
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantiodivergent asymmetric catalysis with the tropos BIPHEP ligand and a proline derivative as chiral selector†
P.
Oczipka
,
D.
Müller
,
W.
Leitner
* and
G.
Franciò
*
Institut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: francio@itmc.rwth-aachen.de; leitner@itmc.rwth-aachen.de
First published on 29th October 2015
Abstract
A catalytic system based on the tropos ligand BIPHEP and (S)-proline methyl ester as chiral selector was studied for Rh-catalysed asymmetric catalysis. By careful control of the catalyst preformation conditions, the enantioselectivity could be completely reversed in asymmetric hydrogenation of prochiral olefins maintaining the same absolute level in favorable cases. The enantiodivergent asymmetric catalysis could be rationalised by the interplay of the dynamic chirality (tropos) of the phosphine ligand and the coordination of the proline selector. Treating a suitable Rh-BIPHEP precursor with the (Sc)-proline-based ionic liquid led to an equimolar mixture of (RaSc)- and (SaSc)-diastereomers that is kinetically stable at 0 °C. At higher temperature, an irreversible diastereomerisation process was observed resulting in the diastereomerically pure (RaSc)-complex [Rh{(Ra)-BIPHEP}{(Sc)-ProlOMe}]. Whereas the use of the pure (RaSc)-complex led to 51% ee (R) in the hydrogenation of methyl 2-acetamidoacrylate, the S-product was formed with almost identical enantioselectivity when the (RaSc)/(SaSc)-mixture was applied under identical conditions. This inversion was associated with the relative stability of the diastereomers in the equilibria forming the catalytically active substrate complex. The possibility to use this different reactivity to control the direction of enantioselectivity was demonstrated for the hydrogenation of different substrates whereby ee's of up to 80% could be achieved. Moreover, the (RaSc)-complex led to high enantioselectivities of up 86% ee in the asymmetric hydroboration of styrene, approaching the performance of the atropos BINAP ligand for this reaction.
Introduction
Asymmetric organometallic catalysis is one of the most useful synthetic tools for producing enantioenriched compounds.1 The stereo-information usually provided by chiral ligands is efficiently “multiplied”2 and high catalytic activities at high levels of stereocontrol can be achieved in a variety of transformations. Some of the most effective ligands with “permanent” chirality are atropisomeric (atropos)3 bidentate disphosphine ligands such as BINAP4 or MeO-BIPHEP.5In contrast, fluxional tropos BIPHEP (1) ligands rapidly interconvert at room temperature between the Ra and Sa conformation (“dynamic chirality”, Scheme 1).
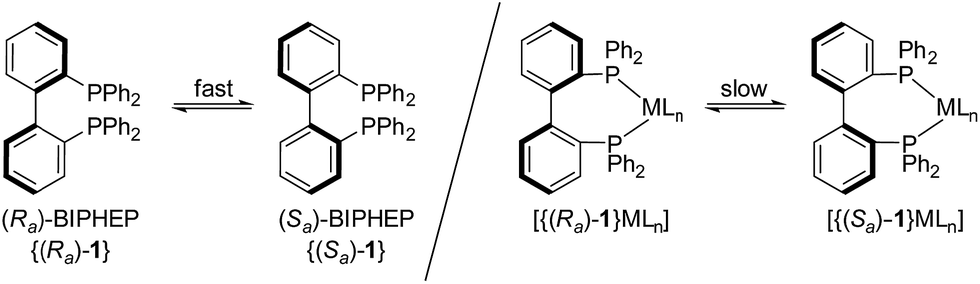 | ||
| Scheme 1 Fluxional behavior of the ligand BIPHEP (1). | ||
Upon coordination to a metal center, however, the rotation about the biaryl axis is much slower and a racemic pair is obtained.6,7 Thus, with the aid of an enantiomeric pure co-ligand3,8 or counteranion,9,10 which act as a chiral template, a dynamic resolution of the BIPHEP (1) backbone can be conveyed and the resulting diastereomeric pure complexes may be isolated.11,12 After displacement of the chiral template molecule (diols, diamins, aminoalcohols, or dienes) the enantiomeric pure BIPHEP-metal fragment can be used in asymmetric catalysis, provided the racemisation is slow at the conditions of catalytic turnover. This approach was first demonstrated by Gagnè13 and applied by Mikami in different catalytic reactions.14Inter alia, high enantioselectivities in the Rh-catalysed hydrogenation have been reported by Brown15 and Mikami.16 All these examples have the following features in common: Complete resolution of the coordinated BIPHEP-ligand (diastereomerisation17) was always accomplished prior to use the complex in catalysis; otherwise no or very poor enantioselectivity was observed.16,18 Only in cases where ligand and selector both remain coordinated to the complex while the substrate is converted, as in Noyori-type catalysts, a complete diastereomerisation was not mandatory due to different reactivity of the two diastereomers in the outer-sphere hydrogen transfer (so called “asymmetric activation”).19
Independently from our studies reported here, the group of Trapp recently developed BIPHEP derivatives that contain covalently bound chiral auxiliaries at the 3,3′-positions. These chiral substituents can also act as chiral selectors for the biphenyl backbone, as demonstrated with “bidirectional enantioselectivity” in Rh-catalysed hydrogenation20 and selective binding for analytical purposes.21
In a complementary approach, research from our laboratories has shown that good enantioselectivities can be achieved with tropos BIPHEP ligands using the chiral ionic liquid (IL) 2 as the reaction medium in the Rh-catalysed hydrogenation.22 The same approach proved successful employing the racemic atropoisomeric ligand BINAP together with the same chiral IL.23 NMR-spectroscopic analysis and kinetic studies furnished evidences that the enantiodifferentiation in this system results from coordination of the proline ester moiety to the chiral cation, effectively blocking one of the two enantiomeric Rh-phosphine units.23 A similar “chiral-poisoning mechanism” was evidenced for cationic racBINAP/Rh-catalysts in the presence of BINOL-based borate anions.24
Here, we report a fundamental investigation of the dynamic interaction of catalytically active fragment and chiral selector responsible for the chirality transfer when [HProlOMe][NTf2] (2) is used as the enantiopure auxiliary in combination with the tropos BIPHEP ligand in Rh-catalysed transformations.22 This knowledge allowed us to set appropriate conditions for the preferred formation of either the R- or S-configured products with almost identical absolute enantioselectivity using the identical catalytic system in the asymmetric hydrogenation of the same substrate. The catalytic system could be successfully applied also in the Rh-catalysed hydroboration demonstrating that the BIPHEP backbone can lead to very significant asymmetric inductions also in this transformation.
