
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guest-dependent directional complexation based on triptycene derived oxacalixarene: formation of oriented rotaxanes†
Han-Xiao Wangac, Zheng Mengac, Jun-Feng Xianga, Yu-Xiang Xiaa, Yihua Sunb, Shu-Zhen Hua, Hui Chen*b, Jiannian Yaob and Chuan-Feng Chen*a
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: cchen@iccas.ac.cn
bCAS Key Laboratory of Photochemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: chenh@iccas.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 8th October 2015
Abstract
The manipulation of supramolecular devices to carry out sophisticated and programmed tasks is bound up with the spatial allocation of their components, especially the threading direction of the guest, which controls the host–guest orientation in the device. However, insights are needed to probe more possibilities for steering the threading direction. We have developed a new system consisting of a three-dimensional nonsymmetric oxacalixarene (H) with a fixed comformation and (bi)pyridinium salts (G1–G3), in which we found that based on the intrinsic discrepancies between the two semi-cavities of H, the electron densities of the axles greatly affect the threading direction. This was unequivocally demonstrated by NMR spectra and single crystal structures. With elaborate design, unidirectional threading was achieved, resulting in an oriented rotaxane. Therefore, we describe a new approach in which the threading direction and final orientation may be finely controlled by adjustment of the structure of the guest.
Introduction
During the last two decades, great progress has been made in the synthesis of mechanically interlocked molecules (MIMs) such as rotaxanes and catenanes,1 with interest arising not only from their aesthetic appeal2 but also their perceived potential to act as sophisticated molecular machines, switches and sensors.3 The general approach to constructing (pseudo)-rotaxanes requires a macrocycle and a linear guest as the wheel and the axle, respectively; if neither of them is in the Dnh point group, two isomers will be obtained via the threading process, resulting in different orientations of components in the final MIMs.4–7 Owing to differences in the spatial arrangement, a pair of orientational isomers may generate quite different properties.8 However, the separation of isomers poses a big problem,4,6,9b so unidirectional threading that leads to an oriented rotaxane is highly appealing. Using cyclodextrin as the wheel, unidirectional threading has been realized due to either kinetic10 or thermodynamic11 stability, resulting in various oriented rotaxanes. Calixarenes with three-dimensional nonsymmetric structures have provided another candidate to form oriented rotaxanes, based on the formation of endo-cavity complexes with organic cations.9,12,13 Using triphenylureido-calix[6]arene as the host and dialkylviologen salt as the guest, Arduini and coworkers succeeded in synthesizing an oriented pseudorotaxane through a kinetically controlled process.9a On that basis, they prepared a pair of isomeric oriented rotaxanes for the first time by reversing the sequence in which two different stopper groups were introduced onto the axle.4 In contrast to the kinetically controlled unidirectional threading strategy, Neri and coworkers developed an “endo-alkyl” rule controlling the threading directionality of alkylbenzylammonium axles with calix[6]arenes.5 Applying the rule, they constructed stereoisomeric (pseudo)[3]rotaxanes13a,b and an oriented handcuff rotaxane.13c Although some important results have been obtained, the formation of oriented rotaxanes based on controllable threading directionality is still emerging as a challenge to be overcome.Heterocalixarenes14 are a kind of calixarenes obtained by replacing conventional methylene bridges with heteroatoms. Introducing bridging heteroatoms makes it possible to finely tune the size, conformation and binding properties of the macrocycle, since heteroatoms can not only adopt different electronic configurations but also form various degrees of conjugation with the neighboring aromatic rings.15 Due to the difficulty of forming “through-the-annulus” complexes with linear guests, the utilization of heterocalixarenes as hosts to realize directional threading and construction of oriented rotaxanes is still a rather unexplored field. Previously, we reported that a triptycene-derived oxacalixarene H16a (Fig. 1a), with an upper semi-cavity encircled by two naphthyridine moieties and a lower semi-cavity encircled by two triptycene moieties, could incorporate various 4,4′-bipyridinium salts to form pseudo-rotaxanes.16b We applied this property to the synthesis of rotaxanes based on H and symmetric 4,4′-bipyridinium salts. Theoretically, if we mixed H with a nonsymmetric axle, a pair of isomeric pseudorotaxanes might be obtained. The crystal structure of H16a disclosed that the portal of the lower semi-cavity was more crowded than that of the upper one (Fig. 1a, 8.53 Å versus 13.57 Å). Moreover, triptycene is known to be more electron-rich than the naphthyridine moiety (the N atoms excluded). Hence, we hypothesized that the discrepancies in size and electron density between the two semi-cavities of H might exert different induction effects upon the threading of guests. Herein, we report a system employing H as the wheel, in which we can control the dominant threading direction by partially changing the structures of the axles, thus tuning their interaction with H in the threading process.
