
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering the electronic structure of two-dimensional subnanopore nanosheets using molecular titanium-oxide incorporation for enhanced photocatalytic activity†
Xiuli
Lu‡
a,
Kun
Xu‡
a,
Shi
Tao
b,
Zewei
Shao
a,
Xu
Peng
a,
Wentuan
Bi
a,
Pengzuo
Chen
a,
Hui
Ding
a,
Wangsheng
Chu
*b,
Changzheng
Wu
*a and
Yi
Xie
a
aHefei National Laboratory for Physical Sciences at the Microscale, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Hefei Science Center (CAS), CAS Key Laboratory of Mechanical Behavior and Design of Materials, University of Science & Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: czwu@ustc.edu.cn
bNational Synchrotron Radiation Laboratory University of Science & Technology of China, Hefei, Anhui 230029, P. R. China. E-mail: chuws@ustc.edu.cn
First published on 11th November 2015
Abstract
Engineering the electronic structure of two-dimensional (2D) nanomaterials endows unique physical and chemical properties. Although developed modification strategies have significantly expanded the applications of 2D nanomaterials, exploring new strategies to regulate the electronic structure of 2D nanomaterials is also expected. Herein, we highlight a new strategy to engineer the electronic structure of 2D subnanoporous nanomaterials. As a proof of concept, based on controllable subnanopore engineering using molecular titanium-oxide incorporation, the electronic band structure of 2D graphitic carbon nitride (CN) nanosheets has been efficiently tuned with the enhancement of visible light absorption as well as separation and the migration rate of photo-excited charge carriers, exhibiting significantly improved photocatalytic activity under visible light irradiation. Our work opens a new door to engineering the intrinsic properties of 2D subnanoporous nanomaterials.
Introduction
Two dimensional (2D) nanomaterials are of great interest due to their 2D confined structure which causes exotic physical properties.1–6 Recently, enormous efforts have been made to regulate the electronic structure of 2D nanomaterials in order to endow them with new functionalities or further enhance their intrinsic physical and chemical properties.7–19 Generally, surface modification, defect engineering, and heteroatom doping are well known approaches to engineer the intrinsic electronic structure of 2D nanomaterials.8,9 For example, the modification of various ions (i.e. F−, H+, Cl− and so on) on the surface of 2D nanomaterials can effectively modulate their carrier concentrations, bringing about new physicochemical properties.10–15 Other modulation strategies such as defect engineering and heteroatom doping, have been widely applied to achieve the synergic advantages of optimized electrical properties and higher chemical activity, holding promise for the enhancement of their catalytic performance.16–20 Although substantial progress has been made, developing new strategies for tuning the electronic structure of 2D nanosheets is also expected.2D polymers with a subnanoporous structure have shown promising application in various fields such as bioimaging, nanoelectronic and catalysis which is attributed to their highly thermally stable and easy-to-adjust electronic structure.21–26 Compared with traditional inorganic 2D materials, 2D microporous polymers (2D-MPs) have unique periodic vacancies (also called as “subnanopores”) in their lattices.23 These unique nanometer-sized pores in 2D-MPs could provide a new opportunity for modifying the electronic structure of 2D nanomaterials as well as retaining the integrity of their matrix. Herein, taking 2D graphitic carbon nitride (CN) as a model material, we develop a controllable subnanopore engineering strategy to regulate the electronic structure of 2D-MP nanosheets while not influencing the pristine matrix structure of the polymer. In our case, the subnanopore was incorporated using a molecular titanium-oxide group, which was highly homogeneously dispersed in the 2D CN framework. And titanium-oxide incorporation in the subnanopores of CN could efficiently tune the electronic band structure of 2D CN, causing the effective improvement in visible light absorption and separation as well as the migration rate of photo-excited charge carriers. As a result, 2D CN with molecular titanium-oxide incorporation shows enhanced photocatalytic activity under visible light irradiation compared with pure 2D g-CN. Our work provides a new way to engineer the intrinsic physical properties of 2D nanomaterials.