Results and discussion
Synthesis, characterisation, and dynamics of Rh-complexes
While the Rh-BIPHEP catalyst was prepared in situ in presence of the chiral selector in our preliminary communication,22 we set out to generate a defined complex of type [Rh(BIPHEP)(ProlOMe)][NTf2] (5). Following the procedure developed for the BINAP system,23 the complex [Rh(BIPHEP)(acac)]25 was treated with a slight excess of the prolininium IL [HProlOMe][NTf2]26 (2) in CH2Cl2. By monitoring this reaction with 1H and 31P-NMR at −30 °C, we found that formation of 5 involves the formal oxidative addition of 2 at the Rh(I)-center followed by reductive elimination of acetylacetone27 leading to the N,O-chelation of the proline ester (Scheme 2).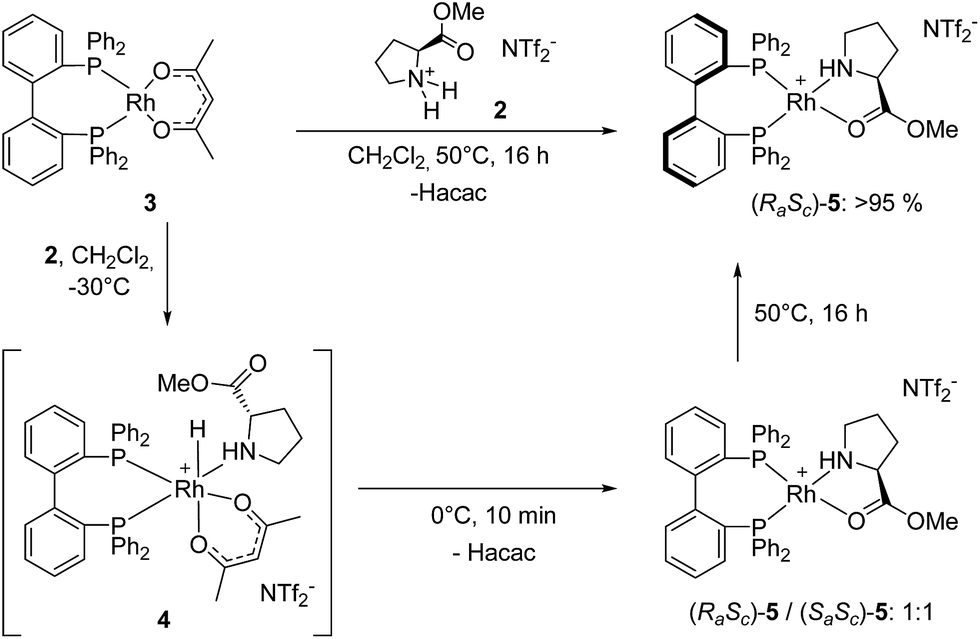 | ||
| Scheme 2 Reaction of Rh-BIPHEP complex 3 with [HProlOMe][NTf2] 2. | ||
The two diastereomers of the intermediate Rh(III)-BIPHEP-hydride species (4) were formed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in solution at −30 °C as deduced from multinuclear NMR spectroscopy (Fig. 1). The structure of 4 was assigned on the basis of following spectroscopic data: (i) in the 31P-NMR, two sets of doublets of doublets for each diastereomer were observed indicating different trans-substituents to each phosphorus; (ii) in the characteristic hydride region of the 1H-NMR, a doublet of triplets for each diastereomer was observed arising from the 103Rh-coupling and two almost identical 31P-coupling, respectively; (iii) distinct signals for the two methyl groups of the acetylacetonato ligand were present in 1H and 13C-NMR; furthermore, cross peaks from both sides of the acetylacetonato group with P-nuclei in the 1H–31P-HMBC spectrum indicated that acetylacetonate is η2-coordinated to the Rh-center and has different substituents in trans-position of each oxygen donor.
