) (adapted with permission from ref. 72. Copyright 2010, Wiley-VCH Verlag GmbH & Co. KGaA). | ||
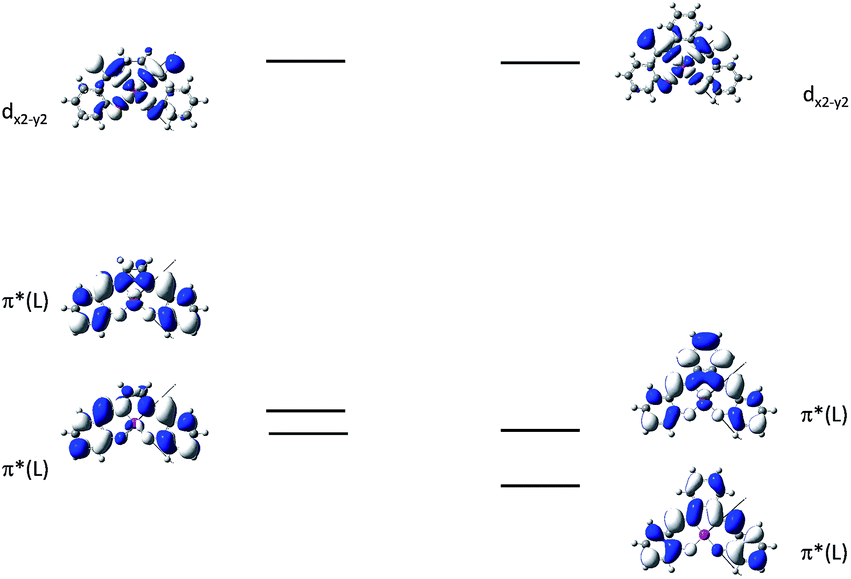 | ||
| Fig. 6 MO diagrams of 16 and 17 showing the 3MLCT–3d–d energy gap change upon ligand modification (adapted with permission from ref. 73. Copyright 2014, Wiley-VCH Verlag GmbH & Co. KGaA). | ||
Che and co-workers previously developed a class of phosphorescent Pt(II) complexes supported by tetradentate bis(pyrrole)-diimine (N4) ligands.76 Complex 22 displays a yellow emission in dilute CH3CN (concentration ∼ 1 × 10−6 M) with an emission quantum yield and lifetime of 0.097 and 4.2 μs, respectively. Interestingly, at elevated concentrations in either solution or CBP (CBP = 4,4’-bis(N-carbazolyl)-1,1’-biphenyl) film, 22 shows a red-shifted emission which has been attributed to the high propensity of 22 for excimer or oligomer formation. Thus, an OLED doped with 22 at 6.0 wt% afforded red electroluminescence with a peak maximum at 620 nm and CIE coordinates of (0.62, 0.38). The maximum EQE, current efficiency, power efficiency, and brightness for this device were 6.5%, 9.0 cd A−1, 4.0 lm W−1, and 11100 cd m−2, respectively. High efficiencies were maintained even at a high brightness of 5000 cd m−2 (5.2%, 7.7 cd A−1, and 2.4 lm W−1). This work represents a proof-of-concept that excimer formation of Pt(II) complexes can be used to achieve efficient low-energy emission. Indeed, NIR OLEDs based on emission from excimeric and aggregate species have been demonstrated by Williams on pincer-type Pt(II) emitters.54,77 However, 22 exhibits a relatively low Td of 288 °C in N2. This is attributed to the significant strain chelate present in 22 as this complex has a fused 5,5,5-membered metallacycle, without the optimal 6-membered rings. Very recently, Chi and co-workers reported a new type of highly emissive Pt(II) complex supported by a tetradentate ligand with four N donor atoms.78 Complex 23 exhibits blue emission (λmax = 461 nm, ϕ = 0.82) in CH2Cl2. This emission has been attributed to a ligand-centred π–π* transition. Complex 24, having an acridine moiety, possesses a dominant charge-transfer character in its lowest triplet excited state. As a result, this complex displays significant solvatochromism. Upon switching the solvent from cyclohexane to CH2Cl2, the emission quantum yield and lifetime change from 0.13 and 20.1 μs to 0.88 and 2.9 μs, respectively. The increased kr is due to the variation of emission character from π–π*/MLCT to ILCT/MLCT. Both 23 and 24 have been used for sky-blue OLED fabrications. The CIE coordinates, peak EQE, and maximum brightness were (0.190, 0.342)/(0.194, 0.391), 12.3%/15.3%, 1924/4121 cd m−2 for 23/24 based OLEDs.
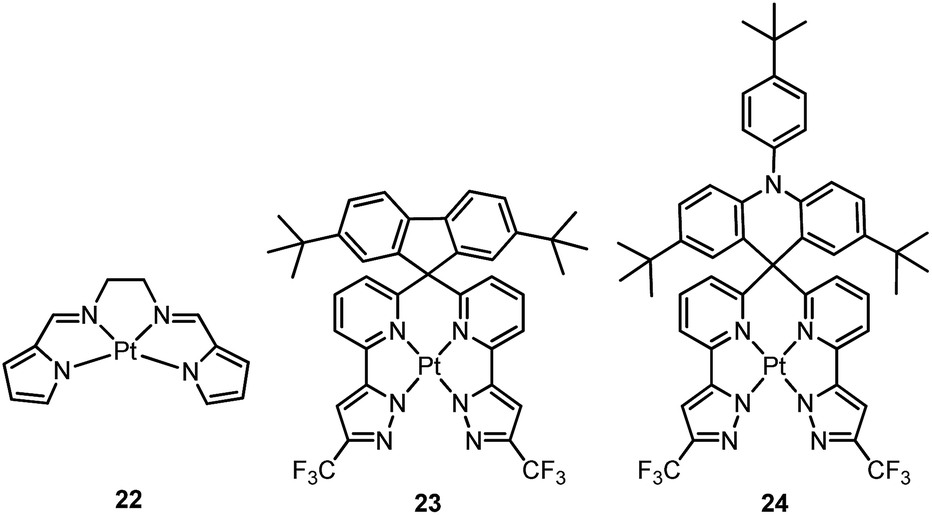
Recently, there has been considerable interest in tetradentate ligand systems containing deprotonated C-atoms and/or NHCs. These ligand systems have been shown to afford superior Pt(II) emitters in terms of both emission efficiency and/or robustness. In 2009, Weck et al. developed complexes containing doubly cyclometalated tetradentate ligands (C^N^N^C).79 Complex 25 shows vibronic-structured phosphorescence in CH2Cl2 with a quantum yield of 0.58 (λmax = 492 nm). In 2010, Huo and co-workers reported a related system in which the cyclometalated C^N motifs are bridged by an amino group as in the case of 26.80 Complex 26 exhibits an intense green (λmax = 512 nm) vibronic structured emission in 2-MeTHF with a quantum yield of 0.74. Upon incorporation of 26 at 4 wt% in a mixed host into an OLED, an excellent device performance with a maximum EQE of 14.7% was observed. Crystal structures reveal that the Pt–C distances of 2.00–2.01 Å for 25 and 26 are close to those of ∼2.00 Å for Pt(ppy)2 (ppy = deprotonated 2-phenylpyridine) while the Pt–N bond lengths of 2.05–2.08 Å are shorter than those of Pt(ppy)2 (∼2.13 Å). In the same work, Huo also reported the photophysical properties of the Pt(II)–N^C^C^N complexes (e.g., 27 and 28) that are isomeric derivatives of the Pt(II)–C^N^N^C ones.80 Similar to the findings for 25 and 26, the formation of a metallacycle results in shorter Pt–C bond distances (∼1.96 Å) for 27, when compared with those of cis-Pt(ppy)2 (∼1.99 Å). Complex 27 emits red light in 2-MeTHF with λmax at 613 nm (ϕ = 0.14). Replacing the pyridyl ring in 27 with the less electron-accepting pyrazolyl ring resulted in a hypsochromic shift of the emission and enhanced emission efficiency, as in the case of 28 (λmax = 486 nm, ϕ = 0.63). Compared to 27, the enhancement of ϕ for 28 is attributed mainly to an increased kr (kr = 1.1 × 105 s−1 for 28vs. 1.8 × 104 s−1 for 27). However, DFT calculations show a comparable metal parentage in the HOMOs of 27 and 28 and, therefore, the metal character of the frontier orbital is not sufficient to account for such a large variation in the kr value. In fact, the factors affecting the radiative decay rate constant are complex. It is necessary to take into account, in addition to spin–orbit coupling (SOC) matrix elements, the singlet–triplet energy gap and the oscillator strength of the transition from S0 to the singlet excited state from which the triplet excited state borrows intensity.31 Fukagawa and co-workers later modified 27 by attaching two tert-butyl groups to the N-pendant phenyl ring to give 29, which emits red light (λmax = 621 nm) with a quantum yield of 0.58 in a bis(benzo[h]quinolin-10-olato-kN,kO)beryllium(II) (Bebq2) film.81 The optimized OLED doped with 29 showed good colour saturation with CIE coordinates of (0.66, 0.34), low driving voltage, high efficiency, and high operational stability. The maximum EQE of over 19% and maximum power efficiency of 30 lm W−1 were comparable to the highest values previously reported for red OLEDs using Ir(III) complexes.82–84 The estimated half-life for the optimized OLED was about 10000 h with an initial brightness of 1000 cd m−2.
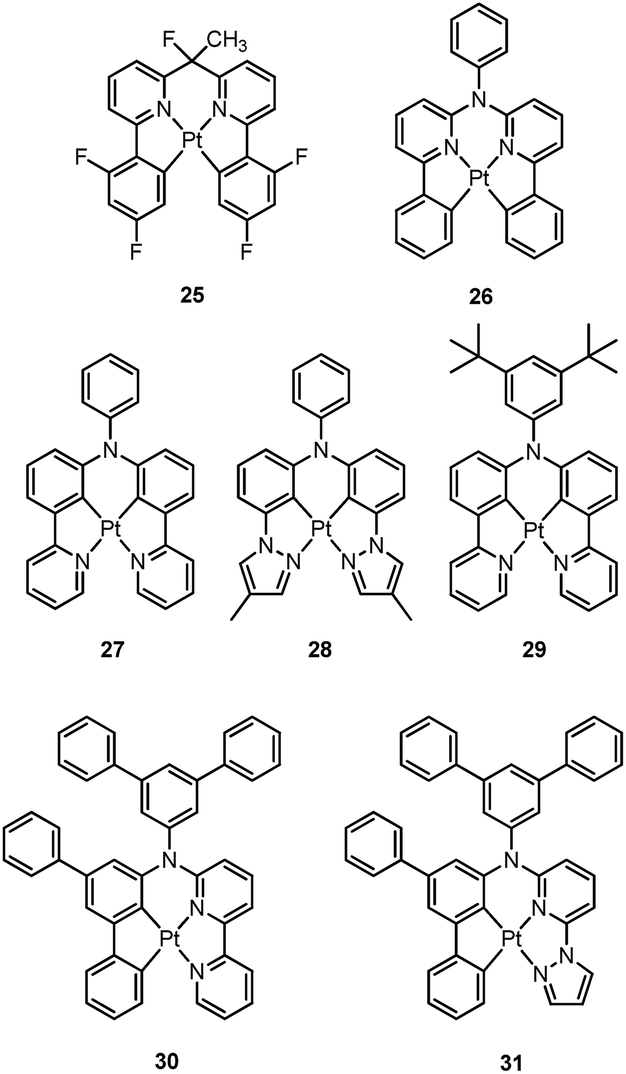
Huo and co-workers later reported the synthesis of Pt(II)–C^C^N^N complexes 30 and 31.85 Complex 30, a closely-related analogue of 25 and 27, is only weakly emissive (ϕ = 0.001) in the deep red region with a λmax of 660 nm. Replacement of the pyridyl ring of 30 with a pyrazolyl ring results in bright yellow phosphorescence at λmax = 550 nm (ϕ = 0.17). The Huang–Rhys factors for 30 and 31 were estimated to be 0.42 and 0.37 respectively, comparable to those found in Pt(II)–C^N^N^C and Pt(II)–N^C^C^N complexes, indicating that these complexes should have similar structural distortion between the emitting triplet excited state and ground state. The much faster non-radiative decay rate of 30 (knr = 1.2 × 106 s−1) is therefore likely to be associated with its lower energy emission, a consequence of the “energy-gap law”.86 On the other hand, although DFT calculations show that the triplet excited states of 30 and 31 are ILCT mixed with MLCT, the distinctly larger kr of 3.9 × 104 s−1 for 31 over 30 (kr = 1.2 × 103 s−1) reflects the significant dependence of kr on subtle ligand modification, again as for that observed for 28 over 27.