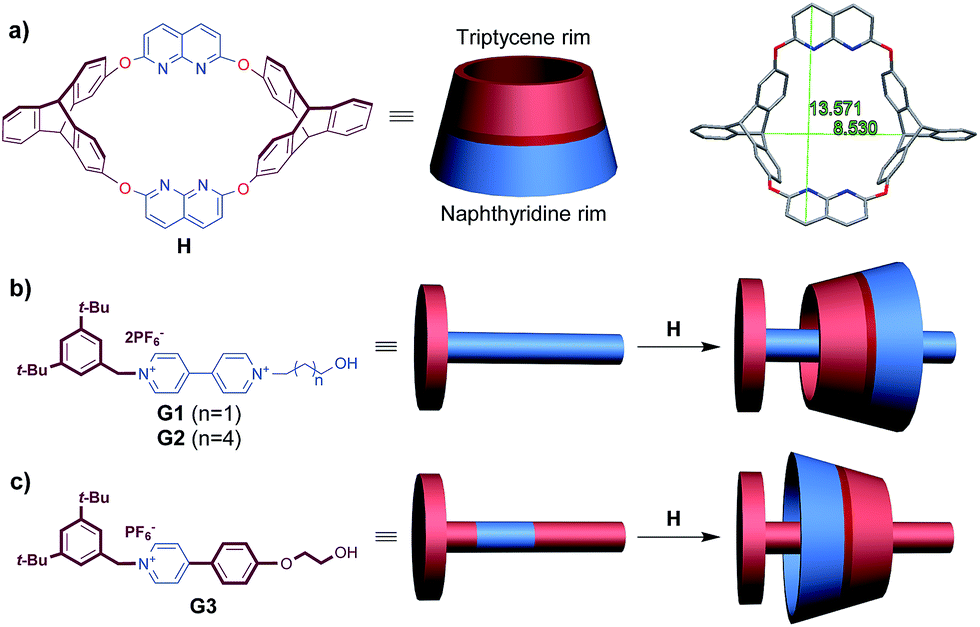 | ||
| Fig. 1 (a) Structure and representation of H and its crystal structure,16a denoting sizes of the portals on both sides. (b) Structure and representation of guests G1–G2 and their directional threading into H. (c) Structure and representation of guest G3 and its directional threading into H. | ||
Results and discussion
Directional complexation between macrocycle H and bipyridinium salts G1 and G2 with alkyl chains of different lengths
We initially designed a linear nonsymmetric bipyridinium salt G1 (Fig. 1b), stoppering one of its ends with a bulky terminal group in order to easily clarify its threading direction into the macrocycle. By comparison of the 1H NMR spectra of H, G1, and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the two in CDCl3/CD3CN solution at room temperature, complexation between the two components was observed (Fig. S31, ESI†). The 1H NMR spectrum of the complex always exhibited only one set of signals, regardless of whether the host or the guest was in excess (Fig. S34, ESI†), indicating a fast equilibrium on the NMR timescale. To acquire information on the relative orientation of the two components, we stoppered the pseudorotaxanes to synthesize the corresponding rotaxanes (Scheme 1).
:1 mixture of the two in CDCl3/CD3CN solution at room temperature, complexation between the two components was observed (Fig. S31, ESI†). The 1H NMR spectrum of the complex always exhibited only one set of signals, regardless of whether the host or the guest was in excess (Fig. S34, ESI†), indicating a fast equilibrium on the NMR timescale. To acquire information on the relative orientation of the two components, we stoppered the pseudorotaxanes to synthesize the corresponding rotaxanes (Scheme 1).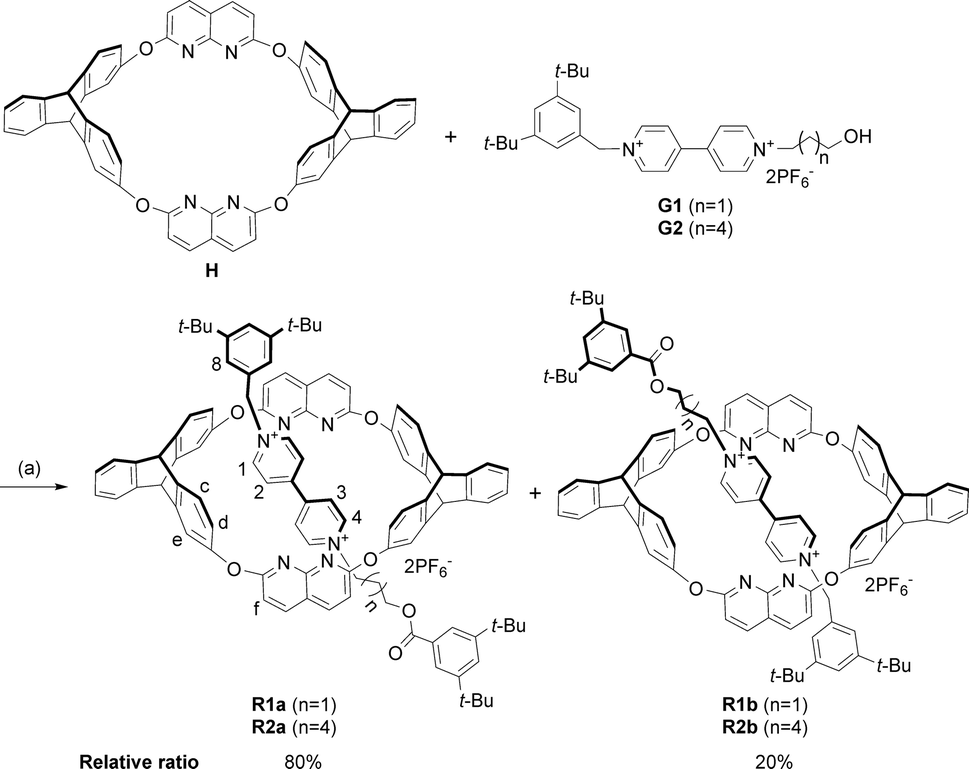 | ||
| Scheme 1 Synthesis of isomeric [2]rotaxanes R1a–b and R2a–b. Conditions: (a) 3,5-di-tert-butylbenzoic anhydride, (n-Bu)3P, CHCl3/CH3CN (2:1, v/v), r.t. | ||