:1 ratio in solution at −30 °C as deduced from multinuclear NMR spectroscopy (Fig. 1). The structure of 4 was assigned on the basis of following spectroscopic data: (i) in the 31P-NMR, two sets of doublets of doublets for each diastereomer were observed indicating different trans-substituents to each phosphorus; (ii) in the characteristic hydride region of the 1H-NMR, a doublet of triplets for each diastereomer was observed arising from the 103Rh-coupling and two almost identical 31P-coupling, respectively; (iii) distinct signals for the two methyl groups of the acetylacetonato ligand were present in 1H and 13C-NMR; furthermore, cross peaks from both sides of the acetylacetonato group with P-nuclei in the 1H–31P-HMBC spectrum indicated that acetylacetonate is η2-coordinated to the Rh-center and has different substituents in trans-position of each oxygen donor.
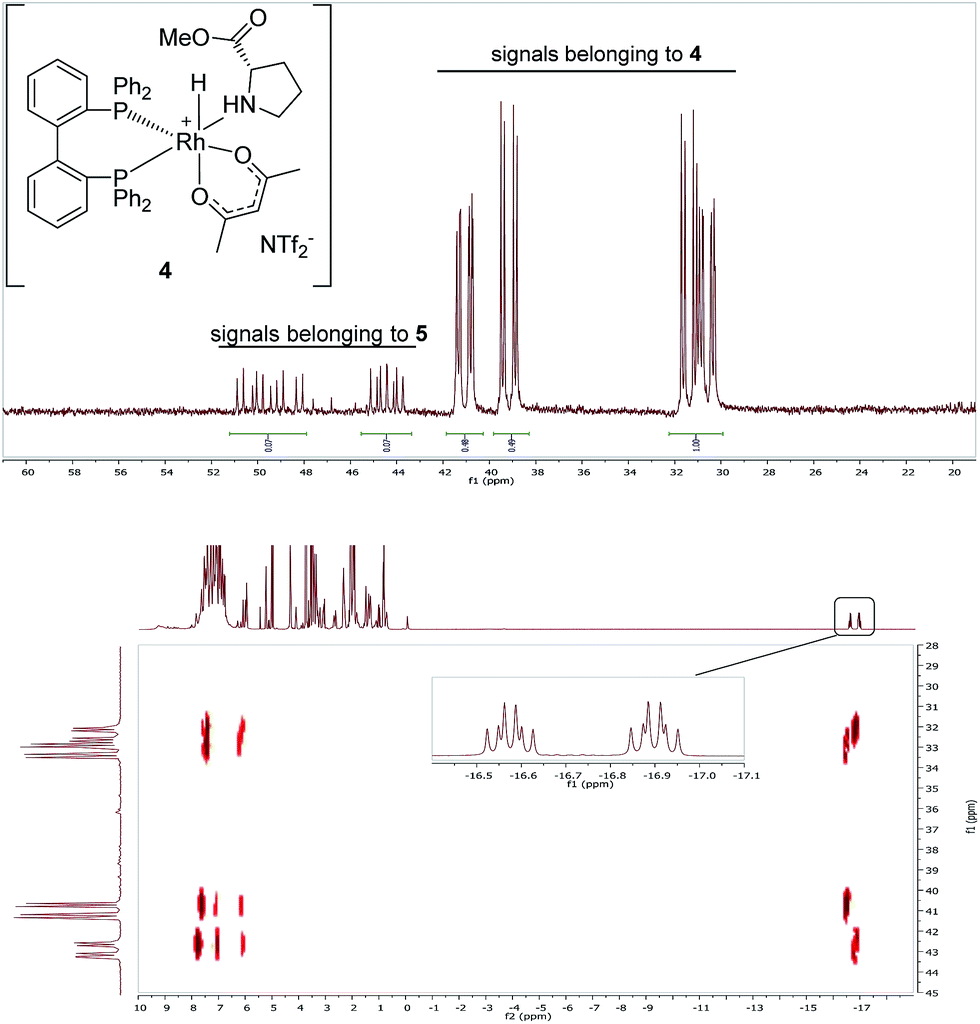 | ||
| Fig. 1 31P{1H}-NMR (CD2Cl2, −30 °C) of Rh complexes 4 (major) and 5 (minor) (upper spectrum) and 2D-NMR-HMBC-31P–1H-NMR of the same sample showing coupling to hydride signals (bottom spectrum). | ||
At 0 °C the octahedral Rh(III) complex 4 rapidly eliminates acetylacetone (Hacac) resulting in the formation of the square planar cationic Rh(I) complex [Rh(BIPHEP)(ProlOMe)][NTf2] 5. As observed for the catalyst system Rh-racBINAP/2,23 the chiral IL [HProlOMe][NTf2] 2 serves upon deprotonation as the source of (S)-proline methylester, a N,O-chelating ligand. The complex is characterised by two sets of doublets of doublets for each diastereomer in the 31P-NMR indicating different donor ligand trans to the bisphosphine. The signal sets centred at δ = 52.1 and 45.8 are assigned to (RaSc)-5, those at δ = 50.44 and 46.3 to the (SaSc)-5 diastereomer, respectively. The assignment of the configuration in the BIPHEP ligand is based on the catalytic results (vide infra) upon comparison with the atropoisomeric ligand BINAP under the plausible assumption that identical axial chirality will lead to the same preferred product configuration.