Che's and Strassner's groups independently reported tetradentate Pt(II) complexes containing dianionic bis(phenolate-NHC) ligands.87,88 The fused 6,6,6-membered metallacycle allows an ideal coordination geometry around the Pt atom with all bite angles being 90 ± 2°.87 As expected, the optimum chelate ring size together with the strong σ-donating C and O donor atoms render these complexes with good thermal stability with a Td of up to ∼410 °C in N2. In poly(methyl methacrylate) (PMMA), both 32 and 33 exhibit deep blue phosphorescence (λmax ∼ 450 nm) with emission efficiencies of ca. 0.3. This is the first report on deep blue emission from Pt(II) complexes supported by tetradentate ligands.87 On the basis of photophysical studies and DFT calculations, the emitting states have been assigned to have mainly ILCT character with MLCT perturbation, similar to the Pt(II) Schiff base complexes. The femtosecond spectroscopic technique has proven useful in providing valuable information to understand the ultrafast excited state dynamics. As depicted in Fig. 7, upon photoexcitation, the S1 state of 33 undergoes an ultrafast ISC process with a fluorescence lifetime of 0.19 ps, revealing that the involved MLCT character is sufficient for promoting the ISC process.89 Recently, Che and co-workers prepared two new complexes 34 and 35, containing para-F and ortho-tBu groups (Scheme S1 in ESI†). These complexes show improved emission quantum yields of 0.43 and 0.37 in THF, respectively (Fig. S3, ESI†). The kr values (4.4–5.1 × 104 s−1) of 32–35 are comparable and the emission variations mainly arising from their differences in non-radiative decay rates (Table 2). It is rational to assume that the ligand-field strength, and thus d–d energy level, is not much affected by the peripheral substitutions on the ligand in the present system. Given their close emission energies (443–465 nm), the non-radiative relaxations for 32–35via thermal population of the 3d–d states are envisaged to be comparably marginal. For the same reason, the extent of the energy-gap law contribution to the non-radiative decay should be similar. Thus, excited state (T1) geometry distortions with respect to the ground state are conceived to be the cause for such differences in the non-radiative decay rate for 32–35. For complexes having the same excited state nature (3LLCT/3MLCT), an indicator of the geometry distortion from S0 to T1 is the absorption–emission Stokes shift. A comparison of the absorption and emission spectra of 32–34 in DMF is shown in Fig. S4 (ESI†). The para-F and ortho-tBu substituents are effective in reducing excited state structural distortions, leading to decreased absorption–emission Stokes shifts from 33 to 32 and 34.
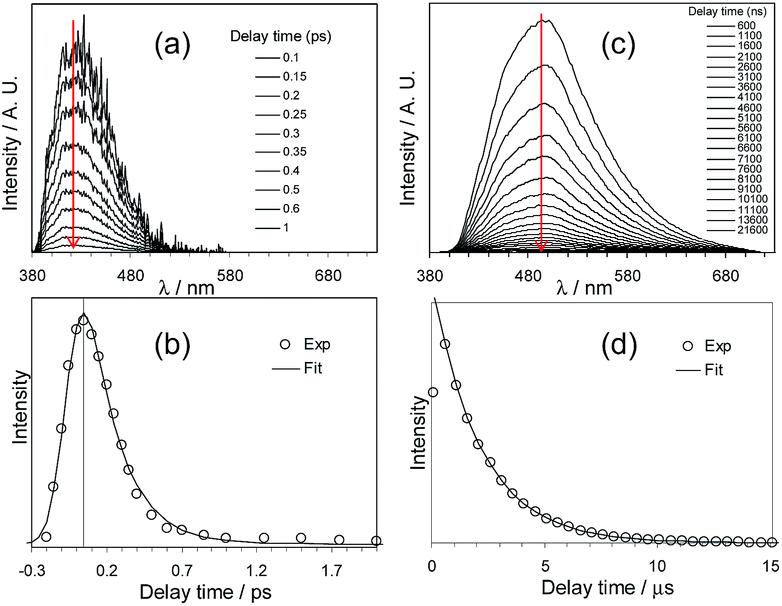 | ||
| Fig. 7 fs-TRF and ns-TRE spectra (a and c) and decay profiles (b and d) recorded at the various denoted delay times after 355 nm excitation of 33 in DMF (reproduced from ref. 89 with permission from the Royal Society of Chemistry). | ||
| Coordination mode | Complex | Medium | λ max/nm | ϕ | τ/μs | k r/104 s−1 | k nr/104 s−1 | T d/°C | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a 6 wt% doped into a thin film of Bebq2. b The lifetime τ0 at infinite dilution determined from the linear variation of the observed emission decay rate constant, kobs, as a function of the concentration of the complex. c Value too low to be reported. d Value not available from the literature. e The work that has not been reported. | |||||||||
| [Pt(O^N^N^O)] | 14 | CH2Cl2 | 586 | 0.6 | 5.3 | 11.3 | 7.5 | 440 | 70 |
| 15 | CH2Cl2 | 595 | 0.1 | 1.9 | 5.3 | 47.4 | 530 | 70 | |
| 16 | Benzene | 546 | 0.27 | 3.9 | 6.9 | 18.7 | 382 | 72 | |
| 17 | Benzene | 628 | 0.26 | 3.1 | 8.4 | 23.9 | 415 | 72 | |
| 18 | CH3CN | 625 | 0.27 | 4.62 | 5.8 | 15.8 | —d | 73 | |
| 19 | CH2Cl2 | 661 | 0.033 | 1.6 | 2.1 | 60.4 | —d | 73 | |
| 20 | CH2Cl2 | 624 | 0.20 | 6.3 | 3.2 | 12.7 | —d | 74 | |
| 21 | CH2Cl2 | 568 | 0.17 | 6.34 | 2.7 | 13.1 | 401 | 75 | |
| [Pt(N^N^N^N)] | 22 | CH3CN | 566, 613 (sh) | 0.097 | 4.2 | 2.3 | 21.5 | 288 | 76 |
| 23 | CH2Cl2 | 461, 487, 521 | 0.82 | 7.8 | 10.5 | 2.3 | —d | 78 | |
| 24 | CH2Cl2 | 520 | 0.88 | 2.9 | 30.3 | 4.1 | —d | 78 | |
| [Pt(C^N^N^C)] | 25 | CH2Cl2 | 492, 520, 564 (sh) | 0.58 | 0.32 | 181 | 131 | —d | 79 |
| 26 | 2-MeTHF | 512, 548 | 0.73 | 7.6b | 9.6 | 3.6 | —d | 80 | |
| [Pt(N^C^C^N)] | 27 | 2-MeTHF | 613 | 0.14 | 7.6 | 1.8 | 11.3 | —d | 80 |
| 28 | 2-MeTHF | 486, 516 | 0.63 | 5.7 | 11.0 | 6.5 | —d | 80 | |
| 29 | Bebq2a | 621 | 0.58 | 5.5 | 10.5 | 7.6 | —d | 81 | |
| [Pt(C^C^N^N)] | 30 | CH2Cl2 | 660 | 0.001 | 0.85 | 0.12 | 118 | —d | 85 |
| 31 | CH2Cl2 | 550 | 0.17 | 4.4 | 3.9 | 18.9 | —d | 85 | |
| [Pt(O^C^C^O)] | 32 | THF | 443, 459 | 0.18 | 3.5 | 5.1 | 23.4 | 410 | 87 |
| 33 | THF | 461 | 0.08 | 1.8 | 4.4 | 51.1 | 400 | 87 | |
| 34 | THF | 463 | 0.43 | 10.0 | 4.3 | 5.7 | 375 | —e | |
| 35 | THF | 465 | 0.37 | 11.5 | 3.2 | 5.5 | —d | —e | |
| [Pt(O^N^C^N)] | 36 | CH2Cl2 | 485, 517, 557 | 0.72 | 12.0 | 6.0 | 2.3 | 414 | 90 |
| 37 | CH2Cl2 | 488, 518 | 0.93 | 13.2 | 7.0 | 0.5 | 406 | 90 | |
| 38 | CH2Cl2 | 515 | 0.99 | 15.5 | 6.4 | —c | —d | 37 | |
| 39 | CH2Cl2 | 528, 568 | 0.74 | 25.3 | 2.9 | 1.0 | —d | 37 | |
| 40 | CH2Cl2 | 503 | 0.73 | 4.7 | 15.5 | 5.7 | 518 | 91 | |
| 41 | CH2Cl2 | 522 | 0.9 | 4.9 | 18.4 | 2.0 | 405 | 91 | |
| 42 | CH2Cl2 | 551 | 0.90 | 4.3 | 20.9 | 2.3 | 423 | 93 | |
| 43 | CH2Cl2 | 517 | 0.80 | 5.1 | 15.7 | 3.9 | 412 | 93 | |
| 44 | CH2Cl2 | 553, 587 | 0.86 | 6.6 | 13.0 | 2.1 | 400 | 92 | |
| 45 | CH2Cl2 | 526 | 0.47 | 5.9 | 8.0 | 9.0 | 410 | 92 | |
| 46 | CH2Cl2 | 527 | 0.49 | 8.8 | 5.6 | 5.8 | 409 | 92 | |
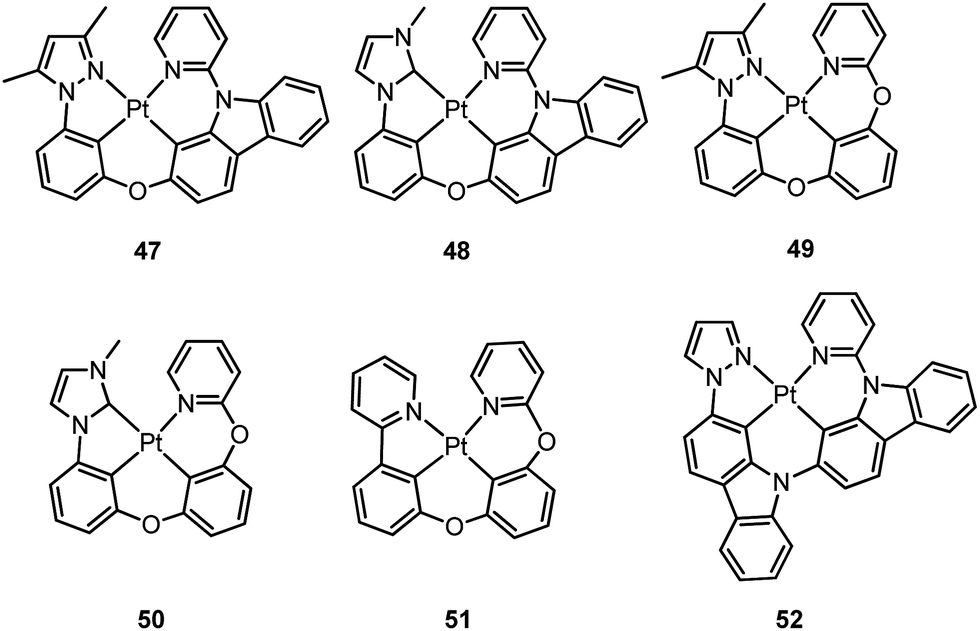
| [Pt(N^C^C^N)] | 47 | CH2Cl2 | 454 (sh), 478 | 0.71 | 3.3 | 21.5 | 8.8 | —d | 94 |
| [Pt(N^C^C^C)] | 48 | CH2Cl2 | 452 | 0.78 | 4.2 | 18.6 | 5.2 | —d | 94 |
| [Pt(N^C^C^N)] | 49 | CH2Cl2 | 430, 456 | 0.39 | 3.0 | 13.0 | 20.3 | —d | 95 |
| [Pt(N^C^C^C)] | 50 | CH2Cl2 | 442 | 0.07 | 0.4 | 17.5 | 232 | —d | 95 |
| [Pt(N^C^C^N)] | 51 | CH2Cl2 | 513 | 0.63 | 2.0 | 31.5 | 18.5 | —d | 95 |
| 52 | CH2Cl2 | 491 | 0.81 | 12.9 | 6.3 | 1.5 | —d | 97 | |
The device doped with 33 at 4 wt% in the host of 9-(4-tert-butylphenyl)-3,6-bis(triphenylsilyl)-9H-carbazole (CzSi) afforded blue phosphorescence with CIE coordinates of (0.19, 0.21).89 The maximum EQE, current efficiency, power efficiency, and brightness were 15%, 23.8 cd A−1, 16.6 lm W−1 and 9500 cd m−2, respectively.89 The EL spectrum was stable in the operating voltage of 6–11 V. In combination with a co-deposited yellow phosphorescent Pt(II) complex, a white light-emitting electro-phosphorescent device was obtained with CIE coordinates, maximum brightness, and current efficiency of (0.32, 0.42), 30000 cd m−2, and 88 cd A−1. This work showed that by including an NHC motif in the tetradentate ligand scaffold, efficient deep blue phosphorescent Pt(II) materials can be generated.