As we demonstrated in our previous work that macrocycle H adopted a fixed 1,3-alternate conformation and would not undergo comformational inversion mainly due to the bulky triptycene moieties,16 the orientation of the obtained rotaxanes could represent the original threading direction of the axle into the wheel on a statistical basis.9b The stoppered products were isolated as a mixture of two orientational isomers, as revealed by both 1H NMR spectra and ESI-HRMS. The integrals in the 1H NMR spectrum (see Fig. S27†) showed that the relative ratio of the two sets of signals (and thus the amounts of the two isomers, R1a‡ and R1b) was approximately 4:1. The ratio calculated from the NMR spectrum revealed that the threading was highly directionally selective, but we could not ascertain the relative orientation of the wheel and axle in the two isomers.
Fortunately, single crystals of both the pseudorotaxane cultivated from the CHCl3/CH3CN solution of the complex (Fig. 2a and b) and rotaxane R1a cultivated from its CHCl3 solution (Fig. 2c and d) were obtained. Both crystal structures showed that the axle had threaded into the wheel from the triptycene rim (the lower rim, Fig. 1b).§ Weak correlations in the 2D ROESY spectrum of R1a (Fig. S22, ESI†) between H2 and Hc, H2 and Hd, and H8 and Hc were also detected, which indicated the same orientation of the host and guest in R1a as was deduced from the crystal structure.
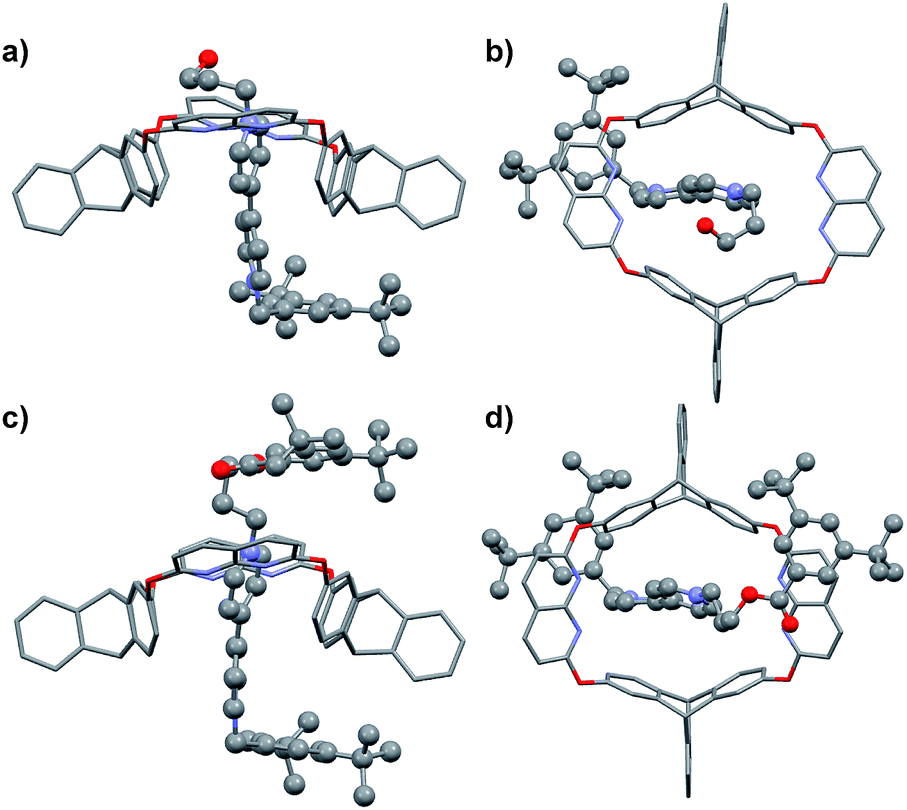 | ||
| Fig. 2 (a) Side and (b) top view of the crystal structure of H@G1. (c) Side and (d) top view of the crystal structure of [2]rotaxane R1a. Hydrogen atoms and PF6− counterions are omitted for clarity. | ||
In order to determine which part of the axle might have exerted a crucial impact on the threading direction,9d we firstly altered the length of the alkyl chain by replacing the hydroxypropyl group with a hydroxyhexyl group (G2, Fig. 1b). We synthesized the corresponding isomeric rotaxanes R2a and R2b (the ratio was also found to be about 4:1, Fig. S28, ESI†) (Scheme 1) and compared the 1H NMR spectrum of the major product R2a with that of R1a. After unambiguous assignment of all signals in the 1H NMR spectrum of R2a on the basis of 2D NMR experiments (Fig. S23 and S24, ESI†), it was found that all the corresponding hydrogens of the two rotaxanes had almost the same chemical shifts (Fig. S9 and S13, ESI†), indicating identical orientation of the wheel and axle in R1a and R2a. Thus it could be summarized that the length of the terminal alkyl chain had a negligible influence on the threading direction, at least when the chain was not very long.