As demonstrated in Fig. 1A, controllable subnanopore engineering in 2D CN using molecular titanium-oxide incorporation (TiO–CN) was achieved by a simple “bottom-up” polycondensation of precursors which contain dicyandiamide (DICY) and titanium oxide ions. In detail, TiCl4 and DICY were firstly dissolved in a cooled ammonium chloride solution, forming a colorless and transparent aqueous solution. During the dissolving process, TiCl4 was rapidly hydrolyzed to TiO2+. It should be noted that the cooled ammonium chloride solution could prevent the TiOCl2 molecules from further hydrolyzing into H2TiO3 nanoparticles. Then, the whole solution was freeze-dried to provide a full mixture of DICY with TiO2+, and finally a white solid powder was obtained. After that, the white powder was annealed at 550 °C for 4 h in a quartz container to get the final TiO–CN samples. Interestingly, CN nanosheets with a varying degree of titanium-oxide incorporation could be easily controlled by tuning the amount of TiCl4. According to the amount of molecular titanium-oxide incorporation in the subnanopores, the TiO–CN samples were denoted as TiO–CN1, TiO–CN2 and TiO–CN3. Pure 2D g-CN was obtained without adding TiCl4, denoted as CN.
 | ||
| Fig. 1 (A) The schematic illustration for the synthesis of TiO–CN from the “bottom-up” polycondensation of the designed precursors. (B) TEM, (C) HAADF-STEM and (D) AFM images of the TiO–CN2 nanosheet. (E) XRD patterns of the CN and TiO–CN2 nanosheets. (F) The solid state 13C NMR spectra of 2D CN and TiO–CN2, and (G) IR spectra of CN and TiO–CN2. | ||
Results and discussion
To unravel the microscopic morphologies of a TiO–CN nanosheet, scanning electron microscopy (SEM), transmission electron microscopy (TEM), and atomic force microscopy (AFM) were carried out. The SEM images of TiO–CN show uniform 2D nanosheets with obvious corrugations and porosities (Fig. S3†). The corresponding TEM image in Fig. 1B also verifies the clean ultrathin nanosheet with a sheet-like morphology as that of CN. The AFM image reveals that the thickness of the TiO–CN2 nanosheet (one of the TiO–CN samples) is about 3–3.3 nm. Furthermore, EDX mapping analyses were performed to identify the distribution of molecular titanium-oxide in the matrix of 2D CN. As exhibited in Fig. S7,† the elemental mapping images reveal the homogeneous distribution of carbon, nitrogen, titanium and oxygen in the whole ultrathin nanosheet. The results indicated that titanium-oxide was highly homogeneously dispersed in the framework of CN. The HAADF-STEM image of the TiO–CN2 nanosheet also confirms that titanium-oxide was homogeneously dispersed in the framework of CN as shown in Fig. 1C and S5.† And at the edge of the TiO–CN nanosheet where the yellow circles are noted, we can clearly identify that the element Ti exists as an isolated atom with atomic size, suggesting that an isolated titanium-oxide species was coordinated in the subnanopores of CN. All of the above characterization results clearly demonstrate that 2D TiO–CN was successfully obtained by subnanopore engineering using homogeneous titanium-oxide incorporation in the framework of CN.In order to investigate whether the C–N framework was maintained after titanium-oxide incorporation, XRD, solid state 13C NMR and FT-IR were performed. As can be seen in the XRD patterns (Fig. S1†), even with the increased amount of molecular titanium-oxide incorporation, there is still no titanium-based compound peaks. Although the overall intensity of the XRD patterns decreases gradually with the increased amount of titanium-oxide incorporation, the two main peaks always exist as they do in pure 2D g-CN (Fig. S1†). The (002) peak at about 27.8° is the characteristic stacking peak of the conjugated aromatic system with an interlayer distance of 0.321 nm. The peak located at about 13.0°, corresponding to 0.681 nm, is derived from the in-plane lattice planes in the g-CN crystal.26 The XRD results clearly illustrate that molecular titanium-oxide incorporation in 2D CN does not destroy the basic building structure of the C–N framework. This result was also confirmed by the solid state 13C NMR spectra in Fig. 1F. The two main peaks in the solid state 13C NMR spectra of 2D CN and TiO–CN2 are similar. The first peak at δ = 164.7 ppm is assigned to the C atoms in CN2(NHX) and the second peak at δ = 164.7 ppm is attributed to the C atoms in CN3.27 Also, the structure of 2D TiO–CN was further characterized using FT-IR spectra (Fig. 1G). All FT-IR spectra of the obtained samples show the feature bands of tri-s-triazine units at about 800 cm−1 and aromatic CN heterocycles between 1600 and 1200 cm−1, similar to those of the pure 2D CN polymer.25 In other words, the CN nanosheet with molecular titanium-oxide successfully incorporating maintains the basic C–N framework.