Based on the comparisons between the above-mentioned tetradentate Pt(II) complexes (Table 2), it is conceived that coordination geometry with short Pt–C bonds and the presence of a fused 6-membered ring may be advantageous in the design of robust, strongly luminescent Pt(II) emitters. Recently Che and co-workers devised an asymmetric O^N^C^N ligand system that features a metallacycle having fused 6,5,5-membered rings (selected examples 36–39).37,90 The decomposition temperature of this series of complexes is higher than 400 °C (Table 2). All complexes exhibit vibronic-structured emission in CH2Cl2 and the emitting state has been assigned as metal-perturbed 3π–π* of the O^N^C^N ligand. The emission quantum yields of this series of Pt–O^N^C^N complexes are higher than 0.65 and the emission energy can be tuned from 485 to 528 nm. Complexes 36–39, bearing rigid fluorenyl-like moieties in their O^N^C^N frameworks have small knr values in the range of 0.06–2.3 × 104 s−1. This is in contrast to the related Pt(II) complexes without the dialkylmethylene bridge, which generally have knr values >2.0 × 104 s−1 (vide infra). Notably, 38 exhibits an emission efficiency of almost one (ϕ = 0.99) in solution at room temperature.37 Attachment of bulky N-carbazolyl groups to the O^N^C^N ligand scaffold to give 38 results in a significant suppression of intermolecular interactions leading to the fabrication of high-efficiency (EQE = 15.6%) monochromic polymer light-emitting diodes (PLEDs).37
Complexes 40 and 41 are related analogues of 36–39, bearing O^N^C^N ligand scaffolds but without a dialkylmethylene bridge.91 The former complexes are also strongly emissive (ϕ = 0.73–0.90 in CH2Cl2) and thermally stable (Td > 400 °C) (Table 2). It is worth noting that the kr and knr values for 40 and 41 are higher than those of 36–39. Consequently, the lifetimes of the former are shortened to fewer than 5 μs while high emission efficiencies are maintained. The bulky norbornene group in 41 was observed to suppress intermolecular interactions, thus disfavouring excimer formation and self-quenching in solution. The high emission efficiency, good thermal stability and ineffective self-quenching property altogether rendered highly phosphorescent OLEDs at a high doping concentration of 41. The device doped with 13 wt% of 41 in 1,3-bis(N-carbazolyl)benzene (mCP) exhibited green phosphorescence with a maximum EQE and peak current efficiency of 18.2% and 66.7 cd A−1, respectively. These values are among the highest values for green OLEDs using Pt(II) emitters. It should be noted that this device showed a very low efficiency roll-off of only 2.4% at 1000 cd m−2 (65.1 cd A−1).
Che has further expanded this O^N^C^N ligand system by inclusion of an additional bridging atom to form fused 6,5,6-membered rings (e.g.42–46).92,93 Complexes 42–46 exhibit strong emission (ϕ = 0.47–0.90 in CH2Cl2) in the yellow-to-green spectral region (Table 2). Contrary to 44 which has a sterically unencumbered ligand scaffold, 42 and 43 have a bulky orthogonal bridging tertiary amine unit and a biphenyl group with a spiro-linkage respectively; these structural motifs efficiently suppress intermolecular interactions, which is in agreement with both the X-ray crystal structure of 43 and the optimized geometry of the dimer of 42 and 43 using DFT calculations.93 As a result, the emission self-quenching rate constants (kq) are as low as 2.0 × 107 and 1.1 × 107 M−1 s−1, respectively. With an optimized device structure, maximum power efficiencies of 118 and 126 lm W−1 have been achieved with the respective yellow-emitting 42- and green-emitting 43-based OLEDs. These values are the highest among the reported Pt(II)-OLEDs; the maximum EQE were respectively 26.0% and 27.6%.93 The emission property of 44 is comparable to that of 42, revealing that the emitting state is localized on the O^N^C^N motif without significant involvement of the tertiary amine linkage.92 The monochromic OLED using 44 as the dopant exhibited a power efficiency of 52.1 lm W−1 which was comparable to those of the best Ir(III)-based yellow OLEDs. In a modified device structure comprising a composite blue host and 44, a WOLED was obtained with an estimated power efficiency of 61 lm W−1. In degassed CH2Cl2, complexes 45 and 46 show moderate emission quantum yields of 0.47 and 0.49, which are relatively lower than those of 42 and 44.
Li and co-workers have developed a series of Pt(II) complexes containing tetradentate ligands of which the conventional cyclometalated fragment C^N is bridged by an O atom to a chelating L^L′ ancillary ligand, resulting in a metallacycle having fused 6,6,5-membered rings (e.g.47–51).94,95 Complexes 47 and 48 having an ancillary pyridyl-carbazole ligand display intense blue phosphorescence in CH2Cl2 with λmax (ϕ) of 478 nm (0.71) and 452 nm (0.78), respectively.94 Interestingly, the full-width at half-maximum (FWHM) of 47 decreases from 85 nm to 20 nm upon attaching an electron-donating tBu group to the 4-position of the pyridyl ring.96 Similar spectral narrowing has also been observed for 48. It was conceived that the highly rigid ancillary carbazolyl pyridine motif serves to suppress structural distortion between the triplet emitting state and the ground state, leading to high efficiency emission for this system of Pt(II) emitters. Moreover, ensuring the localization of the T1 state on the designed lumophore ligand by this design strategy serves to suppress vibrational progressions of the triplet emitting state, leading to emission spectra with small Huang–Rhys factors. In addition to a bridging oxygen atom, Li and co-workers have developed a related system in which pyrazolyl-carbazole was selected to function as the lumophore ligand, as shown in 52.97 This new ligand design imposes further rigidity on the complex because of the conjugated nature of the bridging carbazolyl unit within 52. As a result, a green emission (λmax = 491 nm) with improved efficiency (ϕ = 0.81) and a narrow band (FWHM = 18 nm) was achieved for 52. High-efficiency blue OLEDs with respective EQE of 25.2% and 23.7% were obtained based on 47 and 48.94 Using a tBu-derivative of 48 as the dopant, a highly efficient (EQE = 24.8%) pure blue OLED with CIE coordinates of (0.15, 0.08) was developed.96 A green OLED based on 52 demonstrated a high maximum EQE of 25.6% as well as a very small efficiency roll-off (EQE = 25.5% at 100 cd m−2).97 Complexes 49–51, having more flexible frameworks, show less intense emission in solution.95 In CH2Cl2, the emission quantum yields of 49 and 51 are 0.39 and 0.63 while that of 50 becomes significantly low (0.07). In PMMA, 51 emits with an efficiency of almost one in the green spectral region. A green phosphorescent OLED using 51 as the dopant showed a maximum EQE of 22.3% which was comparable to that of a fac-Ir(ppy)3-based device (EQE = 23.6%) with the same device structure.
4 Self-assembly of luminescent platinum(II) complexes with material applications
Luminescent Pt(II) complexes containing sterically undemanding π-conjugated ligand(s) are prone to associate with each other, driven by intermolecular Pt⋯Pt and/or π–π interactions (Scheme 3). Miskowski et al. pioneered the spectroscopic studies on Pt(II) complexes of aromatic diimines8 and terpyridines98,99 revealing that the absorption and emission energies in the solid state are significantly red-shifted in the presence of intermolecular Pt⋯Pt and/or π–π interactions. The low energy excited states have been termed metal–metal-to-ligand charge transfer (3MMLCT) states. Che and co-workers first reported that a dinuclear Pt(trpy) complex with a rigid guanidine bridge displays a moderately intense absorption at 483 nm and weak phosphorescence at 620 nm in degassed CH3CN; both phenomena were attributed to the MMLCT excited states.100 According to Miskowski and co-workers, the axial 5dz2 orbitals of two Pt(II) ions in close proximity overlap to give bonding dσ and antibonding dσ* orbitals (Scheme 3).101 Similarly, the π and π* orbitals localized on the π-conjugated ligand can also interact with each other, producing bonding σ(π) and σ(π*) and antibonding σ*(π) and σ*(π*) orbitals. As a consequence, a dσ* → σ(π*) (MMLCT) transition (Scheme 3) occurs with a reduced energy gap, accounting for the red-shifted absorption and emission maxima. Other than MMLCT excited states, σ*(π)–σ(π*) transitions (Scheme 3), as a result of intermolecular π–π interactions, may also lead to electronic excited states of relatively low energy.102 Importantly, intermolecular Pt⋯Pt and/or π–π interactions can occur not only between two ground state molecules but also between an excited state molecule and a ground state one, resulting in lower-energy emissive excimeric 3MMLCT or 3[σ*(π)–σ(π*)] (usually simplified as 3π–π*) excited states.44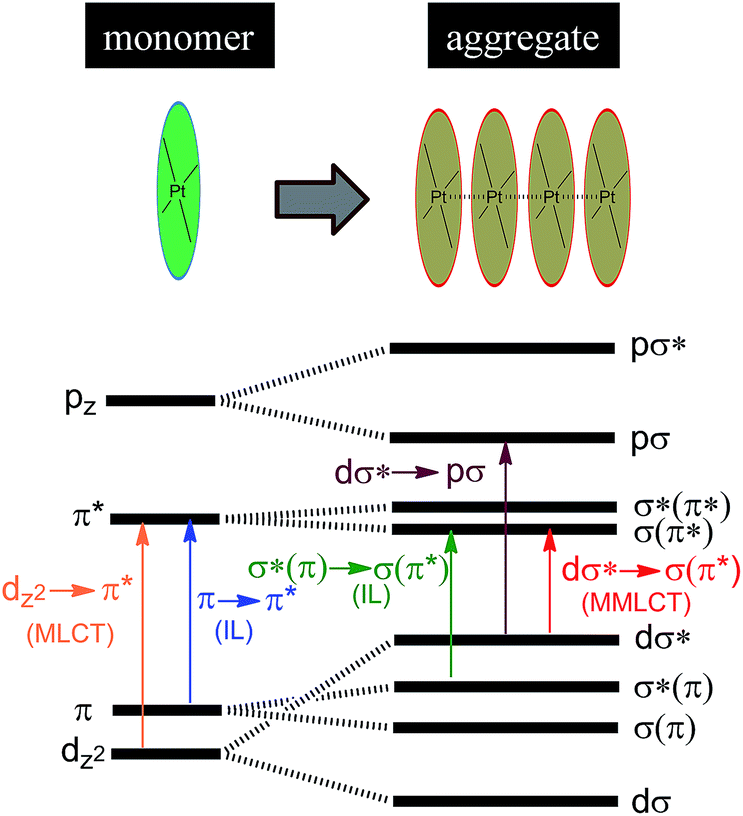 | ||
| Scheme 3 Proposed molecular orbital diagram illustrating d8–d8 and π–π interactions in Pt(II) complexes by Miskowski and co-workers in ref. 101. | ||
The intriguing spectroscopic properties dictated by the unique intermolecular Pt⋯Pt and/or π–π interactions between two ground state molecules or between an excited state molecule and a ground state one can be harnessed for material applications including: (1) excimeric emission for single-dopant WOLEDs; (2) formation of superstructures with optoelectronic applications.