Unidirectional complexation between macrocycle H and pyridinium salt G3
In the dominant threading direction of G1, the process was electrostatically favorable but sterically unfavorable. Therefore, to disclose whether electrostatic effects might have played an important part in the directional selectivity, we replaced the terminal pyridyl ring in G1 with a more electron-rich phenyl ring substituted by an electron-donating alkoxy group (Fig. 1c). Pyridinium salt G3 was encapsulated by H as expected (Fig. S33, ESI†), and similarly, the complex was in fast exchange on the NMR timescale (Fig. S40, ESI†). As the relevant data of the pseudorotaxane provided barely any information about the threading direction, the host–guest complex was stoppered to give the corresponding rotaxane (Scheme 2). To our surprise, the product showed only one set of signals in the 1H NMR spectrum (Fig. S19, ESI†), which meant that only one (R3b) of the isomeric rotaxanes was actually formed and thus the threading had occurred unidirectionally. To clarify the threading direction of G3, we cultivated a single crystal that was suitable for X-ray crystal analysis by slow evaporation of diethyl ether into an acetone solution of R3b (Fig. 3). The crystal structure showed that G3 had threaded into H from the naphthyridine rim (Fig. 1c), the direction of which was completely reversed compared with the dominant threading direction of G1 and G2. This agrees well with the 2D ROESY experiment (Fig. S26, ESI†) in which correlations were observed between H3 and Hc/Hd, H2 and Hc/Hd, H8 and Hf, and H8 and He.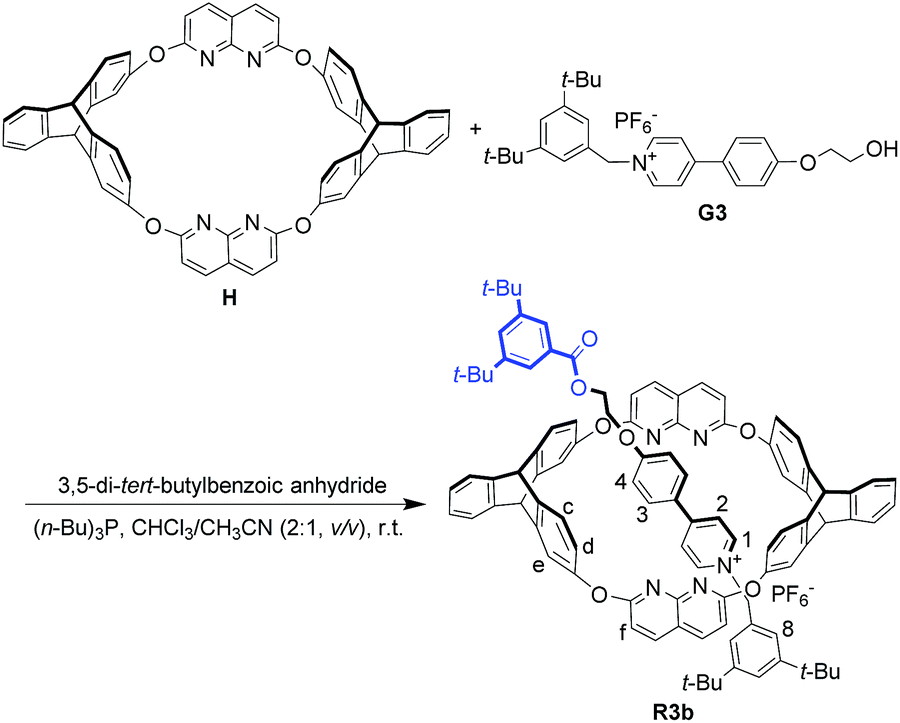 | ||
| Scheme 2 Synthesis of oriented [2]rotaxane R3b. | ||
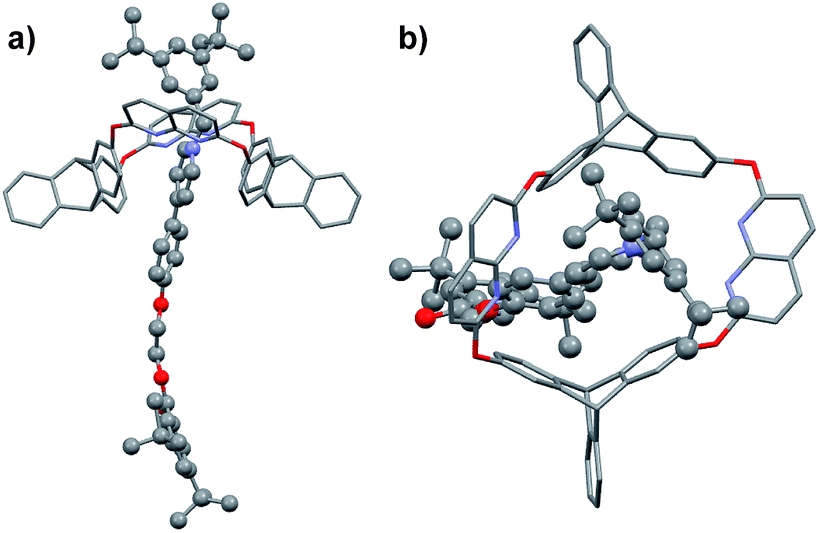 | ||
| Fig. 3 (a) Side and (b) top view of the crystal structure of R3b. Hydrogen atoms and PF6− counterions are omitted for clarity. | ||
Study on the directionally selective mechanism
These results were exciting but not beyond expectation, and could be tentatively explained as follows. In the threading process of G1 (the bipyridinium salt), the electrostatic effect dominated the threading direction while the steric effect posed an opposite influence. As a result, a pair of orientationally isomeric rotaxanes in a ratio of 4:1 was obtained, and in the major product the pre-stoppered end was in proximity to the lower rim of H. However, when axle G3 (the pyridinium salt) threaded into the macrocycle, the electrostatic effect and the steric effect acted in a cooperative way, generating a single compound in which the pre-stoppered end was in proximity to the upper rim of H. Moreover, to