The equimolar ratio of (SaSc)-5 and (RaSc)-5 does not vary even over extended period of time when the temperature is maintained at or below 0 °C. However, raising the temperature to room temperature leads to the increase of one set of signals on the expense of the other pair, indicating that a diastereomerisation process takes place (Fig. 2). The signals of (SaSc)-5 gradually decrease and completely disappear by heating the mixture at 50 °C for 20 h resulting in the clean formation of (RaSc)-5 (>95% by NMR). This process is irreversible, and no reformation of the (SaSc)-diastereomer was observed by temperature variation in any direction once the diastereomerisation was completed.
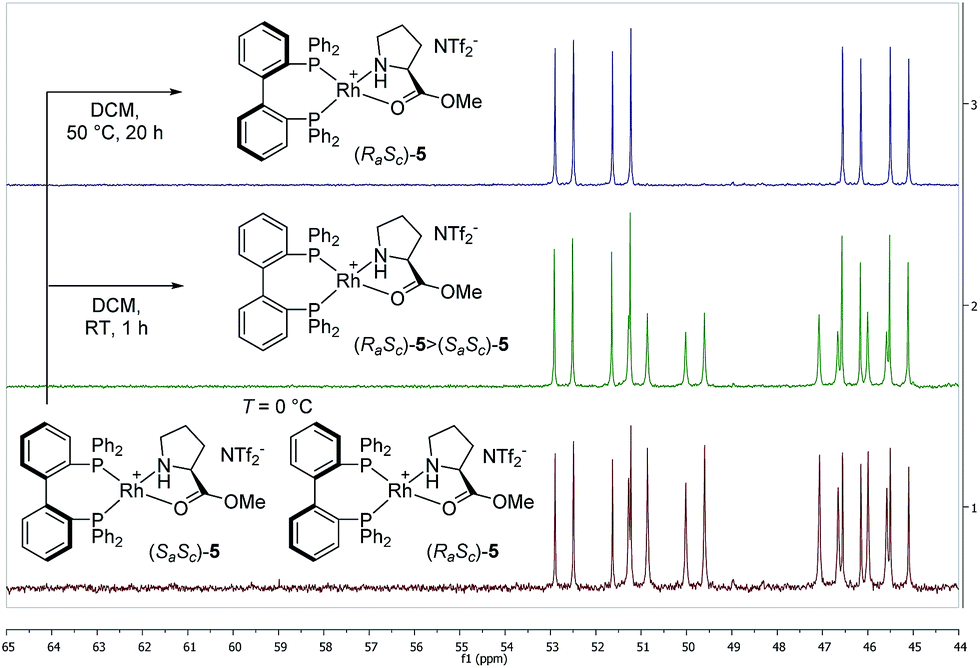 | ||
| Fig. 2 Diastereomerisation of the 1:1 (SaSc)-5:(RaSc)-5 mixture to (RaSc)-5 upon raising the temperature monitored by 31P{1H}-NMR spectra (CD2Cl2). | ||
These results demonstrate that proline-methylester acts as very effective chiral selector that is able to affect the dynamic behaviour of the BIPHEP ligand 1 in the Rh-chelate fragment and eventually to dictate the thermodynamically preferred axial chirality of the tropos-moiety in [Rh(BIPHEP)(ProlOMe)][NTf2] (5). The rate of the diastereomerisation reaction was determined at 25 °C, 30 °C, 35 °C and 50 °C via31P-NMR in dichloroethane-d4 as solvent. From the resulting Arrhenius-plot the rotation barrier for the conversion of (Sa)- to (Ra)-BIPHEP in 5 was determined as 24.3 ± 1 kcal mol−1. This barrier is higher than the energy required for the tropo-inversion of the free ligand (22 ± 1 kcal mol−1)28,29 and in good agreement with the value of 25.0 kcal mol−1 reported by Faller for the racemisation of [{(Sa)-BIPHEP}Rh(CO)2].7 A slightly higher activation energy (26.1 kcal mol−1) was reported for the pseudo-racemisation of the substituted ligand in [{(SaSS)-3,3′-BIPHEPONap*}Rh(cod)]BF4 by Trapp.20
Asymmetric hydrogenation
The diastereomerically pure complex (RaSc)-5 was evaluated first as catalyst for asymmetric hydrogenation (Table 1). Using a solution of (RaSc)-5 prepared as described above, the prototypical substrate methyl 2-acetamidoacrylate (6) could be hydrogenated quantitatively in CH2Cl2 at 0 °C resulting in 51% ee for the (R)-enantiomer (entry 1). Monitoring the hydrogen uptake at a strongly reduced catalyst to substrate ratio of 1:4000 revealed full conversion after 75 min corresponding to a TON of 4000 and an average TOFav of 3200 h−1 while maintaining the same level of asymmetric induction. Addition of 2.5 eq. of the achiral acid HNTf2 to promote the dissociation of the chiral template has no significant impact on the enantioselectivity yielding 53% ee (R) under otherwise identical conditions (entry 2). Rising the reaction temperature to 35 °C resulted in a slightly reduced ee (45% (R), entry 3). This value is still slightly higher than the enantioselectivity (44% ee) reported by Punniyamurthy and Brown using [{(Sa)-BIPHEP}Rh{(S,S)-bicyclo[3.3.1]nona-2,6-diene}]+CF3SO3− as precursor at room temperature.15
| Nr | T [°C] | Solvent | ee [%] |
|---|---|---|---|
|
|
|||
| a See the exp. Section for the procedure. Full conversion achieved in all experiments. b No HNTf2 was added. | |||
| 1b | 0 | DCM | 51 (R) |
| 2 | 0 | DCM | 53 (R) |
| 3 | 35 | DCM | 45 (R) |
| 4 | −20 | DCM | 56 (R) |
| 5 | 0 | THF | 47 (R) |
| 6 | 0 | MeOH | 49 (R) |
| 7 | 0 | 2-PrOH/H2O: 1/1 | 55 (R) |
| 8 | 0 | EtOH/H2O: 1/1 | 57 (R) |
| 9 | 0 | MeOH/H2O: 1/1 | 66 (R) |
Decreasing the reaction temperature to −20 °C led to an increase of enantioselectivity to 56% ee (R) (entry 4). While somewhat lower enantioselectivities were observed upon using THF and methanol as solvents (47% ee, entry 5; and 49% ee entry 6; respectively), a substantial increase could be achieved using alcohol/water mixtures as the reaction medium (entries 7–9). The best results were achieved with a 1:1 methanol/water mixture resulting in 66% ee (R).