4.1 Single-dopant WOLEDs
In principle, when doped at an appropriate concentration into a host material, the high-energy (in the blue to bluish green region) monomeric and low-energy (in the yellow to red region) excimeric emissions from Pt(II) emitters can be produced simultaneously, giving rise to a white-light emission (Fig. 8). In contrast to WOLEDs that typically comprise three primary colours (i.e. red, green, and blue) or two complementary emission colours (blue and yellow) from multiple phosphorescent emitters, single-dopant WOLEDs offer several advantages.103 First, precise control of the relative amount of each dopant for achieving an optimal cascade energy transfer within the emitting layer having multiple dopants is not necessary as only one Pt(II) dopant is used. Satisfactory white-light emission can be reached by adjusting the concentration of the single Pt(II) dopant that displays both highly efficient monomeric and excimeric emissions. Second, EL spectral aging due to different dopant aging processes can be excluded as only one molecular light-emitting species is involved, rendering a stable EL spectrum with long-term operation. In the literature, the first excimer-based single-dopant phosphorescent WOLED was demonstrated using 53.104 This complex was reported to display a blue monomeric and orange excimeric emission when doped into a host matrix.104,105 Later, pincer-type Pt(II) complexes 54 and 55, containing N^C^N ligands, were examined for this purpose.58,106 Complexes 54 and 55 show efficient blue phosphorescence (λmax = 453–472 nm) with quantum yields of 0.60–0.80 in dilute CH2Cl2, which is in stark contrast to 53 (ϕ = 0.02 in CH2Cl2).42 However, for all the complexes 53–55, white EL with a high efficiency and satisfactory CIE as well as CRI were not obtained due to either inefficient monomer emission or inappropriate excimer emission. In 2013, Li and co-workers reported the first high-performance single-dopant WOLED with an EQE > 20%, a satisfactory CIE of (0.33, 0.33), and a CRI of 80 using the pincer-type 56 supported by an NHC-based cyclometalated ligand.107 Recently, the EQE of a single-dopant WOLED was boosted to over 25% by Li's group and Che's group by using 57, 58 or 60 as the emitter.93,108 Complex 57 displays efficient blue emission in CH2Cl2 at λmax = 471 nm with a quantum yield and lifetime of 0.77 and 3.2 μs, respectively.108 This complex also shows very efficient excimeric emission at elevated concentrations when doped into a host material. As a consequence, the WOLED singly doped with 57 exhibited a peak EQE of 25.7%. A series of related Pt(II) emitters with sterically un-congested ligands, with 58 as an example, have been prepared; these complexes show both monomeric and excimeric emissions.109 Complex 59 exhibits a 3π–π*(O^N^C^N) centred bluish-green emission (λmax = 482, 512 nm; ϕ = 0.75) in dilute CH2Cl2.90 At elevated concentrations (up to 1 × 10−4 M), the emission intensity at 480–520 nm decreases with a concomitant increase in excimeric emission with a maximum at 620 nm. An efficient WOLED was fabricated using 59 as a single dopant. The peak EQE, current efficiency, and power efficiency were 16.5%, 71.0 cd A−1, and 55.8 lm W−1, respectively.90 Since steric bulkiness of the alkyl chains on the fluorene moiety may affect the intermolecular interactions, the butyl chains in 59 have been replaced with ethyl chains to facilitate intermolecular interactions in 60.37 Very recently, the EQE and power efficiency of the vacuum-deposited WOLED singly doped with 60 were reported to be 25.1% and 55.5 lm W−1 respectively in an optimized device structure.93 Che and co-workers also examined the EL performances of 59 and 60 in solution-processed white polymer OLEDs. A peak EQE, current efficiency, and power efficiency of 9.7%, 17.0 cd A−1, and 9.1 lm W−1, respectively, were observed for 59.90 For 60, the EQE was 12.73%, which slightly decreased to 11.51% at a high brightness of 1000 cd m−2, revealing a very low efficiency roll-off.37 The high EQE obtained for vacuum-deposited or solution-processed WOLEDs based on 57–60 suggests that Pt(II) emitters with highly efficient monomeric and excimeric emissions at appropriate energies are highly promising candidate materials for WOLEDs. However, for all WOLEDs singly doped with 57–60, the CIE (y > 0.41) and CRI < 80 revealed an unsatisfactory quality of the white light output. These could be attributed to an insufficiently high energy of the blue monomer emission. Hence, the development of highly efficient deep blue emitting Pt(II) complexes with structures that favour highly emissive excimer formation is crucial for attaining high-quality single-dopant WOLEDs.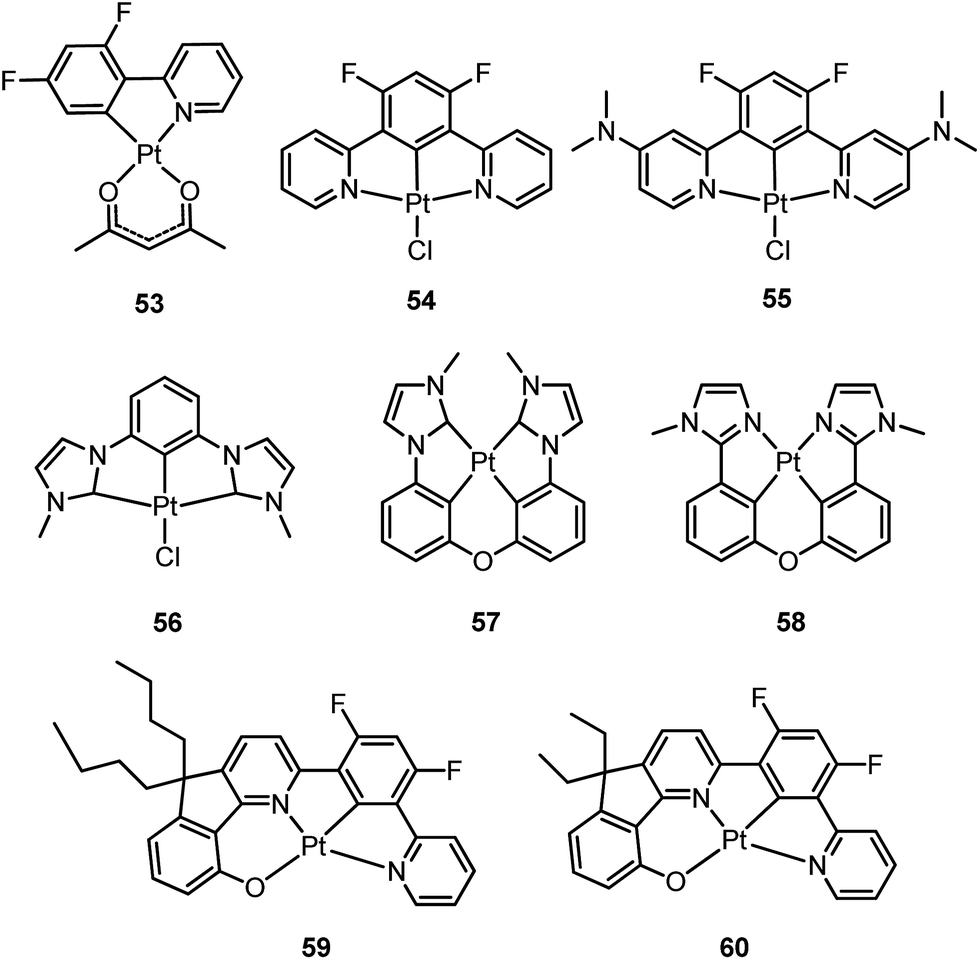
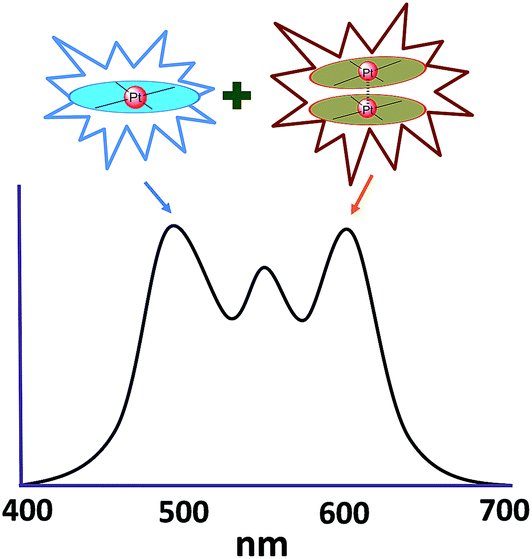 | ||
| Fig. 8 Schematic illustration of the combination of monomeric emission and excimeric emission to produce white light. | ||
4.2 Self-assembled functional molecular materials
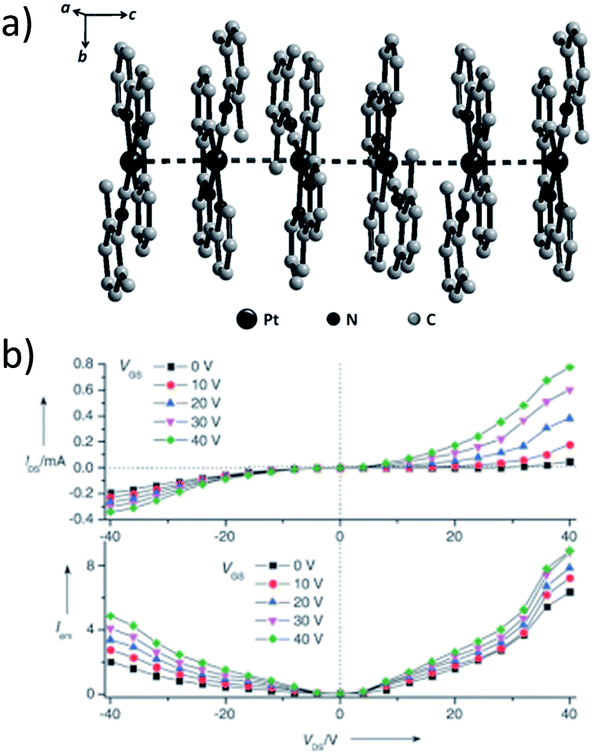
A subject of growing interest is the self-assembly of phosphorescent Pt(II) complexes into functional molecular materials. This bottom-up approach is illustrated in Scheme 4: (A) self-assembly of Pt(II) complexes into polynuclear aggregates, and (B) further aggregation into quasi-1D nano- or micro-structures (nanowire, nanorod, nanofiber, etc.),110–116 which can, in certain cases, (C) organize into soft materials such as gels.115,117–122 In comparison to the self-assembly of pure organic systems, the presence of phosphorescent Pt(II) units can provide a spectroscopic handle to follow the self-assembling process based on the high sensitivity of the emission of Pt(II) complexes to Pt⋯Pt and π–π interactions. Furthermore, the presence of Pt⋯Pt and Pt(II)–organic ligand interactions can enhance the stability of the resultant soft material. Due to the luminescent properties and highly ordered stacking arrangements, the resulting Pt(II)-superstructures with optoelectronic properties can be used for potential applications in materials science. In this context, we and others have explored the self-assembly behavior of a series of luminescent pincer-type Pt(II) complexes. For instance, we showed that cationic Pt(II) complex 61 can pack in a highly ordered manner with alternate Pt⋯Pt distances of 3.382 and 3.344 Å. The nanowires self-assembled from 61 display semiconducting properties and phosphorescence, permitting the fabrication of organic light-emitting field-effect transistors (OLEFETs) (Fig. 9).112 Che and Lu recently reported the synthesis of a strongly emissive Pt(II) allenylidene complex 63, which was found to form nanorods via self-assembly in CH3CN/H2O.113 Interestingly, transformation from nanorods to nanorings upon increasing the water fraction in the solvent mixture was observed.113 Charge transport is conceived to be facilitated along highly ordered one-dimensional Pt⋯Pt and π–π packing chains. A high field-effect electron mobility of up to 20 cm2 V−1 s−1 was recorded with a transistor fabricated from a single microcrystal of 64.114 De Cola and co-workers developed a neutral Pt(II) emitter, 65, supported by dianionic tridentate terpyridine-like ligand. This complex is non-emissive in dilute solution and undergoes self-assembly to give gelating nanowires accompanied by a striking phosphorescence switch-on (ϕ = 0.90).115 Apart from the 1D nano- and micro-structures, Che and co-workers showed that the neutral Pt(II) complex 66 underwent self-assembly into quasi-2D nanosheets driven by the orthogonal Pt⋯Pt and C–H⋯π(CC) interactions. These nano-sheets showed NIR phosphorescence and visible light-modulated electronic conductivity (Fig. 10).123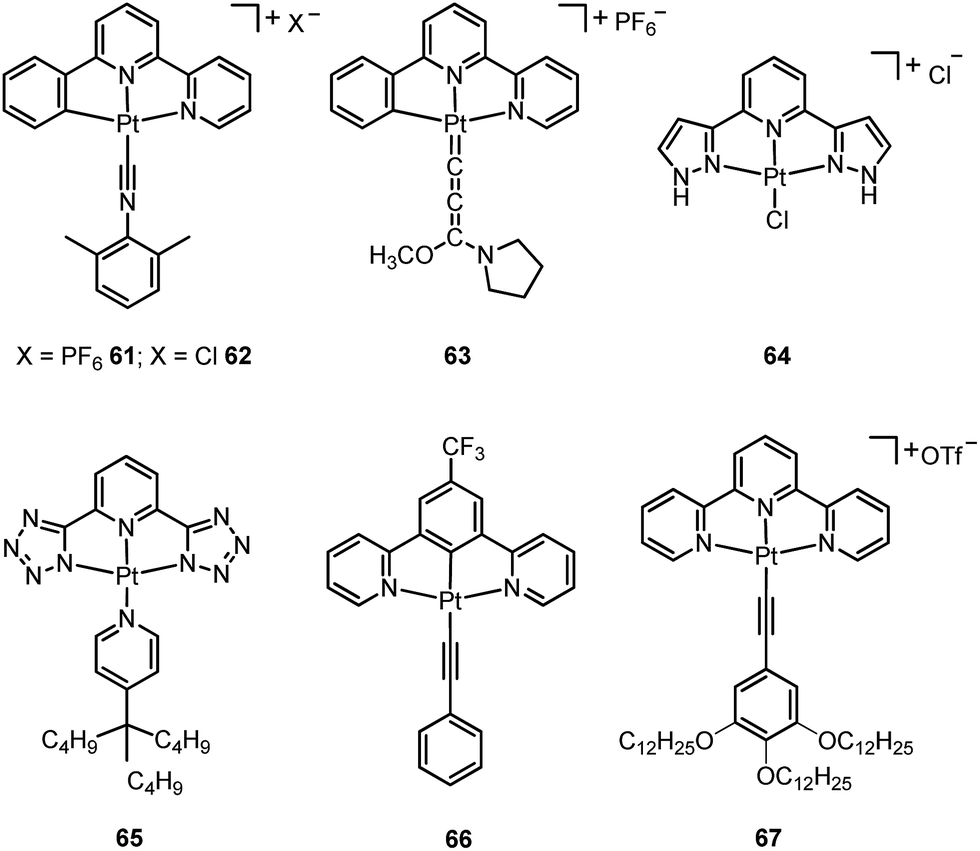