manifest the high selectivity in the threading direction of G3 into H in an in situ manner, a variable-temperature 1H NMR experiment on the CDCl3/CD3CN solution of the pseudorotaxane based on G3 and H (G3:H = 1.5:1) was conducted (Fig. S43–45, ESI†). From the spectra, we observed that when the temperature was brought down to approximately 233 K, the peaks of the free guest started to appear, indicating that the threading and dethreading had become a slow exchange process on the NMR timescale. Meanwhile, it was found that the pseudorotaxane still appeared as a single set of signals, which meant that at and below this temperature, the 1H NMR spectra clearly showed that the threading of G3 into H occurred unidirectionally.¶DFT theoretical modeling was carried out in order to establish whether, for each axle, the orientational outcome is dictated by kinetic factors or by the different thermodynamic stability of the two isomers.† We calculated the relative Gibbs free energies of the two orientational isomers of pseudorotaxane H@G1 (PR1a and PR1b), and found that the free energy of PR1a (corresponding to the orientation of R1a) was lower than that of its isomer PR1b by only 0.9 kcal mol−1. However, for the pseudorotaxanes based on G3 and H (PR3a and PR3b), the calculations indicated that the relative Gibbs free energy of PR3b (corresponding to the orientation of rotaxane R3b) was lower than that of PR3a by a sizable 3.6 kcal mol−1. Thus, the DFT calculations verified the relative stabilities of the two pairs of isomers: both dominant isomers (PR1a and PR3b) were the relatively more stable ones, though PR1a only had a small advantage over PR1b energetically.
The ESPs of H in the absence of guest molecules were also calculated, as depicted in Fig. 4a and b. Two features were notable; one was the electron-rich character of the phenyl rings of the triptycene semi-cavity, and the other was the polarity of the naphthyridine moieties, characterized by the electron-rich N atoms and the electron-poor remaining part. The difference in the electron densities of guests G1 and G3 was visualized by comparing their ESPs (Fig. 4c and d). G3 had a clearly higher electron density than G1, especially with respect to the unstoppered end (the phenyl ring). Therefore, the structures revealed the electrostatic repulsion of the electron-poor pyridinyl ring (the one further from the pre-stopper) in G1 and the attraction of the electron-rich phenyl ring in G3 exerted by the upper semi-cavity, and also the repulsion of the phenyl ring in G3 by the lower semi-cavity, in the threading processes of the two guests.