Analysis of the reaction mixture after complete hydrogenation of the substrate by 31P{1H}-NMR spectroscopy revealed that the complex (RaSc)-5 was the major species present in solution (>90%, see ESI†). Only very small signals of other BIPHEP–Rh-species were observed, but no signals resulting from the diastereomeric complex (SaSc)-5 could be detected. This observation further corroborates the high stability of the (RaSc)-configuration pair. Under the reasonable assumption that the hydrogenation under these conditions follows a typical Halpern-Brown mechanism,30 this observation is consistent with a release/catch mechanism where (RaSc)-5 serves as pre-catalyst for the catalytically active complex [{(Ra)-BIPHEP}Rh(6)]+. The chirality of the chelating phosphine unit is stabilised and prevented from racemisation by re-coordination of the proline ester once the substrate is converted and product is released.
A striking different result was obtained when the distereomeric mixture (RaSc)/(SaSc)-5 was used as pre-catalyst. Under identical condition as in Table 1 entry 1, the hydrogenation of 6 resulted in preferential formation of the (S)-product with 51% ee. Thus, the same absolute value of enantioselectivity was achieved but towards the opposite enantiomer as with the (RaSc)-5 complex (Fig. 3).31 As the racemisation of the BIPHEP-Rh unit is very slow at 0 °C, this indicates that product formation now occurs practically exclusively via (Sa)-configured phosphine–rhodium complexes.
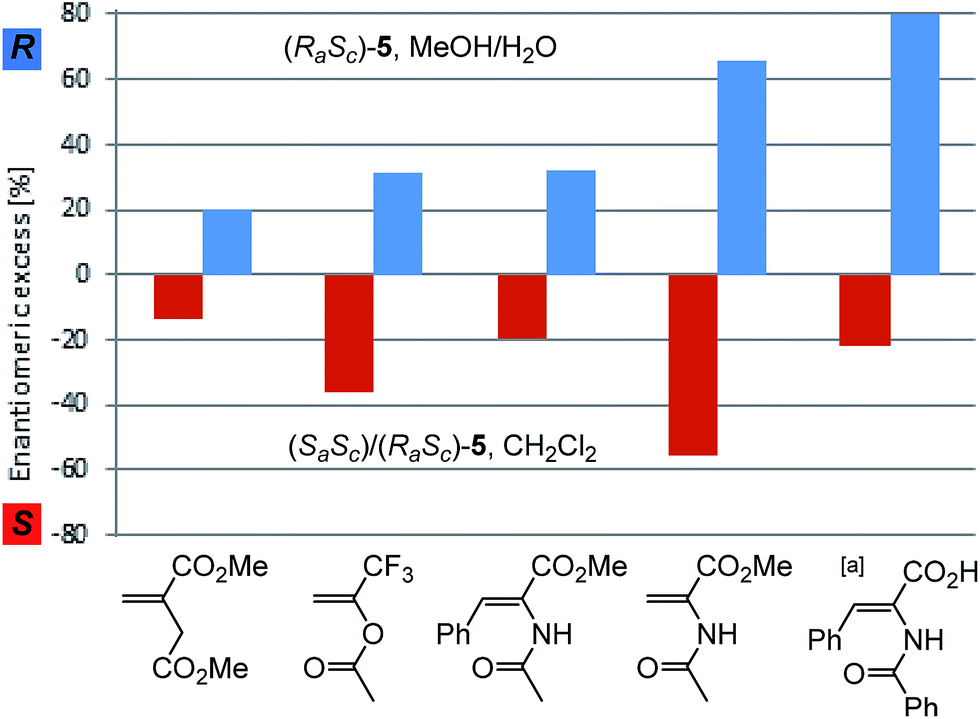 | ||
| Fig. 3 Enantiodivergent asymmetric hydrogenation of prochiral substrates with (RaSc)-5 and (RaSc)/(SaSc)-5. Full conversion was obtained in all experiments. [a] MeOH/H2O was used also with the diastereomeric mixture (RaSc)/(SaSc)-5. | ||
The 31P{1H}-NMR spectra of the mixture recorded after the catalysis support this assumption. The relative concentration of (RaSc)-5 had not changed significantly indicating that only a small fraction of (SaSc)-5 was potentially converted to the more stable diastereomer (see ESI†). The signals of (SaSc)-5 were, however, considerably broadened and partially converted to new species with signals in the similar region, most probably associated with the corresponding π-phenyl bridged dimer.32 Thus, the “inverse” enantioselectivity obtained with the (RaSc)/(SaSc)-5 mixture can be explained by a chiral poisoning mechanism.23 While the proline methyl ester is very readily displaced by the incoming substrate from the less stable (SaSc)-5, the thermodynamically preferred (RaSc)-5 remains “catalytically silent” under competitive conditions. Thus, the enantiodivergent asymmetric hydrogenation with 5 can be rationalised by the relative stability of the two diastereomeric complexes in the pre-equilibrium to form the catalytically active substrate complex.