It is well known that XAFS is one of the most powerful tools to probe the local electronic and geometrical structures around a selected element.28–30 To further study the local geometrical structure of Ti–O in the subnanopores of the CN nanosheets, we performed XAS measurements of TiO–CN at the Ti K-edge and chose anatase TiO2 as the reference sample. Fig. 2A shows the Ti K-edge XANES of the Ti–O structure in CN compared with the spectrum of the standard sample. The XANES of anatase TiO2 presents a well-defined triple pre-edge feature before the main crest, this is the same as the spectra reported in other literature.31,32 This triple pre-edge feature can be assigned to the distorted TiO6 configuration. That is, Ti is coordinated by six oxygen atoms. By contrast, the Ti–O structure in CN presents an intense single pre-edge feature. It is a typical “fingerprint” pre-edge feature of non-central symmetry.32 This means the titanium should coordinate with only one oxygen atom out of the CN layer. In order to confirm this configuration of TiO–CN, the XANES spectra at the Ti K-edge of TiO2 and the prepared sample were calculated using the Feff8 code in the framework of full-potential multiple scattering theories.33 It can be seen that most features, especially the pre-edge peaks, of the XANES of both anatase TiO2 and the TiO–CN sample, using the local structure obtained from the EXAFS fit as discussed below, have been reproduced successfully as shown in Fig. 2B. Furthermore, Fig. 2C gives the Fourier transformation (FT) of the EXAFS oscillation of the prepared sample, compared with standard anatase TiO2. In contrast to anatase TiO2, the FT magnitude of the Ti-oxide structure in CN presents only a broad phase-shift peak. The dismissal of the FT peaks of the second (Ti–Ti pair) and higher shells in TiO–CN were clear evidence that only isolated Ti-oxide species were integrated into g-CN. A phase shift to the larger direction in the r-space of the first peak means a longer Ti–O or Ti–N bond length. This is consistent with the proposed configuration that there is a TiO pair in the hole of the g-CN network, in which the Ti–N bond length is about 2.38 Å. In fact, the first FT peak of the EXAFS only can be simulated successfully by the configuration of double Ti–N pairs, rather than that of only one Ti–N set. The quality of the EXAFS simulation is also shown in Fig. S11† and the fit structure parameters are given in Table 1. This result from EXAFS quantitative analysis also confirmed that the isolated Ti-oxide species, Ti only coordinating to an O atom, is indeed located in the hole of the g-CN sample. Based on the above analyses, the XAFS results clearly verified that molecular titanium-oxide was successfully incorporated into the subnanopores of CN coordinated by six nitrogen atoms and one oxygen atom as shown in Fig. 2D, which is also consistent with the results from XPS (see ESI in S4†) and HAADF-STEM.