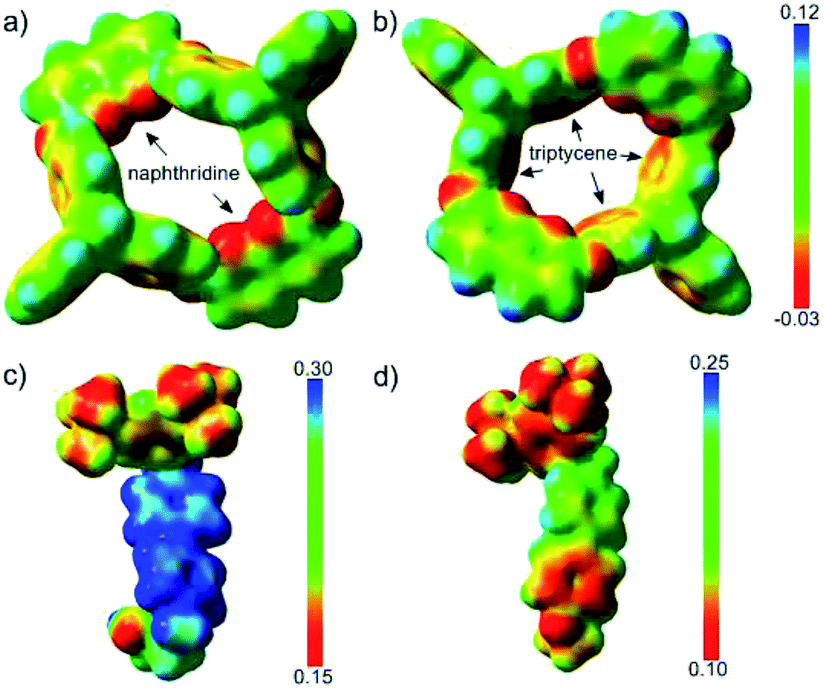 | ||
| Fig. 4 ESPs mapped onto electron density isosurfaces (ρ = 0.01) for H viewed from the lower rim (a) and upper rim (b), and for G1 (c) and G3 (d). | ||
From the above calculations, we concluded that the two isomers of H@G1 had similar stability, but in forming PR1b from the less crowded upper rim, the barrier posed by the electrostatic effect might be higher than that in forming PR1a, while the subordinate factor, the steric effect, acted oppositely; as a result, two isomers in a ratio of 4:1 were obtained. However, with regard to H@G3, on the one hand PR3b was more stable than its isomer by 3.6 kcal mol−1, and on the other hand, threading from the upper rim was favorable in terms of both electrostatic and steric effects; thus, unidirectional threading was attained.
As we hypothesized that the directional selectivity in the H@G1 system resulted mainly from the difference in energy barriers in the threading processes in each direction, increasing the temperature ought to narrow the gap between them. To verify this consideration, rotaxanes based on G1 and H were synthesized at raised temperatures (313 K and 333 K), and the ratios of the isomers were determined via1H NMR analysis (Fig. S29–30, ESI†). In agreement with our prediction, the directional selectivity in the threading process of G1 decreased with the increase in temperature (Table 1). However, in the same experiments on G3 and H, the orientational isomer of R3b could not even be traced. The results further demonstrated that at ambient temperature the threading of G1 was kinetically controlled.
Conclusions
In this work, we successfully demonstrated that the threading direction of (bi)pyridinium salts (G1–G3) into a triptycene-derived oxacalixarene (H) could be effectively controlled by tuning the electron densities of the guests, and the single crystal structures of (pseudo)rotaxanes exclusively manifested the orientation of the host and the guests. As the directional selectivity could be varied by tuning the roles played by steric and electrostatic effects, this work potentially provides a new approach towards the construction of oriented and well-aligned hierarchical assemblies. This is the first report on selective or unidirectional threading based on oxacalixarenes, and high directional selectivity was attained without either the use of special counteranions5,13 for the guest or further functionalization of the calixarene.4,9 The design of new guests and modification of the macrocycle by replacing the naphthyridine moieties with other electron-deficient moieties to obtain more precise control of directional selectivity from both sides, and the construction of high-order mechanically interlocked structures capable of performing unidirectional motion, are currently under investigation in our laboratory.Experimental
General remarks
The synthesis of precursors of the rotaxanes is described in the ESI.† All the other reagents and solvents were bought from commercial suppliers and used as received.Synthesis of R1a and R1b
A mixture of H (62 mg, 0.075 mmol) and G1 (53 mg, 0.075 mmol) in chloroform (10 mL) and acetonitrile (5 mL) was stirred at ambient temperature for 12 h. 3,5-Di-tert-butylbenzoic anhydride (135 mg, 0.3 mmol) and (n-Bu)3P (3 mg, 0.015 mmol) were then added to the mixture. The reaction mixture was stirred under argon for 24 h at ambient temperature. The solvent was removed under vacuum, and the residue was purified by column chromatography (CH2Cl2/acetone, 100:1 v/v) to give R1a and R1b as a mixture (73 mg) in 56% total yield. The macrocycle in the unreacted pseudorotaxanes could be recycled nearly quantitatively. The mixture was further separated carefully by preparative thin-layer chromatography to give pure R1a and R1b, both as yellow powders, for characterization.Synthesis of R2a and R2b
A mixture of H (62 mg, 0.075 mmol) and G2 (56 mg, 0.075 mmol) in chloroform (10 mL) and acetonitrile (5 mL) was stirred at ambient temperature for 12 h. 3,5-Di-tert-butylbenzoic anhydride (135 mg, 0.3 mmol) and (n-Bu)3P (3 mg, 0.015 mmol) were then added to the mixture. The reaction mixture was stirred under argon for 24 h at ambient temperature. The solvent was removed under vacuum, and the residue was purified by column chromatography (CH2Cl2/acetone, 100:1 v/v) to give R2a and R2b as a mixture (71 mg) in 53% total yield. The macrocycle in the unreacted pseudorotaxanes could be recycled nearly quantitatively. Then, the mixture was carefully separated by preparative thin-layer chromatography to give a fraction of pure R2a as a yellow powder, but it turned out to be difficult to obtain pure R2b for further characterization.Synthesis of R3b