The catalytic system was then evaluated in the asymmetric hydrogenation of structurally different prochiral olefins (Fig. 3). In all cases the enantiodivergent formation of either the (S)- or the (R)-product was observed using the (RaSc)/(SaSc)-5 mixture or the pure (RaSc)-5 complex, respectively. The enantioselectivity achieved with the single diastereomer (RaSc)-5 as pre-catalyst can be regarded as the intrinsic maximum asymmetric induction of the enantiomerically pure BIPHEP ligand in the Rh-catalysed hydrogenation under the given conditions. Moderate enantioselectivities were achieved in the hydrogenation using dimethylitaconate, 1-(trifluoromethyl)vinyl acetate, and methyl Z-2-acetamido-3-phenylacrylate. Z-2-benzamido-3-phenylacrylic acid could be hydrogenated with 80% ee in MeOH/H2O using (RaSc)-5, thus even surpassing the ee of the standard substrate 6.
The magnitude of absolute enantioselectivity obtained towards the opposite product enantiomer with the diastereomeric mixture (RaSc)/(SaSc)-5 in CH2Cl2 as solvent is generally in the same range as with the pure diastereomer, except for Z-2-benzamido-3-phenylacrylic acid where both reactions were carried out in MeOH/H2O. The strong influence of substrate and solvent on the absolute value of the inversion may reflect the influence of the equilibria preluding the catalytic cycle.
Remarkably, the enantioselectivites obtained with this system are in a similar range than those observed with the enantiopure atropos ligand BINAP in rhodium-catalysed hydrogenation of these substrates. Whereas higher ee's are observed with BINAP for Z-2-benzamido-3-phenylacrylic acid (96%)32 and dimethylitaconate (67%),24 they are lower for methyl Z-2-acetamido-3-phenylacrylate (16%)33 and methyl 2-acetamidoacrylate (25%).23 For the latter substrate, very high ee's were reported by Mikami (95%)16 and Brown (81%)15 using substituted BIPHEP ligands, showing a clear path for future developments.
Asymmetric hydroboration
Next, the use of tropos BIPHEP together with the chiral selector 2 was evaluated in the rhodium-catalysed hydroboration of styrene. Atropoisomeric ligands like BINAP belong to the most efficient diphosphines for this transformation.34,35 Recently, Faller et al. used a diastereomerically pure (Sa)-BIPHEP–Rh complex bearing an enantiomerically pure diene as chiral auxiliary in this reaction. However, only a very low enantioselectivity of 12% ee was obtained with this pre-catalyst.7Using the diastereomeric mixture (RaSc)/(SaSc)-5 as pre-catayst for the hydroboration of styrene with catecholborane under various conditions resulted only in racemic product. This indicates that the substrates are able to displace the proline-methylester from both complexes with equal efficiency, resulting in two enantiomeric catalytic cycles. In contrast, an enantioselectivity of 55% ee at full conversion was achieved using the diastereomerically pure pre-catalyst (RaSc)-5 in THF within 1 h at room temperature (Table 2, entry 1). Lowering the temperature to 0 °C, both the regio- (97%) and the enantioselectivity (70% ee) could be enhanced (entry 2). A temperature reduction to −50 °C resulted in a further increase of the enantioselectivity (77% ee) albeit at the expenses of strongly reduced conversion (entry 3). Using 1,2-dimethoxyethane (1,2-DME) as solvent gave similar enantioselectivities, but conversion now remained high even at the very low temperatures (entries 4–5). Most gratifyingly, the use of 2-methyl-tetrahydrofurane (2-Me-THF), which is recommended as most sustainable ether-type solvent by solvent selection guides,36 allowed to achieve full conversion, perfect regioselectivity, and an ee of 86% (entry 6).
|
|
||||||
|---|---|---|---|---|---|---|
| Nr | solvent | t [h] | T [°C] | Conv. [%] | 7/8 [%] | ee [%] |
| 1 | THF | 1 | r.t. | >99 | 93 | 55 (R) |
| 2 | THF | 3 | 0 | >99 | 97 | 70 (R) |
| 3 | THF | 12 | −50 | 8 | 95 | 77 (R) |
| 4 | 1,2-DME | 3 | 0 | >99 | 97 | 70 (R) |
| 5 | 1,2-DME | 12 | −50 | 91 | >99 | 79 (R) |
| 6 | 2-Me-THF | 12 | −50 | >99 | >99 | 86 (R) |
The performance of the tropos BIPHEP together with the proline-based selector 2 compares well with that obtained with the enantiomerically pure atropos ligand (Ra)-BINAP under optimised comparable conditions (88% ee (R), 54% conversion, 1 h, −50 °C, 1,2-DME).35 This demonstrates that the chirality adjusted in the flexible ligand framework during the diasteromerisation is maintained stable throughout the catalytic cycle. Together with the previous results of Faller et al.,7 this suggests that the proline-based auxiliary is again not only responsible for the selection, but also the stabilisation of the chiral fragment under catalytic conditions.