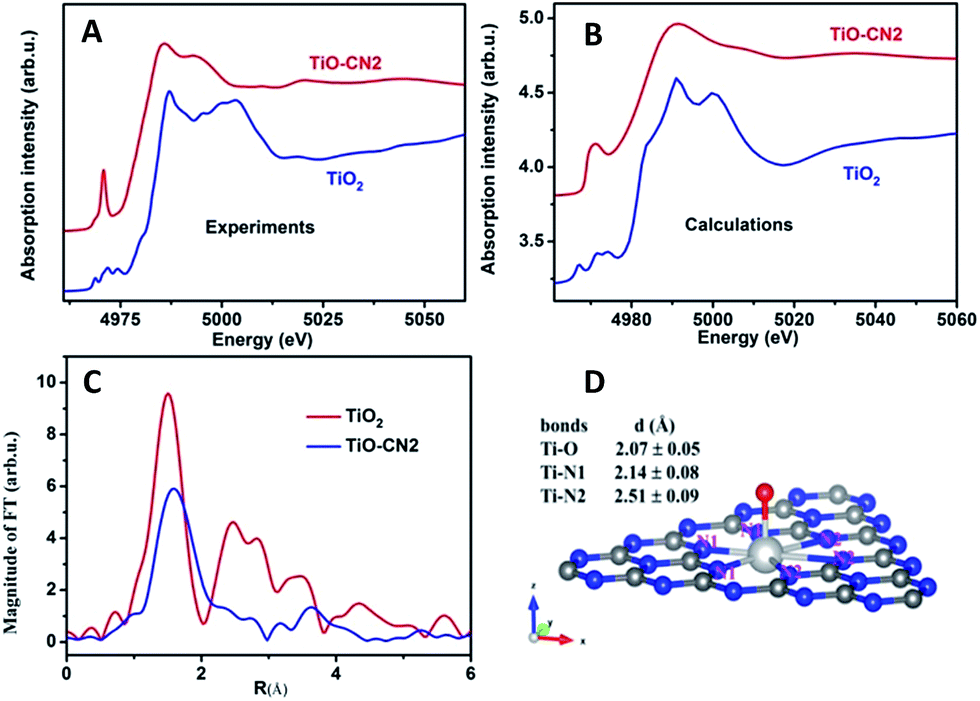 | ||
| Fig. 2 (A and B) Experimental and calculated Ti K-edge XANES spectra of prepared TiO–CN2 compared with the anatase TiO2 powder. (C) FT-EXAFS comparison between the prepared sample and standard anatase TiO2. (D) The illustration of the detailed TiO–CN structure. | ||
| Samples | Bonds | N | R (Å) | σ 2 (×10−3 Å2) | E 0 (eV) |
|---|---|---|---|---|---|
| a Note: (1) S02 = 0.8 from the fit of the TiO2 powder was used in the fit of the sample; (2) the coordination number of the first shell of the sample was fixed in order to reduce the varied parameters. | |||||
| TiO2 | Ti–O | 6.0 ± 0.3 | 1.96 ± 0.02 | 5.8 ± 0.6 | −0.3 |
| TiO–CN | Ti–O | 1 | 2.07 ± 0.05 | 1.0 ± 0.1 | −8.0 |
| Ti–N(1) | 3 | 2.14 ± 0.08 | 5.0 ± 0.5 | −5.0 | |
| Ti–N(2) | 3 | 2.51 ± 0.09 | 6.0 ± 0.6 | −5.0 | |
Controllable subnanopore engineering with molecular titanium-oxide incorporation into the “nitrogen pots” of the CN nanosheets provided the opportunity to finely tune the electronic band structure of 2D CN. The electronic band structure of CN with different amounts of titanium-oxide incorporation was evaluated using the UV-visible absorption spectra and room temperature photoluminescence (PL) spectra. As demonstrated in Fig. 3A, when molecular titanium-oxides are incorporated into the “nitrogen pots” of CN, the molecular titanium-oxide ions contribute more electrons into the CN framework, causing enhanced π-electron delocalization in the conjugated system with a more narrowed bandgap, showing red shift of the absorption band edge. Taking 2D CN and TiO–CN2 for example, the bandgap narrowed from 2.83 eV to 2.23 eV with an obvious color change from light yellow to crimson (Fig. 3B). The VB of both 2D CN and TiO–CN2 were measured using the XPS valence spectra (Fig. S12†), revealing nearly the same valence band edge. In this case, based on the bandgap of 2D CN and TiO–CN2, the conduction band edge of TiO–CN2 down-shifts by about 0.6 eV with respect to 2D CN as displayed in Fig. 3C. Additionally, the intensity of the PL spectra decreased gradually when increasing the titanium-oxide incorporation amount, illustrating the effective restraint of the recombination rates of photogenerated carriers after subnanopore engineering.25 To further evaluate the photo-excited charge carriers migration, the electrochemical impedance spectra (EIS) and photocurrent responses of the CN and TiO–CN2 electrodes were obtained, revealing that the mobility of the photo-excited charge carriers was promoted after molecular titanium-oxide incorporation (Fig. 3E and F). In general, the 2D carbon nitride nanosheets with titanium-oxide incorporation could realize a narrower band gap with optimized separation/recombination rates of photogenerated carriers.