A mixture of H (62 mg, 0.075 mmol) and G3 (43 mg, 0.075 mmol) in a solution of chloroform (10 mL) and acetonitrile (5 mL) was stirred at ambient temperature for 12 h. 3,5-Di-tert-butylbenzoic anhydride (135 mg, 0.3 mmol) and (n-Bu)3P (3 mg, 0.015 mmol) were then added to the mixture. The reaction mixture was stirred under argon for 24 h at ambient temperature. The solvent was removed under vacuum, and the residue was purified by column chromatography (CH2Cl2/acetone, 100:1 v/v) to give R3b (63 mg, 52% yield) as a white powder. The macrocycle in the unreacted pseudorotaxanes could be recycled nearly quantitatively. Mp: 211–213 °C. 1H NMR (400 MHz, CDCl3): δ 8.08 (d, J = 8.6 Hz, 4H), 8.03 (d, J = 8.5 Hz, 2H), 8.00 (s, 2H), 7.79 (d, J = 6.2 Hz, 2H), 7.66 (s, 1H), 7.53 (d, J = 6.2 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 7.23 (s, 1H), 7.17–7.23 (m, 4H), 7.02 (d, J = 8.6 Hz, 4H), 6.98 (brs, 4H), 6.87–6.89 (m, 4H), 6.79–6.85 (m, 8H), 6.55 (s, 2H), 5.15 (s, 2H), 4.95 (s, 2H), 4.86 (brs, 4H), 4.61 (t, J = 4.2 Hz, 2H), 1.35 (s, 18H), 1.08 (s, 18H). 13C NMR (75 MHz, CDCl3): δ 167.3, 164.1, 162.1, 153.8, 153.0, 151.9, 151.2, 150.1, 146.2, 145.3, 144.6, 141.9, 141.0, 140.0, 130.5, 129.9, 129.3, 127.4, 126.9, 125.3, 125.0, 124.0, 123.7, 123.55, 123.50, 123.4, 117.1, 117.0, 115.7, 112.3, 66.6, 62.7, 53.5, 52.3, 35.0, 34.6, 31.4, 31.1, 29.3. ESI-HRMS calcd for C99H88N5O7 [M − PF6−]+ 1458.6678; found 1458.6679.Acknowledgements
The authors thank the National Natural Science Foundation of China (21332008, 21202172, and 21290194), the National Basic Research Program (2011CB932501), and the Strategic Priority Research Program of Chinese Academy of Sciences (XDB12010400) for financial support.Notes and references
- (a) A. Harada, Acc. Chem. Res., 2001, 34, 456–464 CrossRef CAS PubMed; (b) K. Kim, Chem. Soc. Rev., 2002, 31, 96–107 RSC; (c) M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC; (d) J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC; (e) K. D. Hänni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC; (f) Z. Zhang, C. Han, G. Yu and F. Huang, Chem. Sci., 2012, 3, 3026–3031 RSC; (g) S. Dong, J. Yuan and F. Huang, Chem. Sci., 2014, 5, 247–252 RSC; (h) G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef PubMed.
- (a) K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed; (b) J.-P. Sauvage and C. Dietrich-Buchecker, Molecular Catenanes, Rotaxanes and Knots: A Journey Through the World of Molecular Topology, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (c) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC; (d) D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978–982 CrossRef CAS PubMed; (e) J.-F. Ayme, G. Gil-Ramírez, D. A. Leigh, J.-F. Lemonnier, A. Markevicius, C. A. Muryn and G. Zhang, J. Am. Chem. Soc., 2014, 136, 13142–13145 CrossRef CAS PubMed; (f) J. E. Beves, J. J. Danon, D. A. Leigh, J.-F. Lemonnier and I. J. Vitorica-Yrezabal, Angew. Chem., Int. Ed., 2015, 54, 7555–7559 CrossRef CAS PubMed; (g) G. Zhang, G. Gil-Ramírez, A. Markevicius, C. Browne, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2015, 137, 10437–10442 CrossRef CAS PubMed.
- (a) A. Credi, V. Balzani, S. J. Langford and J. F. Stoddart, J. Am. Chem. Soc., 1997, 119, 2679–2681 CrossRef CAS; (b) B. L. Feringa, Molecular Switches, Wiley-VCH, Weinheim, Germany, 2001 CrossRef PubMed; (c) V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines: A Journey into the Nano World, Wiley-VCH, Weinheim, Germany, 2003 CrossRef PubMed; (d) X. Ma and H. Tian, Chem. Soc. Rev., 2010, 39, 70–80 RSC; (e) N. H. Evans and P. D. Beer, Chem. Soc. Rev., 2014, 43, 4658–4683 RSC; (f) S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC; (g) E. R. Kay and D. A. Leigh, Angew. Chem., Int. Ed., 2015, 54, 10080–10088 CrossRef CAS PubMed; (h) Y.-X. Xia, T. Xie, Y. Han and C.-F. Chen, Org. Chem. Front., 2014, 1, 140–147 RSC.
- A. Arduini, F. Ciesa, M. Fragassi, A. Pochini and A. Secchi, Angew. Chem., Int. Ed., 2005, 44, 278–281 CrossRef CAS PubMed.