 | ||
| Scheme 4 The illustration of self-assembly of Pt(II) complexes into 1D nanostructures and soft materials. | ||
 | ||
| Fig. 9 (a) Crystal packing diagram of 61·H2O. The PF6− and water molecules are omitted for clarity. (b) Output characteristics (IDSvs. VDS) and electroluminescence intensity (I) of an OLEFET device with nanowires of 61 after annealing at 350 K (adapted with permission from ref. 112. Copyright 2008, Wiley-VCH Verlag GmbH & Co. KGaA). | ||
 | ||
| Fig. 10 (a) Output characteristics (IDSvs. VDS) of a FET device with nanosheets of 66. (b) Transient measurement (VDS = VG = ± 40 V) of the FET device. The transient channel current was recorded with a 40 mW cm−2 light switching on and off every five seconds in a 90 second period. IDS = drain-source current, VDS = drain-source voltage, VG = gate voltage (adapted with permission from ref. 123. Copyright 2009, Wiley-VCH Verlag GmbH & Co. KGaA). | ||
Yam and co-workers have extensively studied supramolecular soft materials based on luminescent alkynylplatinum(II) complexes with tridentate N-donor ligands, particularly terpyridine.124 It was reported that 67 can form a metallo-gel in DMSO with a low critical gelation concentration of 4.4 mg mL−1.118 Drastic colour and emission changes were observed upon the sol-to-gel transition (Fig. 11a and b), revealing that Pt⋯Pt and π–π interactions were involved in the gel formation. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images of the xerogel showed a network of fibrous structures with a diameter and length of approximately 470 nm and 10 μm, respectively (Fig. 11c and d). Unlike the supramolecular soft materials formed from organic molecules bearing H-bonding motifs and/or long soft alkyl chains,125 the Pt⋯Pt and π–π interactions between organoplatinum(II) units provide the driving force for self-assembly. On the other hand, the anion of cationic Pt(II) complexes has been found to play a crucial role in determining both the extent of intermolecular interactions and the interactions with the solvent medium, thereby providing an entry for tuning the softness of the self-assembled structures.126,127 As a result, simple and easy-to-synthesize Pt(II) molecules, not containing H-bonding motifs and long alkyl chains, can be used as building blocks for supramolecular soft materials. For example, 62, an analogue of 61 with a Cl− anion, is soluble in water and was observed to form a chromic meso-phase in water with a critical concentration of 1.5 wt%.119
 | ||
| Fig. 11 Photographs of 67 in DMSO in (a) the gel form at room temperature, (b) the sol form at elevated temperature. (c) TEM and (d) SEM images of the xerogel of 67 formed in DMSO (adapted from ref. 118 with permission from the Royal Society of Chemistry). | ||
The manipulation of the molecular packing arrangement in the solid state by external mechanical stimuli renders Pt(II) emitters127–131 as promising mechanoluminescent (ML) candidates for the development of sensory and data storage materials. We synthesized another new type of luminescent Pt(II) complex, 68, with tetradentate O^N^N^C ligands (Scheme S2, ESI†). Complex 68 is an analogue of the Pt(II)–O^N^C^N complex 36. Yellow and orange crystal polymorphs of 68 in monoclinic and triclinic forms, respectively, were obtained by slow evaporation of a CH2Cl2 solution (Fig. 12a and b). The intermolecular distances between adjacent molecules in the monoclinic and triclinic crystal forms are 3.524 and 3.468 Å, respectively, revealing the presence of weak π–π interactions. There are no Pt⋯Pt interactions in either case, since the determined closest Pt⋯Pt distance is longer than 4.8 Å. The yellow crystalline solid (triclinic form) precipitated from CH2Cl2/hexane displays a vibronic-structured emission (λmax = 541, 571, 621 (sh) nm). Upon grinding, the emission peak first shifted to 588 nm and then a broad emission band at 681 nm was obtained (Fig. 12c and d). This red-shift of emission is probably due to enhanced intermolecular Pt⋯Pt and/or π–π interactions. Addition of Et2O or another solvent to the ground solid restored the colour and emission spectrum of the yellow crystalline solid. Interestingly, soaking the ground sample with Et2O for about 2 h resulted in an orange crystalline solid, of which the PXRD pattern was in line with the simulated pattern of the monoclinic crystal form. The interconversions between different forms of 68 were confirmed by PXRD studies (Fig. S5, ESI†). To the best of our knowledge, 68 represents the first example of a Pt(II) complex supported by a tetradentate ligand that shows polymorphic and mechanochromic luminescence properties.
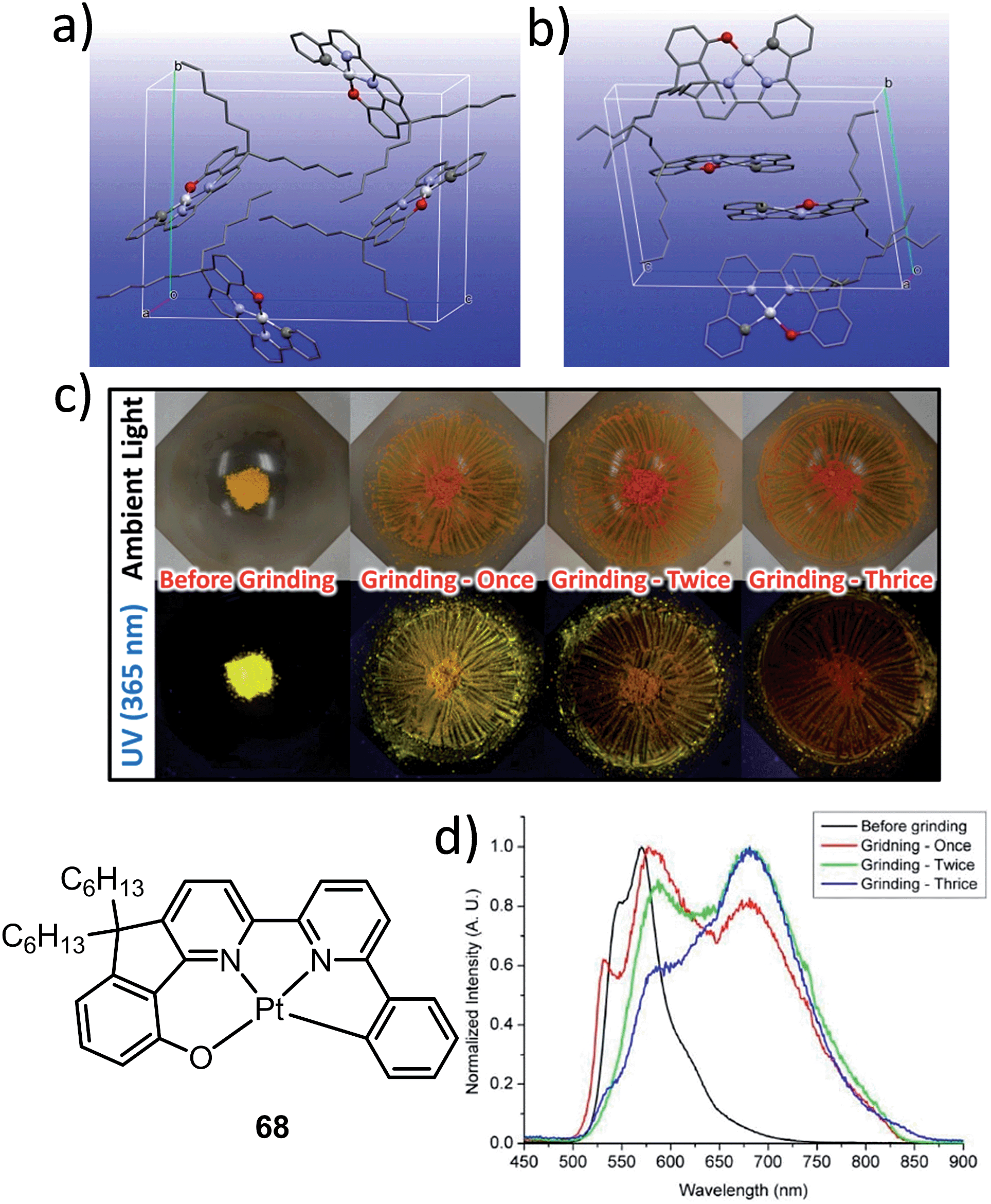 | ||
| Fig. 12 Crystal packing of 68 in (a) monoclinic form and (b) triclinic form. (c) Photographs of the yellow crystalline solid of 68 before and after grinding under ambient and UV light irradiation (365 nm). (d) Emission spectra of 68 (yellow triclinic form) before and after grinding. | ||
The self-organization of d8 transition metal complexes, such as those of Rh(I), into luminescent functional assemblies constitutes a fascinating topic within supramolecular chemistry because of their unique photophysical properties imparted by metal⋯metal interactions.132,133 The main driving forces have been generally attributed to the interplay of intermolecular metal⋯metal and/or π–π interactions (Scheme 3).134 In this context, the nature of the intermolecular interactions and the contribution of each interaction component, such as metallophilicity, to the stabilization of aggregates has been an ongoing debate due to the difficulty in accessing relatively high-level theory for computational studies.135–139 Recently, a dispersion-corrected DFT method has been shown to be a viable approach to provide a quantitative assessment of the contribution of each interaction component to the intermolecular stabilizing energy of d8 Rh(I) and d10 Au(I) dimers.139,140 It was shown that the d8⋯d8 interactions of Rh(I) dimers only account for a small fraction (10–15%) of the dispersion contribution to the total binding energy when π-conjugated ligands are present.139 In the case of relatively large Au(I) systems, the weak ligand–ligand interaction dominates dimer formation.140 The role of dispersion interactions in driving the self-assembly of luminescent Pt(II) systems has not been studied and its interplay with other weak non-covalent interactions in determining the morphology of the self-assembly structures has not been defined. This prompted us to examine the nature and weight of Pt⋯Pt interactions by studying the dimer of pincer-type Pt(II) complex 61 which has a strong tendency to aggregate (Fig. 9).112 As depicted in Fig. 13, at a Pt⋯Pt distance of 3.28 Å, the dispersive Pt⋯Pt, Pt⋯ligand and ligand⋯ligand interactions amount to 0.62, 6.98, and 24.81 kcal mol−1, respectively, revealing that the London dispersive attraction dominates the dimer formation of the Pt(II) molecules.