Conclusions
The combination of the simple tropos BIPHEP ligand and the readily accessible proline-based IL [(Sc)-HProlOMe][NTf2] (2) as chiral selector provides an effective system for high levels of enantioselectivity in rhodium catalysed reactions. In asymmetric hydrogenation, it is one of the few systems that can generate both enantiomeric products from a prochiral olefin using the same catalyst components under identical reaction conditions. The key to this enantiodivergent asymmetric catalysis lies in the temperature control during the formation of the pre-catalyst by reaction of the [Rh(BIPHEP)(acac)] with 2, providing selective access to the kinetic product mixture (RaSc)/(SaSc)-[Rh(BIPHEP)(ProlOMe)][NTf2] (5) or the thermodynamically preferred (RaSc)-5 as single diastereomer (Fig. 4).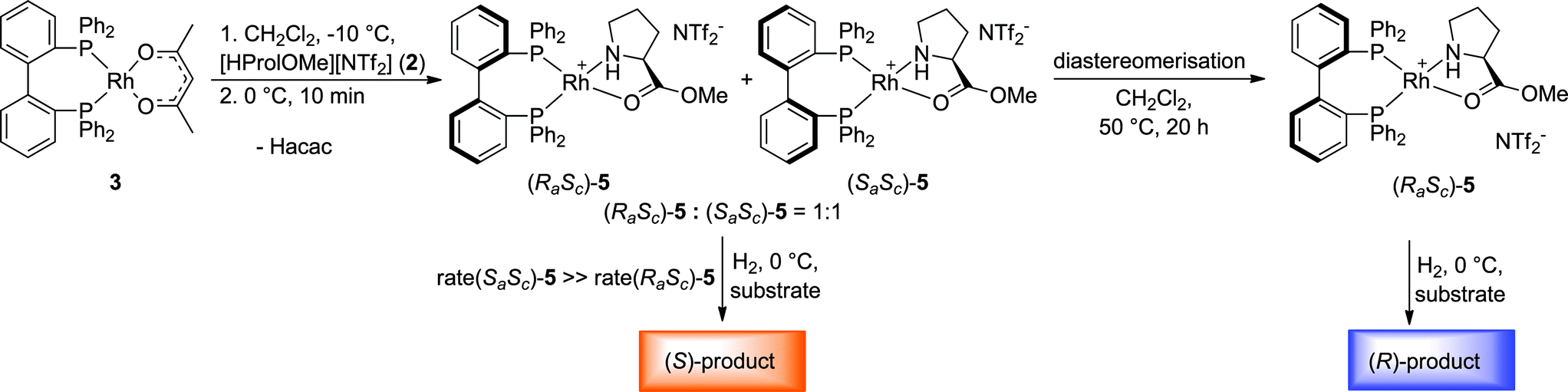 | ||
| Fig. 4 Overall scheme for enantiodivergent asymmetric catalysis using BIPHEP and the prolin-based selector 2via the diastereomeric complexes 5. | ||
The diastereomeric mixture and the pure diastereomer constitute two sides of a Janus-faced catalytic system yielding the two product configurations with similar or even identical absolute levels of ee depending on the nature of the substrate. When the diastereomeric mixture is used, the chiral selector is readily displaced from (SaSc)-diastereomer and the resulting catalytically active (Sa)-BIPHEP/Rh species leads to the preferential formation of the S-hydrogenation products. Whereas the more stable (RaSc)-5 diastereomer remains silent under these competitive conditions, it acts as effective pre-catalyst and stabilizer for the (Ra)-BIPHEP/Rh fragment, resulting in up to 80% ee in hydrogenation reactions. The diastereomerically pure (RaSc)-5 could be also successfully used in the asymmetric hydroboration resulting in full conversion, perfect regioselectivity, and an enantioselectivity of 86% ee, competitive with the best results obtained with the enantiopure atropos BINAP ligand in this transformation.
The results of this study fosters the increasing evidence that the strategy of combining a latent chiral tropos BIPHEP ligand and a chiral selector opens new opportunities in the asymmetric catalysis. It demonstrates that adaptive chiral systems can be steered into different enantiomeric reaction channels on basis of the understanding of the underlying mechanism. Using cheap and readily available proline as the chiral selector is of course attractive from a practical point of view. At the same time, the modular approach of the present systems allows exploitation of the structural diversity of amino acid esters and other chelating structures as the selector as well as the use of ligands with different fluxional backbones,37 opening a broad range of possibilities for further optimisation and application.
Acknowledgements
We thank the company W. C. Heraeus for generous supply of precious metal precursors, Christel Dittmer (IOC, RWTH Aachen) for HR-MS measurements, Wolfgang Falter for ESI-MS measurements, Julia Wurlitzer and Hannelore Eschmann for GC- and HPLC-analysis and Ines Bachmann-Remy for NMR-measurements. This work was supported by the priority program 1191 of the Deutsche Forschungsgemeinschaff (SPP 1191: Schwerpunktsprogramm ionic liquids).Notes and references
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999 Search PubMed.
- H. Brunner, J. Organomet. Chem., 1986, 300, 39 CrossRef CAS; R. Noyori, Chem. Soc. Rev., 1989, 18, 187 RSC.