 | ||
| Fig. 3 (A) The UV-visible absorption spectra of 2D CN and TiO–CN. (B) The plot of transformed Kubelka–Munk function vs. photon energy for 2D CN and TiO–CN2. (C) Band structure diagram of 2D CN and TiO–CN2. (D) Fluorescence emission spectra of 2D CN and TiO–CN samples. (E) EIS and (F) photocurrent responses of the CN and TiO–CN2 electrodes. | ||
As expected, the electronic structure of the 2D CN nanosheets has been effectively tuned using molecular titanium-oxide incorporation and has brought about encouraging results such as improved optical absorption in the visible range, and the enhancement of the separation and migration rate of the photo-excited charge carriers, benefiting the photocatalytic reaction. To evaluate the photocatalytic activities of the CN and TiO–CN nanosheets, we first monitored an important active species for the decomposition of organic molecules, the amount of ˙OH radicals, by detecting 2-hydroxyterephthalic acid in water/terephthalic acid solution.26 As shown in Fig. 4A, the TiO–CN2 nanosheets show the highest reactivity among all the investigated samples in generating ˙OH radicals. In order to better understand the capability of the generation of ˙OH radicals, DMPO spin-trapping ESR measurements were performed for the CN and TiO–CN samples.34,35 As we know, DMPO spin-trapping ESR is a more effective technique for detecting reactive radicals. All the ESR spectra exhibited in Fig. 4B show exclusively a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 quartet signal due to the DMPO-˙OH adducts formed upon trapping of the ˙OH radicals by the DMPO molecules. Obviously, as exhibited in Fig. 4B, the ESR spectra show that the TiO–CN2 nanosheets exhibited the best capability in generating ˙OH radicals, corresponding well with the PL results as shown in Fig. 4A. Furthermore, the superior photocatalytic performance of the TiO–CN nanosheets was demonstrated by decomposing rhodamine B under light irradiation. As exhibited in Fig. 4C, after visible light irradiation for 5 minutes, the percentage of degraded rhodamine B by the TiO–CN2 nanosheets could reach as high as 83.7% which is much higher than that of the pure CN nanosheets (21.3%). To fully decompose rhodamine B under visible light, TiO–CN2 only needed about 10 minutes while the CN sample needed more than 45 minutes. Moreover, TiO–CN2 shows no obvious change after the photocatalytic reaction suggesting its good stability as a photocatalyst. The results of the photocatalytic reaction clearly illustrate the important role of subnanopore engineering in CN nanosheets which could significantly promote photocatalytic activity.