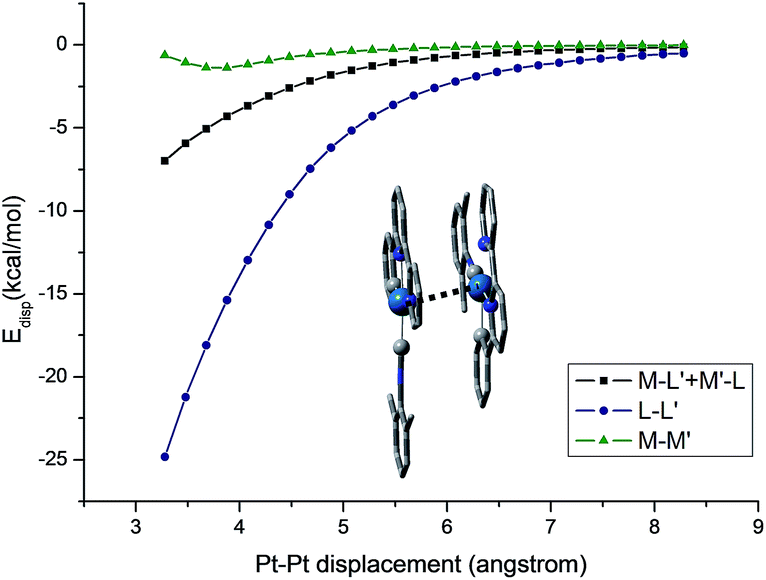 | ||
| Fig. 13 Dispersion contributions of the metal atoms and ligand fragments to the dimer [61–PF6]22+. The Pt⋯Pt′ distance was used as a reaction coordinate. | ||
Inclusion of spin–orbit coupling (SOC) is also essential to understanding the photophysical properties of d8–d8 diplatinum complexes. Recently, spin–orbit TDDFT (SO-TDDFT) calculations were performed on [Pt2(μ-P2O5H2)4]4− (abbreviated as Pt(pop)) and its perfluoroborated derivative, [Pt2(μ-P2O5(BF2)2)4]4− (abbreviated as Pt(pop-BF2)) to shed light on the ISC mechanism in these complexes.141 It was highlighted that the 1dσ*pσ → 3dσ*pσ ISC is facilitated by second-order spin–orbit coupling with the high-lying ligand-to-metal–metal charge transfer (LMMCT) dπpσ and pπpσ excited states. Owing to the calculation results that Pt(pop-BF2) has a larger energy gap between the dσ*pσ and the LMMCT excited states, Pt(pop-BF2) has a slower ISC rate than Pt(pop). It was also proposed that the structural flexibility of Pt(pop) facilitates transient distortions which allows enhanced spin–orbit coupling (SOC) between the 1dσ*pσ and 3dσ*pσ excited states, leading to a much faster ISC rate in Pt(pop). As a result, Pt(pop) shows dominant phosphorescent emissions but dual fluorescence-phosphorescence is observed for Pt(pop-BF2).
5 Conclusion and outlook
Multidentate ligands containing strong σ-donor atoms are advantageous for the construction of robust, phosphorescent Pt(II) emitters. The strong σ-donor strength and high rigidity of the ligand scaffold can push the d–d states well above the triplet emitting state thereby minimizing excited state structural distortion and non-radiative decay. Due to strong chelate effect, the stability of Pt(II) complexes can be significantly improved with the use of multidentate ligands. Following these design principles, a number of phosphorescent Pt(II) complexes that are photo-stable, kinetically inert and chemically stable under various conditions have been developed. Using terdentate or tetradentate cyclometalating ligands, tremendous progress has been made in tuning the emission energy and emission quantum yield as well as excited state lifetime of Pt(II) complexes. OLEDs based on luminescent Pt(II) complexes emanating red, yellow, green, or blue light with maximum EQEs higher than 20% have been reported. The unique excimeric emission from luminescent Pt(II) complexes has been harnessed in the fabrication of efficient single-dopant WOLEDs. Luminescent Pt(II) complexes have also been shown to have rich photochemistry; for example, they act as efficient photocatalysts for organic transformations. Intermolecular Pt⋯Pt and π–π interactions provide directional driving forces for the anisotropic growth of 1D or 2D nano- or microstructures with interesting optoelectronic properties.The planar coordination geometry also endows the phosphorescent Pt(II) complexes with axial coordination sites for metal–substrate interactions, forming an operating principle for luminescent sensory applications such as cellular imaging. Numerous Pt(II) terpyridyl (trpy) complexes have been recognized to be effective DNA and RNA intercalators, owing to the presence of a planar Pt-trpy moiety that favours π–π stacking interactions with the base pairs of nucleic acids.142,143 However, Pt(II) terpyridyl complexes usually suffer from inferior luminescence properties. To circumvent the d–d state-induced emission quenching of Pt(II) terpyridyl complexes, physiologically stable luminescent Pt(II) complexes supported by a cyclometalated ligand or tetradentate ligand were developed and studied.144–146 The binding of these complexes with bio-molecules were observed to induce elevations of emission intensity and/or shifts of emission maxima, as a result of the reduced excited state geometry distortion and/or the formation of new emissive adducts. In this context, the good kinetic stability of Pt(II) complexes with tetradentate ligands in solution, together with their luminescent properties, makes them highly promising bio-sensors.
Che and co-workers described their molecular design studies of a Pt(II) Schiff base complex 69 containing peripheral amine side chains as a c-myc G-quadruplex DNA binder.147 Based on UV-Vis absorption and NMR spectroscopic measurements, this complex is stable in aqueous solution for 72 h at room temperature. Complex 69 is weakly emissive in aqueous Tris/KCl buffer solution. Upon addition of the G4A1 quadruplex DNA, an intense emission at 652 nm developed and the emission intensity increased 8-fold at a [G-quadruplex]/[69] ratio ≥ 20 (Fig. 14). By absorption titration experiments, the binding constant was determined to be 1.72 ± 0.26 M−1 at 20 °C, which is approximately ten-fold of that with non-quadruplex double-stranded DNA molecules. An external end-stacking mode between 69 and G-quadruplex DNA was suggested based on the findings from NMR titration experiments and molecular modeling studies. To the best of our knowledge, this represents the first system of a Pt(II) complex supported by tetradentate ligands as a luminescent bio-probe.
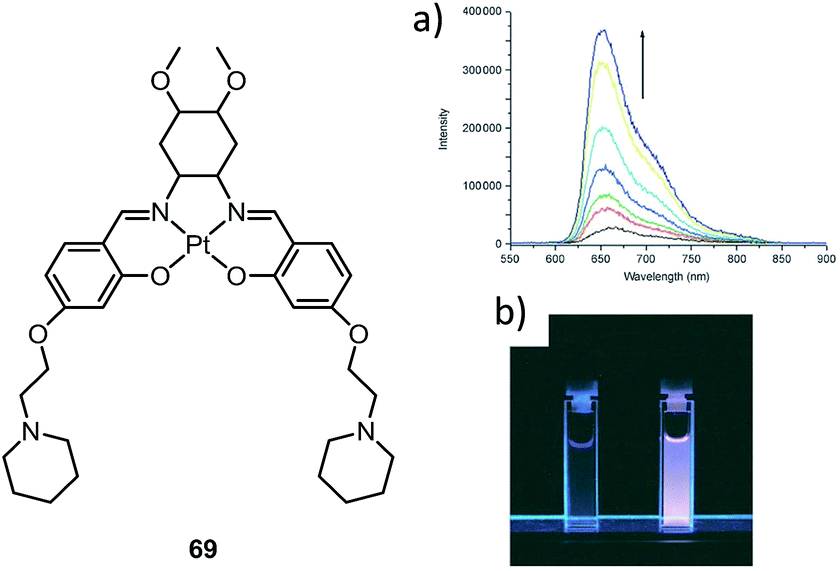 | ||
| Fig. 14 (a) Emission spectral traces of 69 (50 μM) in Tris/KCl buffer with increasing concentrations of G-quadruplex DNA at 20 °C. (b) Photographs of 69 in the absence or the presence of G-quadruplex DNA (adapted with permission from ref. 147. Copyright 2009, Wiley-VCH Verlag GmbH & Co. KGaA). | ||
Complex 70 has been reported to display an intense bluish-green emission at low concentrations (<10−5 M in CH2Cl2) (λmax = 479, 510 nm) while giving an intense excimeric red emission (λmax = 624 nm) at elevated concentrations close to 10−4 M.37 Complex 70 was found to aggregate into micron sized rod-like structures in both organic and aqueous media (Fig. S6, ESI†). These rods showed a strong red emission under ambient conditions. We examined the cell-imaging properties of 70. Cervical epithelioid carcinoma (HeLa) cells treated with 70 initially displayed a strong orange emission, revealing the presence of both monomeric and excimeric emissions from 70. Interestingly, the formation of an emissive rod-aggregate was observed in the cellular environment after a 12 h treatment with 70. No obvious morphological change was identified after a 24 h treatment at 10 μM, suggesting 70 is of low cytotoxicity. The co-staining analysis revealed that the complex selectively accumulates in the endoplasmic reticulum (ER) prior to aggregate formation (Fig. 15). Hence, our results show that 70 localizes in ER and subsequently self-assembles into a rod-like morphology. The self-assembled superstructures of luminescent Pt(II) complexes have been recently recognized to afford advantages in bioimaging applications because of their stable and persistent emission by shielding oxygen- and solvent molecule-induced emission quenching, and their more biocompatible low-energy emission.148–150 Herein, we have demonstrated the application of a highly luminescent Pt(II) complex with a tetradentate ligand in cell-imaging and, more importantly, the control of the self-assembly behaviour of exogenous molecules in a cellular environment.
 | ||
| Fig. 15 Fluorescence imaging of cells treated with 70. (a) Imaging of HeLa cells treated with 70 (5 μM) for 4 and 24 h (λex = 440 nm; λem > 590 nm). (b) Co-localization of 70 and ER-RFP (red fluorescent protein) revealed that the complex selectively accumulates in the ER. Cells were treated with 70 (5 μM) for 2 h. | ||
Despite the striking advances that have been made in the development of robust, efficient Pt(II) emitters, there are fundamental issues that need to be resolved prior to their practical applications. These issues include: (1) the limited number of Pt(II) complexes that display efficient deep blue emission. In view of their importance in full-colour display and white-light illumination and in the design of new strongly oxidizing and reducing excited state species for visible light-driven organic transformations, a deep understanding of the structure–photophysics relationship of, and further development of deep blue emitting Pt(II) complexes are required; (2) the pursuit of stable NIR emitting Pt(II) complexes for medical applications because of the biocompatibility and high transmittance of NIR photons; (3) the small number of Pt(II) complexes reported to display efficient excimeric emissions. As the structural features affecting the photophysical properties of excimeric emissions are not well understood, the rational design and exploitation of excimeric emissions of luminescent Pt(II) complexes remains a formidable challenge. Nevertheless, robust and highly phosphorescent Pt(II) complexes would have tremendous applications in diverse disciplines. In addition to OLED applications, the practical utilization of luminescent Pt(II) complexes, supported by tetradentate ligands, as robust cell imaging agents, new photocatalysts, and building blocks for self-assembled functional molecular materials has a bright future.