- C. Gaeta, F. Troisi and P. Neri, Org. Lett., 2010, 12, 2092–2095 CrossRef CAS PubMed.
- (a) R. Isnin and A. E. Kaifer, J. Am. Chem. Soc., 1991, 113, 8188–8190 CrossRef CAS; (b) J. W. Park and H. J. Song, Org. Lett., 2004, 6, 4869–4872 CrossRef CAS PubMed.
- C. Casati, P. Franchi, R. Pievo, E. Mezzina and M. Lucarini, J. Am. Chem. Soc., 2012, 134, 19108–19117 CrossRef CAS PubMed.
- For selected examples in biological systems and materials science in which a difference in spatial arrangement caused a change in properties, see: (a) E. Tajkhorshid, P. Nollert, M. Ø. Jensen, L. J. W. Miercke, J. O'Connell, R. M. Stroud and K. Schulten, Science, 2002, 296, 525–530 CrossRef CAS PubMed; (b) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- (a) A. Arduini, F. Calzavacca, A. Pochini and A. Secchi, Chem.–Eur. J., 2003, 9, 793–799 CrossRef CAS PubMed; (b) A. Arduini, R. Bussolati, A. Credi, G. Faimani, S. Garaudée, A. Pochini, A. Secchi, M. Semeraro, S. Silvi and M. Venturi, Chem.–Eur. J., 2009, 15, 3230–3242 CrossRef CAS PubMed; (c) A. Arduini, R. Bussolati, A. Credi, S. Monaco, A. Secchi, S. Silvi and M. Venturi, Chem.–Eur. J., 2012, 18, 16203–16213 CrossRef CAS PubMed; (d) A. Arduini, R. Bussolati, A. Credi, A. Secchi, S. Silvi, M. Semeraro and M. Venturi, J. Am. Chem. Soc., 2013, 135, 9924–9930 CrossRef CAS PubMed.
- (a) T. Oshikiri, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2005, 127, 12186–12187 CrossRef CAS PubMed; (b) T. Oshikiri, Y. Takashima, H. Yamaguchi and A. Harada, Chem.–Eur. J., 2007, 13, 7091–7098 CrossRef CAS PubMed; (c) T. Oshikiri, H. Yamaguchi, Y. Takashima and A. Harada, Chem. Commun., 2009, 5515–5517 RSC.
- Q.-C. Wang, X. Ma, D.-H. Qu and H. Tian, Chem.–Eur. J., 2006, 12, 1088–1096 CrossRef CAS PubMed.
- M. Xue, Y.-S. Su and C.-F. Chen, Chem.–Eur. J., 2010, 16, 8537–8544 CrossRef CAS PubMed.
- (a) C. Talotta, C. Gaeta, T. Pierro and P. Neri, Org. Lett., 2011, 13, 2098–2101 CrossRef CAS PubMed; (b) C. Talotta, C. Gaeta and P. Neri, Org. Lett., 2012, 14, 3104–3107 CrossRef CAS PubMed; (c) R. Ciao, C. Talotta, C. Gaeta, L. Margarucci, A. Casapullo and P. Neri, Org. Lett., 2013, 15, 5694–5697 CrossRef CAS PubMed; (d) C. Gaeta, C. Talotta, S. Mirra, L. Margarucci, A. Casapullo and P. Neri, Org. Lett., 2013, 15, 116–119 CrossRef CAS PubMed; (e) C. Gaeta, C. Talotta and P. Neri, Chem. Commun., 2014, 50, 9917–9920 RSC; (f) C. Talotta, C. Gaeta, Z. Qi, C. A. Schalley and P. Neri, Angew. Chem., Int. Ed., 2013, 52, 7437–7441 CrossRef CAS PubMed.
- S. Ibach, V. Prautzsch and F. Vögtle, Acc. Chem. Res., 1999, 32, 729–740 CrossRef CAS.
- (a) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed; (b) W. Maes and W. Dehaen, Chem. Soc. Rev., 2008, 37, 2393–2402 RSC; (c) C.-F. Chen, Chem. Commun., 2011, 47, 1674–1688 RSC.
- (a) S.-Z. Hu and C.-F. Chen, Chem. Commun., 2010, 46, 4199–4201 RSC; (b) S.-Z. Hu and C.-F. Chen, Chem.–Eur. J., 2011, 17, 5424–5431 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Further details of synthesis, characterization, 1H and 2D NMR experiments and computational modeling. CCDC 1009829, 1009830, and 1419743. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03511b |
| ‡ In this paper, isomers of pseudorotaxanes and rotaxanes are denoted such that a indicates that the corresponding guest is threaded from the triptycene rim, while b indicates that the corresponding guest is threaded from the naphthyridine rim. |
| § The absence of the pseudorotaxane in which the macrocycle and axle are oriented the other way (PR1b) in the crystal might be caused by the difference in the amounts of the two isomeric pseudorotaxanes and the selective crystallization of the dominant one. |
| ¶ As the signals of the pseudorotaxane changed gradually with decreasing temperature without saltation of each chemical shift, we could safely deduce that the threading direction did not change during the cooling process. |
| This journal is © The Royal Society of Chemistry 2016 |