:2:2:1 quartet signal due to the DMPO-˙OH adducts formed upon trapping of the ˙OH radicals by the DMPO molecules. Obviously, as exhibited in Fig. 4B, the ESR spectra show that the TiO–CN2 nanosheets exhibited the best capability in generating ˙OH radicals, corresponding well with the PL results as shown in Fig. 4A. Furthermore, the superior photocatalytic performance of the TiO–CN nanosheets was demonstrated by decomposing rhodamine B under light irradiation. As exhibited in Fig. 4C, after visible light irradiation for 5 minutes, the percentage of degraded rhodamine B by the TiO–CN2 nanosheets could reach as high as 83.7% which is much higher than that of the pure CN nanosheets (21.3%). To fully decompose rhodamine B under visible light, TiO–CN2 only needed about 10 minutes while the CN sample needed more than 45 minutes. Moreover, TiO–CN2 shows no obvious change after the photocatalytic reaction suggesting its good stability as a photocatalyst. The results of the photocatalytic reaction clearly illustrate the important role of subnanopore engineering in CN nanosheets which could significantly promote photocatalytic activity.
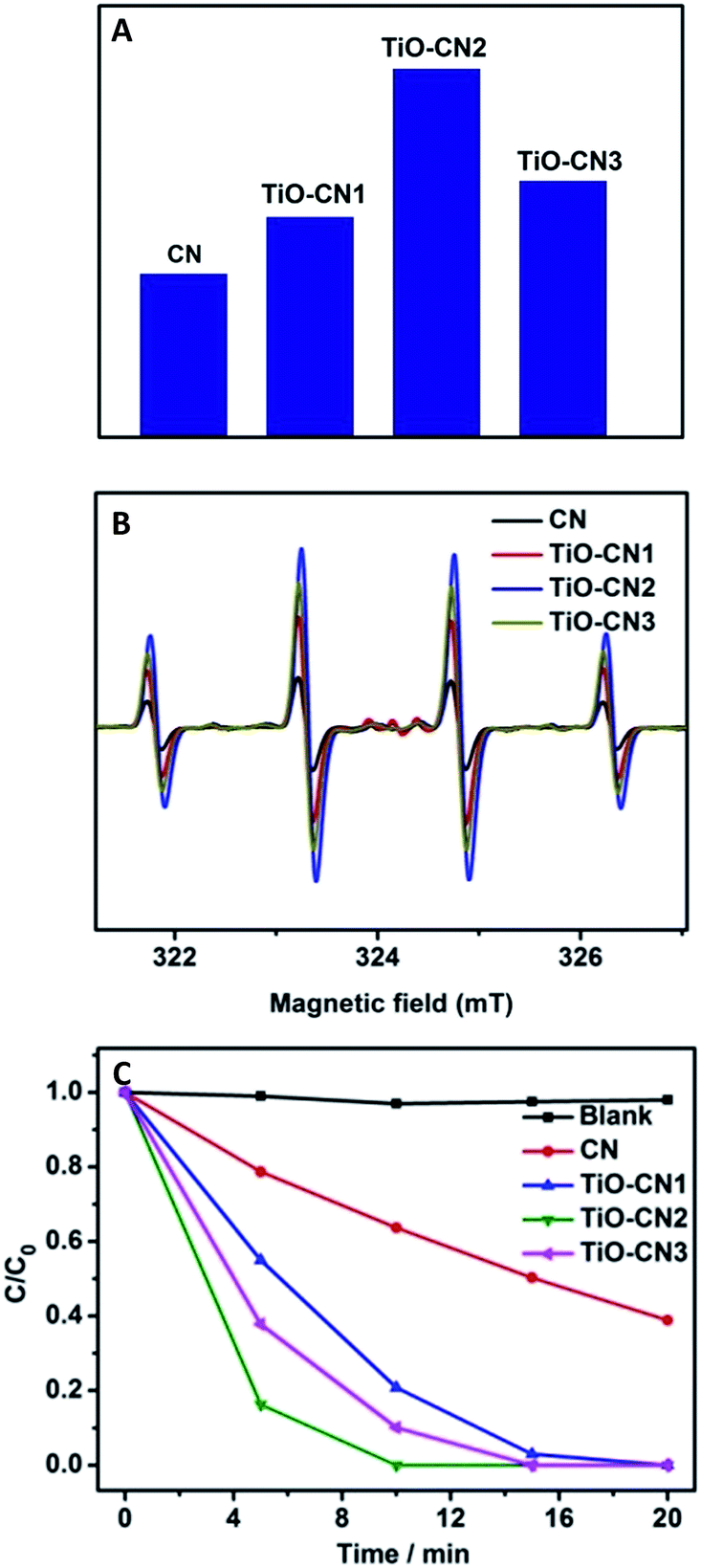 | ||
| Fig. 4 (A) Comparison of the PL relative intensity of 2-hydroxyterephthalic generated by reacting terephthalic acid with ˙OH radicals in the suspension of CN and TiO–CN nanosheets under visible light (λ > 420 nm) for 25 min. (B) DMPO spin-trapping ESR spectra of samples in aqueous dispersions for DMPO-˙OH. (C) Time-dependent photocatalytic degradation of rhodamine B by the CN and TiO–CN samples under visible light irradiation (λ > 420 nm). | ||
In our case, the valance band edge of CN is unchanged (Fig. S12†) and the band gap exhibits a gradual narrowing (Fig. S13†) after molecular titanium-oxide incorporation, suggesting gradual decreasing of the conduction band edge with increasing amount of molecular titanium-oxide incorporation in the CN nanosheets. The successful incorporation of molecular titanium-oxide into the subnanopores has effectively tuned the electronic band structure of the 2D carbon nitride nanosheets with band gap narrowing (increasing the visible light absorption). Also, the radiative recombination of photo-excited electrons and holes greatly decreased as exhibited in Fig. 3D. Both of these features are favorable for ˙OH generation (improving the photocatalytic activity). However, bandgap narrowing would also induce a decrease of the conduction band edge of the CN nanosheets (Fig. 3C). For the photocatalysts, a lower conduction band gap edge means a weaker reduction