Note added after first publication
This article replaces the version published on 7th January 2016, which contained errors in the caption for Fig. 5.Acknowledgements
This work was supported by the National Key Basic Research Program of China (No. 2013CB834802), the University Grants Committee (Area of Excellence Scheme AoE/P-03/08), and the Innovation and Technology Commission of the HKSAR Government (ITS/084/14). This work was also supported by the Guangdong Special Project of the Introduction of Innovative R & D Teams.Notes and references
- A. F. Rausch, H. H. H. Homeier and H. Yersin, Top. Organomet. Chem., 2010, 29, 193 CrossRef CAS.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef CAS.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638 RSC.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926 CrossRef CAS PubMed.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259 RSC.
- V. H. Houlding and V. M. Miskowski, Coord. Chem. Rev., 1991, 111, 145 CrossRef CAS.
- S.-W. Lai and C.-M. Che, Top. Curr. Chem., 2004, 241, 27 CrossRef CAS.
- V. Guerchais and J.-L. Fillaut, Coord. Chem. Rev., 2011, 255, 2448 CrossRef CAS.
- K. M.-C. Wong and V. W.-W. Yam, Coord. Chem. Rev., 2007, 251, 2477 CrossRef CAS.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC.
- J. E. McGarrah, Y.-J. Kim, M. Hissler and R. Eisenberg, Inorg. Chem., 2001, 40, 4510 CrossRef CAS PubMed.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726 CrossRef CAS PubMed.
- D. Zhang, L.-Z. Wu, L. Zhou, X. Han, Q.-Z. Yang, L.-P. Zhang and C.-H. Tung, J. Am. Chem. Soc., 2004, 126, 3440 CrossRef CAS PubMed.
- J.-J. Zhong, Q.-Y. Meng, G.-X. Wang, Q. Liu, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Chem.–Eur. J., 2013, 19, 6443 CrossRef CAS PubMed.
- F. Guo and W. Sun, J. Phys. Chem. B, 2006, 110, 15029 CrossRef CAS PubMed.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762 CrossRef CAS.
- D.-L. Ma, H.-Z. He, K.-H. Leung, D. S.-H. Chan and C.-H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666 CrossRef CAS PubMed.
- M. P. Coogan and V. Fernández-Moreira, Chem. Commun., 2014, 50, 384 RSC.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401 CrossRef CAS.
- E. Baranoff, S. Suàrez, P. Bugnon, C. Barolo, R. Buscaino, R. Scopelliti, L. Zuppiroli, M. Graetzel and M. K. Nazeeruddin, Inorg. Chem., 2008, 47, 6575 CrossRef CAS PubMed.
- J. M. Fernández-Hernández, C.-H. Yang, J. I. Beltrán, V. Lemaur, F. Polo, R. Fröhlich, J. Cornil and L. De Cola, J. Am. Chem. Soc., 2011, 133, 10543 CrossRef PubMed.
- E. Baranoff, H. J. Bolink, F. De Angelis, S. Fantacci, D. Di Censo, K. Djellab, M. Grätzel and M. K. Nazeeruddin, Dalton Trans., 2010, 39, 8914 RSC.
- S. Schmidbauer, A. Hohenleutner and B. König, Adv. Mater., 2013, 25, 2114 CrossRef CAS PubMed.
- S. Archer and J. A. Weinstein, Coord. Chem. Rev., 2012, 256, 2530 CrossRef CAS.
- L. De Cola, F. Barigelletti, V. Balzani, P. Belser, A. von Zelewsky, F. Voegtle, F. Ebmeyer and S. Grammenudi, J. Am. Chem. Soc., 1988, 110, 7210 CrossRef CAS.
- F. Barigelletti, L. De Cola, V. Balzani, P. Belser, A. von Zelewsky, F. Voegtle, F. Ebmeyer and S. Grammenudi, J. Am. Chem. Soc., 1989, 111, 4662 CrossRef CAS.
- A. M. Prokhorov, T. Hofbeck, R. Czerwieniec, A. F. Suleymanova, D. N. Kozhevnikov and H. Yersin, J. Am. Chem. Soc., 2014, 136, 9637 CrossRef CAS PubMed.
- G. S. M. Tong and C.-M. Che, Chem.–Eur. J., 2009, 15, 7225 CrossRef CAS PubMed.
- P.-K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084 CrossRef CAS PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic ChemistryWiley and Sons, New York, 6th edn, 1999 Search PubMed.
- R. F. Beeston, S. L. Larson and M. C. Fitzgerald, Inorg. Chem., 1989, 28, 4187 CrossRef CAS.
- H. Duerr, R. Schwarz, C. Andreis and I. Willner, J. Am. Chem. Soc., 1993, 115, 12362 CrossRef CAS.
- A. Ruggi, M. Berenguel Alonso, D. N. Reinhoudt and A. H. Velders, Chem. Commun., 2010, 46, 6726 RSC.
- G. Cheng, P.-K. Chow, S. C. F. Kui, C.-C. Kwok and C.-M. Che, Adv. Mater., 2013, 25, 6765 CrossRef CAS PubMed.
- L. Chassot, E. Mueller and A. von Zelewsky, Inorg. Chem., 1984, 23, 4249 CrossRef CAS.
- L. Chassot and A. von Zelewsky, Inorg. Chem., 1987, 26, 2814 CrossRef CAS.
- D. Sandrini, M. Maestri, V. Balzani, L. Chassot and A. von Zelewsky, J. Am. Chem. Soc., 1987, 109, 7720 CrossRef CAS.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055 CrossRef CAS PubMed.
- S. Huo, J. Carroll and D. A. K. Vezzu, Asian J. Org. Chem., 2015, 4, 1210 CrossRef CAS.
- S.-Y. Chang, Y.-M. Cheng, Y. Chi, Y.-C. Lin, C.-M. Jiang, G.-H. Lee and P.-T. Chou, Dalton Trans., 2008, 6901 RSC.
- S.-W. Lai, M. C.-W. Chan, T.-C. Cheung, S.-M. Peng and C.-M. Che, Inorg. Chem., 1999, 38, 4046 CrossRef CAS.
- W. Lu, B.-X. Mi, M. C. W. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, J. Am. Chem. Soc., 2004, 126, 4958 CrossRef CAS PubMed.
- S. C. F. Kui, I. H. T. Sham, C. C. C. Cheung, C.-W. Ma, B. Yan, N. Zhu, C.-M. Che and W.-F. Fu, Chem.–Eur. J., 2007, 13, 417 CrossRef CAS PubMed.
- B.-P. Yan, C. C. C. Cheung, S. C. F. Kui, H.-F. Xiang, V. A. L. Roy, S.-J. Xu and C.-M. Che, Adv. Mater., 2007, 19, 3599 CrossRef CAS.
- C.-L. Ho, H. Li and W.-Y. Wong, J. Organomet. Chem., 2014, 751, 261 CrossRef CAS.
- Q. Xu, W.-f. Fu, G. Zhang, Z. Bian, J. Zhang, X. Han and W. Xu, Catal. Commun., 2008, 10, 49 CrossRef CAS.
- D. Ravindranathan, D. A. K. Vezzu, L. Bartolotti, P. D. Boyle and S. Huo, Inorg. Chem., 2010, 49, 8922 CrossRef CAS PubMed.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609 CrossRef CAS PubMed.
- D. J. Cárdenas, A. M. Echavarren and M. C. Ramírez de Arellano, Organometallics, 1999, 18, 3337 CrossRef.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690 CrossRef CAS PubMed.
- E. Rossi, L. Murphy, P. L. Brothwood, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, M. Cocchi and J. A. G. Williams, J. Mater. Chem., 2011, 21, 15501 RSC.
- E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, F. Demartin, M. Cocchi, P. Brulatti, V. Fattori and J. A. G. Williams, Chem. Commun., 2012, 48, 3182 RSC.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312 CrossRef CAS PubMed.
- W. Mroz, C. Botta, U. Giovanella, E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, A. Valore and J. A. G. Williams, J. Mater. Chem., 2011, 21, 8653 RSC.
- L. Murphy, P. Brulatti, V. Fattori, M. Cocchi and J. A. G. Williams, Chem. Commun., 2012, 48, 5817 RSC.
- M. Maestri, C. Deuschel-Cornioley and A. von Zelewsky, Coord. Chem. Rev., 1991, 111, 117 CrossRef CAS.
- V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong, X.-X. Lu, K.-K. Cheung and N. Zhu, Chem.–Eur. J., 2002, 8, 4066 CrossRef CAS.
- W. Lu, M. C. W. Chan, K.-K. Cheung and C.-M. Che, Organometallics, 2001, 20, 2477 CrossRef CAS.
- S. C. F. Kui, S. S.-Y. Chui, C.-M. Che and N. Zhu, J. Am. Chem. Soc., 2006, 128, 8297 CrossRef CAS PubMed.
- J. R. Berenguer, E. Lalinde and J. Torroba, Inorg. Chem., 2007, 46, 9919 CrossRef CAS PubMed.
- S. Fuertes, S. K. Brayshaw, P. R. Raithby, S. Schiffers and M. R. Warren, Organometallics, 2011, 31, 105 CrossRef.
- S. C. F. Kui, F.-F. Hung, S.-L. Lai, M.-Y. Yuen, C.-C. Kwok, K.-H. Low, S. S.-Y. Chui and C.-M. Che, Chem.–Eur. J., 2012, 18, 96 CrossRef CAS PubMed.
- M. Ikai, F. Ishikawa, N. Aratani, A. Osuka, S. Kawabata, T. Kajioka, H. Takeuchi, H. Fujikawa and Y. Taga, Adv. Funct. Mater., 2006, 16, 515 CrossRef CAS.
- C. Borek, K. Hanson, P. I. Djurovich, M. E. Thompson, K. Aznavour, R. Bau, Y. Sun, S. R. Forrest, J. Brooks, L. Michalski and J. Brown, Angew. Chem., Int. Ed., 2007, 46, 1109 CrossRef CAS PubMed.
- J. R. Sommer, A. H. Shelton, A. Parthasarathy, I. Ghiviriga, J. R. Reynolds and K. S. Schanze, Chem. Mater., 2011, 23, 5296 CrossRef CAS.
- K. R. Graham, Y. Yang, J. R. Sommer, A. H. Shelton, K. S. Schanze, J. Xue and J. R. Reynolds, Chem. Mater., 2011, 23, 5305 CrossRef CAS.
- Y.-Y. Lin, S.-C. Chan, M. C. W. Chan, Y.-J. Hou, N. Zhu, C.-M. Che, Y. Liu and Y. Wang, Chem.–Eur. J., 2003, 9, 1263 CrossRef CAS PubMed.
- C.-M. Che, S.-C. Chan, H.-F. Xiang, M. C. W. Chan, Y. Liu and Y. Wang, Chem. Commun., 2004, 1484 RSC.
- C.-M. Che, C.-C. Kwok, S.-W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Zhu and H. Yersin, Chem.–Eur. J., 2010, 16, 233 CrossRef CAS PubMed.
- G. S. M. Tong, P. K. Chow, W.-P. To, W.-M. Kwok and C.-M. Che, Chem.–Eur. J., 2014, 20, 6433 CrossRef CAS PubMed.
- L. Zhou, C.-L. Kwong, C.-C. Kwok, G. Cheng, H. Zhang and C.-M. Che, Chem.–Asian J., 2014, 9, 2984 CrossRef CAS PubMed.
- J. Zhang, F. Zhao, X. Zhu, W.-K. Wong, D. Ma and W.-Y. Wong, J. Mater. Chem., 2012, 22, 16448 RSC.
- H.-F. Xiang, S.-C. Chan, K. K.-Y. Wu, C.-M. Che and P. T. Lai, Chem. Commun., 2005, 1408 RSC.
- F. Nisic, A. Colombo, C. Dragonetti, D. Roberto, A. Valore, J. M. Malicka, M. Cocchi, G. R. Freeman and J. A. G. Williams, J. Mater. Chem. C, 2014, 2, 1791 RSC.
- K.-Y. Liao, C.-W. Hsu, Y. Chi, M.-K. Hsu, S.-W. Wu, C.-H. Chang, S.-H. Liu, G.-H. Lee, P.-T. Chou, Y. Hu and N. Robertson, Inorg. Chem., 2015, 54, 4029 CrossRef CAS PubMed.
- K. Feng, C. Zuniga, Y.-D. Zhang, D. Kim, S. Barlow, S. R. Marder, J. L. Brédas and M. Weck, Macromolecules, 2009, 42, 6855 CrossRef CAS.
- D. A. K. Vezzu, J. C. Deaton, J. S. Jones, L. Bartolotti, C. F. Harris, A. P. Marchetti, M. Kondakova, R. D. Pike and S. Huo, Inorg. Chem., 2010, 49, 5107 CrossRef CAS PubMed.
- H. Fukagawa, T. Shimizu, H. Hanashima, Y. Osada, M. Suzuki and H. Fujikake, Adv. Mater., 2012, 24, 5099 CrossRef CAS PubMed.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS PubMed.
- D. H. Kim, N. S. Cho, H.-Y. Oh, J. H. Yang, W. S. Jeon, J. S. Park, M. C. Suh and J. H. Kwon, Adv. Mater., 2011, 23, 2721 CrossRef CAS PubMed.
- C.-H. Fan, P. Sun, T.-H. Su and C.-H. Cheng, Adv. Mater., 2011, 23, 2981 CrossRef CAS PubMed.
- S. Huo, C. F. Harris, D. A. K. Vezzu, J. P. Gagnier, M. E. Smith, R. D. Pike and Y. Li, Polyhedron, 2013, 52, 1030 CrossRef CAS.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952 CrossRef CAS.
- K. Li, X. Guan, C.-W. Ma, W. Lu, Y. Chen and C.-M. Che, Chem. Commun., 2011, 47, 9075 RSC.
- A. Meyer, Y. Unger, A. Poethig and T. Strassner, Organometallics, 2011, 30, 2980 CrossRef CAS.
- K. Li, G. Cheng, C. Ma, X. Guan, W.-M. Kwok, Y. Chen, W. Lu and C.-M. Che, Chem. Sci., 2013, 4, 2630 RSC.
- S. C. F. Kui, P. K. Chow, G. S. M. Tong, S.-L. Lai, G. Cheng, C.-C. Kwok, K.-H. Low, M. Y. Ko and C.-M. Che, Chem.–Eur. J., 2013, 19, 69 CrossRef CAS PubMed.
- S. C. F. Kui, P. K. Chow, G. Cheng, C.-C. Kwok, C. L. Kwong, K.-H. Low and C.-M. Che, Chem. Commun., 2013, 49, 1497 RSC.
- S.-L. Lai, W.-Y. Tong, S. C. F. Kui, M.-Y. Chan, C.-C. Kwok and C.-M. Che, Adv. Funct. Mater., 2013, 23, 5168 CrossRef CAS.
- G. Cheng, S. C. F. Kui, W.-H. Ang, M.-Y. Ko, P.-K. Chow, C.-L. Kwong, C.-C. Kwok, C. Ma, X. Guan, K.-H. Low, S.-J. Su and C.-M. Che, Chem. Sci., 2014, 5, 4819 RSC.
- X.-C. Hang, T. Fleetham, E. Turner, J. Brooks and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6753 CrossRef CAS PubMed.
- E. Turner, N. Bakken and J. Li, Inorg. Chem., 2013, 52, 7344 CrossRef CAS PubMed.
- T. Fleetham, G. Li, L. Wen and J. Li, Adv. Mater., 2014, 26, 7116 CrossRef CAS PubMed.
- G. Li, T. Fleetham, E. Turner, X.-C. Hang and J. Li, Adv. Opt. Mater., 2015, 3, 390 CrossRef CAS.
- J. A. Bailey, V. M. Miskowski and H. B. Gray, Inorg. Chem., 1993, 32, 369 CrossRef CAS.
- J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34, 4591 CrossRef CAS.
- H.-K. Yip, C.-M. Che, Z.-Y. Zhou and T. C. W. Mak, J. Chem. Soc., Chem. Commun., 1992, 1369 RSC.
- V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446 CrossRef CAS.
- V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529 CrossRef CAS.
- B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585 CrossRef.
- V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D'Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, New J. Chem., 2002, 26, 1171 RSC.
- E. L. Williams, K. Haavisto, J. Li and G. E. Jabbour, Adv. Mater., 2007, 19, 197 CrossRef CAS.
- X. Yang, Z. Wang, S. Madakuni, J. Li and G. E. Jabbour, Adv. Mater., 2008, 20, 2405 CrossRef CAS.
- T. Fleetham, J. Ecton, Z. Wang, N. Bakken and J. Li, Adv. Mater., 2013, 25, 2573 CrossRef CAS PubMed.
- G. Li, T. Fleetham and J. Li, Adv. Mater., 2014, 26, 2931 CrossRef CAS PubMed.
- T. Fleetham, L. Huang and J. Li, Adv. Funct. Mater., 2014, 24, 6066 CrossRef CAS.
- Y. Sun, K. Ye, H. Zhang, J. Zhang, L. Zhao, B. Li, G. Yang, B. Yang, Y. Wang, S.-W. Lai and C.-M. Che, Angew. Chem., Int. Ed., 2006, 45, 5610 CrossRef CAS PubMed.
- W. Lu, V. A. L. Roy and C.-M. Che, Chem. Commun., 2006, 3972 RSC.
- M.-Y. Yuen, V. A. L. Roy, W. Lu, S. C. F. Kui, G. S. M. Tong, M.-H. So, S. S.-Y. Chui, M. Muccini, J. Q. Ning, S. J. Xu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9895 CrossRef CAS PubMed.
- X.-S. Xiao, W.-L. Kwong, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem.–Eur. J., 2013, 19, 9457 CrossRef CAS PubMed.
- C.-M. Che, C.-F. Chow, M.-Y. Yuen, V. A. L. Roy, W. Lu, Y. Chen, S. S.-Y. Chui and N. Zhu, Chem. Sci., 2011, 2, 216 RSC.
- C. A. Strassert, C.-H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946 CrossRef CAS PubMed.
- M. Mauro, A. Aliprandi, C. Cebrian, D. Wang, C. Kubel and L. De Cola, Chem. Commun., 2014, 50, 7269 RSC.
- F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J.-P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659 CrossRef CAS PubMed.
- A. Y.-Y. Tam, K. M.-C. Wong, G. Wang and V. W.-W. Yam, Chem. Commun., 2007, 2028 RSC.
- W. Lu, Y. Chen, V. A. L. Roy, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 7621 CrossRef CAS PubMed.
- X.-S. Xiao, W. Lu and C.-M. Che, Chem. Sci., 2014, 5, 2482 RSC.
- N. K. Allampally, C. A. Strassert and L. De Cola, Dalton Trans., 2012, 41, 13132 RSC.
- J. Wang, Y. Chen, Y.-C. Law, M. Li, M.-X. Zhu, W. Lu, S. S.-Y. Chui, N. Zhu and C.-M. Che, Chem.–Asian J., 2011, 6, 3011 CrossRef CAS PubMed.
- Y. Chen, K. Li, W. Lu, S. S.-Y. Chui, C.-W. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909 CrossRef CAS PubMed.
- A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540 RSC.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS PubMed.
- V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and N. Zhu, Chem.–Eur. J., 2005, 11, 4535 CrossRef PubMed.
- M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952 CrossRef CAS PubMed.
- J. Ni, X. Zhang, Y.-H. Wu, L.-Y. Zhang and Z.-N. Chen, Chem.–Eur. J., 2011, 17, 1171 CrossRef CAS PubMed.
- L.-M. Huang, G.-M. Tu, Y. Chi, W.-Y. Hung, Y.-C. Song, M.-R. Tseng, P.-T. Chou, G.-H. Lee, K.-T. Wong, S.-H. Cheng and W.-S. Tsai, J. Mater. Chem. C, 2013, 1, 7582 RSC.
- V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., Int. Ed., 2008, 47, 6286 CrossRef CAS PubMed.
- X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376 RSC.
- K. R. Mann, J. G. Gordon and H. B. Gray, J. Am. Chem. Soc., 1975, 97, 3553 CrossRef CAS.
- Y. Chen, K. Li, H. O. Lloyd, W. Lu, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2010, 49, 9968 CrossRef CAS PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed.
- P. Pyykkö and Y. Zhao, Angew. Chem., Int. Ed., 1991, 30, 604 CrossRef.
- P. Pyykkö and F. Mendizabal, Chem.–Eur. J., 1997, 3, 1458 CrossRef.
- L. Magnko, M. Schweizer, G. Rauhut, M. Schutz, H. Stoll and H.-J. Werner, Phys. Chem. Chem. Phys., 2002, 4, 1006 RSC.
- J. Muñiz, C. Wang and P. Pyykkö, Chem.–Eur. J., 2011, 17, 368 CrossRef PubMed.
- S. Grimme and J.-P. Djukic, Inorg. Chem., 2011, 50, 2619 CrossRef CAS PubMed.
- M. Andrejić and R. A. Mata, Phys. Chem. Chem. Phys., 2013, 15, 18115 RSC.
- S. Záliš, Y.-C. Lam, H. B. Gray and A. Vlček, Inorg. Chem., 2015, 54, 3491 CrossRef PubMed.
- D. R. McMillin and J. J. Moore, Coord. Chem. Rev., 2002, 229, 113 CrossRef CAS.
- S. D. Cummings, Coord. Chem. Rev., 2009, 253, 1495 CrossRef CAS.
- P. Wu, E. L.-M. Wong, D.-L. Ma, G. S. M. Tong, K.-M. Ng and C.-M. Che, Chem.–Eur. J., 2009, 15, 3652 CrossRef CAS PubMed.
- T. Zou, J. Liu, C. T. Lum, C. Ma, R. C.-T. Chan, C.-N. Lok, W.-M. Kwok and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 10119 CrossRef CAS PubMed.
- D.-L. Ma, C.-M. Che and S.-C. Yan, J. Am. Chem. Soc., 2009, 131, 1835 CrossRef CAS PubMed.
- P. Wu, D.-L. Ma, C.-H. Leung, S.-C. Yan, N. Zhu, R. Abagyan and C.-M. Che, Chem.–Eur. J., 2009, 15, 13008 CrossRef CAS PubMed.
- C.-N. Lok, T. Zou, J.-J. Zhang, I. W.-S. Lin and C.-M. Che, Adv. Mater., 2014, 26, 5550 CrossRef CAS PubMed.
- D. Septiadi, A. Aliprandi, M. Mauro and L. De Cola, RSC Adv., 2014, 4, 25709 RSC.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03766b |
| This journal is © The Royal Society of Chemistry 2016 |