- K. Mikami, K. Aikawa, Y. Yusa, J. J. Jodry and M. Yamanaka, Synlett, 2002, 1561 CrossRef CAS.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS.
- R. Schmid, E. A. Broger, M. Cereghetti, Y. Crameri, J. Foricher, M. Lalonde, R. K. Müller, M. Scalone, G. Schoettel and U. Zutter, Pure Appl. Chem., 1996, 68, 131 CrossRef CAS.
- M. D. Tudor, J. J. Becker, P. S. White and M. R. Gagné, Organometallics, 2000, 19, 4376 CrossRef CAS.
- J. W. Faller and J. C. Wilt, J. Organomet. Chem., 2006, 691, 2207 CrossRef CAS.
- K. Mikami and S. Matsukawa, Nature, 1997, 385, 613 CrossRef CAS.
- J. Lacour and V. Hebbe-Viton, Chem. Soc. Rev., 2003, 32, 373 RSC.
- M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed.
- N. Kurono, T. Katayama and T. Ohkuma, Bull. Chem. Soc. Jpn., 2013, 86, 577 CrossRef CAS.
- For diastereomerisation of BIPHEP-type phosphine oxides over chiral solid supports and their use as organocatalysts in asymmetric aldol condensation, see: F. Maier and O. Trapp, Angew. Chem., Int. Ed., 2014, 53, 8756 CrossRef CAS PubMed.
- J. J. Becker, P. S. White and M. R. Gagnè, J. Am. Chem. Soc., 2001, 123, 9478 CrossRef CAS PubMed.
- K. Aikawa and K. Mikami, Chem. Commun., 2012, 48, 11050 RSC.
- T. Punniyamurthy, M. Mayr, A. S. Dorofeev, C. J. R. Bataille, S. Gosiewska, B. Nguyen, A. R. Cowley and J. M. Brown, Chem. Commun., 2008, 5092 RSC.
- K. Aikawa, Y. Takabayashi, S. Kawauchi and K. Mikami, Chem. Commun., 2008, 5095 RSC.
- C. Wolff, Dynamic Stereochemistry of Chiral Compounds, RSC Publishing, Cambridge, 2008 Search PubMed.
- K. Mikami, K. Aikawa and Y. Yusa, Org. Lett., 2002, 4, 95 CrossRef CAS PubMed.
- K. Mikami, T. Korenaga, M. Terada, T. Ohkuma, T. Pham and R. Noyori, Angew. Chem., Int. Ed., 1999, 38, 495 CrossRef CAS.
- G. Storch and O. Trapp, Angew. Chem., Int. Ed., 2015, 54, 3580 CrossRef CAS PubMed.
- G. Storch, M. Siebert, F. Rominger and O. Trapp, Chem. Commun., 2015, 51, 15665 RSC.
- M. Schmitkamp, D. Chen, W. Leitner, J. Klankermayer and G. Franciò, Chem. Commun., 2007, 4012 RSC.
- D. Chen, M. Schmitkamp, G. Franciò, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2008, 47, 7339 CrossRef CAS PubMed.
- D. Chen, B. Sundararaju, R. Krause, J. Klankermayer, P. H. Dixneuf and W. Leitner, ChemCatChem, 2010, 2, 55 CrossRef CAS.
- [Rh(BIPHEP)(acac)] was synthesised from [Rh(CO)2(acac)] and one equivalent of BIPHEP: see ESI† for details.
- G.-H. Tao, L. He, N. Sun and Y. Kou, Chem. Commun., 2005, 3562 RSC.
- A. Jegorov, J. Podlaha, J. Podlahová and F. Tureček, J. Chem. Soc., Dalton Trans., 1990, 3259 RSC.
- O. Desponds and M. Schlosser, Tetrahedron Lett., 1996, 37, 47 CrossRef CAS.
- For the quantification of the inversion barrier of several 3,3′-substituted BIPHEP and BIPHEPO derivatives, see: F. Maier and O. Trapp, Angew. Chem., Int. Ed., 2012, 51, 2985 CrossRef CAS PubMed.
- J. M. Brown, Hydrogenation of functionalised carbon-carbon double bonds, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, vol. 1, pp. 121–182 Search PubMed.
- For recent examples of enantioinversion see: R. J. Chew, X.-R. Li, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem.–Eur. J., 2015, 21, 4800 CrossRef CAS PubMed; M. K. Ilg, L. M. Wolf, L. Mantilli, C. Farès, W. Thiel and A. Fürstner, Chem.–Eur. J., 2015, 21, 12279 CrossRef PubMed.
- For instance the π-phenyl bridged Rh–BINAP dimer displays in 31P-NMR a doublet at δ 46.41 ppm (JRhP = 199.2 Hz): A. Miyashita, H. Takaya, T. Souchi and R. Noyori, Tetrahedron, 1984, 40, 1245 CrossRef CAS.
- A. Preetz, H.-J. Drexler, S. Schulz and D. Heller, Tetrahedron: Asymmetry, 2010, 21, 1226 CrossRef CAS.
- C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695 CrossRef CAS.
- T. Hayashi, Y. Matsumoto and Y. Ito, Tetrahedron: Asymmetry, 1991, 2, 601 CrossRef CAS.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517 CrossRef CAS.
- See for instance: K. Mikami, K. Wakabayashi, Y. Yusa and K. Aikawa, Chem. Commun., 2006, 2365 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03465e |
| This journal is © The Royal Society of Chemistry 2016 |