ability of the electrons which are generated from the conduction band.36 Obviously, the weaker reduction ability of the electrons, which are generated from the conduction band, is unfavorable for ˙OH generation (decreasing the photocatalytic activity). Thus, to get better photocatalytic performance, there should be a compromise between the narrowing of the band gap of the TiO–CN samples and the reduction ability of their photo-excited electrons. Of note, TiO–CN2 presents the best photocatalytic ability in our investigated system due to its good balance between wide light absorption and strong reduction ability.
Conclusions
In conclusion, controllable subnanopore engineering, as a new strategy to regulate the electronic structure of 2D subnanoporous nanomaterials, has been realized in 2D carbon nitride ultrathin nanosheets using molecular titanium-oxide incorporation. The successful incorporation of titanium-oxide into the subnanopores has effectively tuned the electronic band structure of the 2D carbon nitride nanosheets with band gap narrowing and the improvement of their photocatalytic activity under visible light. The introduction of foreign molecular groups into subnanopores could pave a new way to regulate the electronic structure of 2D nanomaterials catering for energy and catalytic applications.Acknowledgements
This work was financially supported by the National Basic Research Program of China (2015CB932302), National Natural Science Foundation of China (No. 21222101, U1432133, 11132009, 21331005, 11321503, J1030412), Chinese Academy of Science (XDB01020300), the Fok Ying-Tong Education Foundation, China (Grant No. 141042), and the Fundamental Research Funds for the Central Universities (No. WK2060190027). WS Chu acknowledges NSFC project (11275227, 11179006), the MSnano project, and a European-funded network working on the modelling of spectroscopies within the multiple scattering framework.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed
- M. Chhowalla, Z. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584–2586 RSC
- K. Xu, P. Chen, X. Li, C. Wu, Y. Guo, J. Zhao, X. Wu and Y. Xie, Angew. Chem., Int. Ed., 2013, 52, 10477–10481 CrossRef CAS PubMed
- Y. Du, Z. Yin, J. Zhu, X. Huang, X.-J. Wu, Z. Zeng, Q. Yan and H. Zhang, Nat. Commun., 2012, 3, 1177 CrossRef PubMed
- Z. Fan, X. Huang, C. Tan and H. Zhang, Chem. Sci., 2015, 6, 95–111 RSC
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed
- Y. Guo, K. Xu, C. Wu, J. Zhao and Y. Xie, Chem. Soc. Rev., 2015, 44, 637–646 RSC
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed
- C. Lin, X. Zhu, J. Feng, C. Wu, S. Hu, J. Peng, Y. Guo, L. Peng, J. Zhao, J. Huang, J. Yang and Y. Xie, J. Am. Chem. Soc., 2013, 135, 5144–5151 CrossRef CAS PubMed
- M. Du, X. Li, A. Wang, Y. Wu, X. Hao and M. Zhao, Angew. Chem., Int. Ed., 2014, 53, 3645–3649 CrossRef CAS PubMed
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610–613 CrossRef CAS PubMed
- D. Voiry, A. Goswami, R. Kappera, C. de Carvalho Castro e Silva, D. Kaplan, T. Fujita, M. Chen, T. Asefa and M. Chhowalla, Nat. Chem., 2015, 7, 45–49 CrossRef CAS PubMed
- D. Kiriya, M. Tosun, P. Zhao, J. S. Kang and A. Javey, J. Am. Chem. Soc., 2014, 136, 7853–7856 CrossRef CAS PubMed
- S. W. Han, Y. H. Hwang, S.-H. Kim, W. S. Yun, J. D. Lee, M. G. Park, S. Ryu, J. S. Park, D.-H. Yoo, S.-P. Yoon, S. C. Hong, K. S. Kim and Y. S. Park, Phys. Rev. Lett., 2013, 110, 247201 CrossRef PubMed
- C. Backes, N. C. Berner, X. Chen, P. Lafargue, P. LaPlace, M. Freeley, G. S. Duesberg, J. N. Coleman and A. R. McDonald, Angew. Chem., Int. Ed., 2015, 54, 2638–2642 CrossRef CAS PubMed
- I.-Y. Jeon, M. Choi, H.-J. Choi, S.-M. Jung, M.-J. Kim, J.-M. Seo, S.-Y. Bae, S. Yoo, G. Kim, H. Y. Jeong, N. Park and J.-B. Baek, Nat. Commun., 2015, 6, 7123 CrossRef PubMed
- J. Xie, S. Li, R. Wang, H. Zhang and Y. Xie, Chem. Sci., 2014, 5, 1328–1335 RSC
- K. Xu, X. Li, P. Chen, D. Zhou, C. Wu, Y. Guo, L. Zhang, J. Zhao, X. Wu and Y. Xie, Chem. Sci., 2015, 6, 283–287 RSC
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed
- L. Hao, J. Ning, B. Luo, B. Wang, Y. Zhang, Z. Tang, J. Yang, A. Thomas and L. Zhi, J. Am. Chem. Soc., 2015, 137, 219–225 CrossRef CAS PubMed
- J. Mahmood, E. K. Lee, M. Jung, D. Shin, I.-Y. Jeon, S.-M. Jung, H.-J. Choi, J.-M. Seo, S.-Y. Bae, S.-D. Sohn, N. Park, J. H. Oh, H.-J. Shin and J.-B. Baek, Nat. Commun., 2015, 6, 6486 CrossRef CAS PubMed
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814–11818 CrossRef CAS PubMed
- T. G. Yin, M. Nishikawa, Y. Nosaka, N. Srinivasan, D. Atarashi, E. Sakai and M. Miyauchi, ACS Nano, 2015, 9, 2111–2119 CrossRef PubMed
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed
- L. A. Grunes, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 2111–2131 CrossRef CAS
- F. Farges, G. E. Brown and J. J. Rehr, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1809–1819 CrossRef CAS
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed
- K. Zhao, L. Zhang, J. Wang, Q. Li, W. He and J. J. Yin, J. Am. Chem. Soc., 2013, 135, 15750–15753 CrossRef CAS PubMed
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03551a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |