
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold(III) tetraarylporphyrin amino acid derivatives: ligand or metal centred redox chemistry?†
Sebastian
Preiß
,
Jascha
Melomedov
,
Anica
Wünsche von Leupoldt
and
Katja
Heinze
*
Institute of Inorganic Chemistry and Analytical Chemistry, Johannes Gutenberg-University of Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: katja.heinze@uni-Mainz.de; Fax: +49-6131-39-27277; Tel: +49-6131-39-25886
First published on 26th October 2015
Abstract
Meso tetraarylporphyrinato gold(III) cations bearing different substituents at the aryl substituents (COOMe, COOH, NO2, NH2, NHAc, H, OnBu, CF3) were prepared and characterised. Their reversible one-electron reductions were studied by (spectro)electrochemical means as well as by selective chemical one-electron reduction using cobaltocene. The preferred location of the spin density, namely gold centred or porphyrin centred, was probed by electron paramagnetic resonance spectroscopy (g values, 197Au hyperfine coupling) as well as by density functional theory calculations (spin densities). In all cases studied experimentally and theoretically, the gold(II) valence isomer (5d9 electron configuration) is preferred over the porphyrin π radical anion. In the hexafluorophosphate salt of the nitro derivative a further nitro π radical anion valence isomeric species is significantly populated. In the presence of chloride ions this nitro π radical anion/AuII valence isomeric equilibrium evolves towards the porphyrin π radical anion. The electronic structures of the nitro π radical and the AuII σ radical valence isomers (5dx2−y2 orbital) could be calculated by DFT methods. The electron transfer pathway between the nitro π radical anion and the AuII valence isomer is well described by the location of the hexfluorophosphate counterion, the Au–N distances (corresponding to the totally symmetric stretching vibration), the symmetric stretching mode of the NO2 substituent and a meso-nitrophenyl rotation. The specific geometric and electronic properties of the favoured gold(II) σ radical valence isomer, namely counterion dislocation and σ symmetry of the redox orbital, might stabilise charge-shifted states [(gold(II) porphyrin)-donor˙+] by retarding the back electron transfer to give the ground state (gold(III) porphyrin)-donor. This will guide the design of (photo-induced) electron transfer pathways with tetraarylporphyrinato gold(III) complexes as electron acceptors.
Introduction
Synthetic metallo porphyrins are of increasing interest due to their suitability as chromophores, as well as electron and hole acceptors in artificial photosynthetic systems,1 due to their catalytic and sensing properties,2 due to their medical applications3 as well as due to their propensity to stabilise unusual metal oxidation states. Specifically, porphyrinato gold(III) complexes have evolved as efficient anticancer drugs.4 Furthermore, they are catalysts for the cycloisomerization of allenones.5 Recently, gold clusters with face-on coordinated free-base porphyrins have been reported.6 Finally, gold(III) porphyrins are suitable ingredients in photoinduced electron transfer chains with the gold(III) porphyrin acting as electron acceptor.7The site of gold(III) porphyrin reduction, namely ligand or metal centred, has been discussed controversially. Based on early UV/Vis spectroscopic and theoretical studies the products of the reduction of gold(III) porphyrins had been described as porphyrin-centred π radical anions.8 In a seminal paper, Kadish, Fukuzumi and Crossley provided compelling EPR spectroscopic evidence that the one-electron reduction of [AH]+ to AH is metal centred giving gold(II) porphyrins (Scheme 1).9 Only a few ligand types, such as thiolates or thioethers, are capable to stabilise mononuclear gold in the oxidation state +II.10 Further outstanding examples are the fluorosulfate11 and xenon complexes12 of AuII. Nitrogen donor ligands such as porphyrinato ligands have been reported to stabilise AuII with respect to disproportionation and dimerization9,13 to [AuII2] species14 as well (Scheme 1, A, B, [C+]*). Disproportionation and dimerisation of [AuII(en)2]2+D2+ has been suppressed by encapsulation in the pores of a zeolite (en = ethylenediamine).15
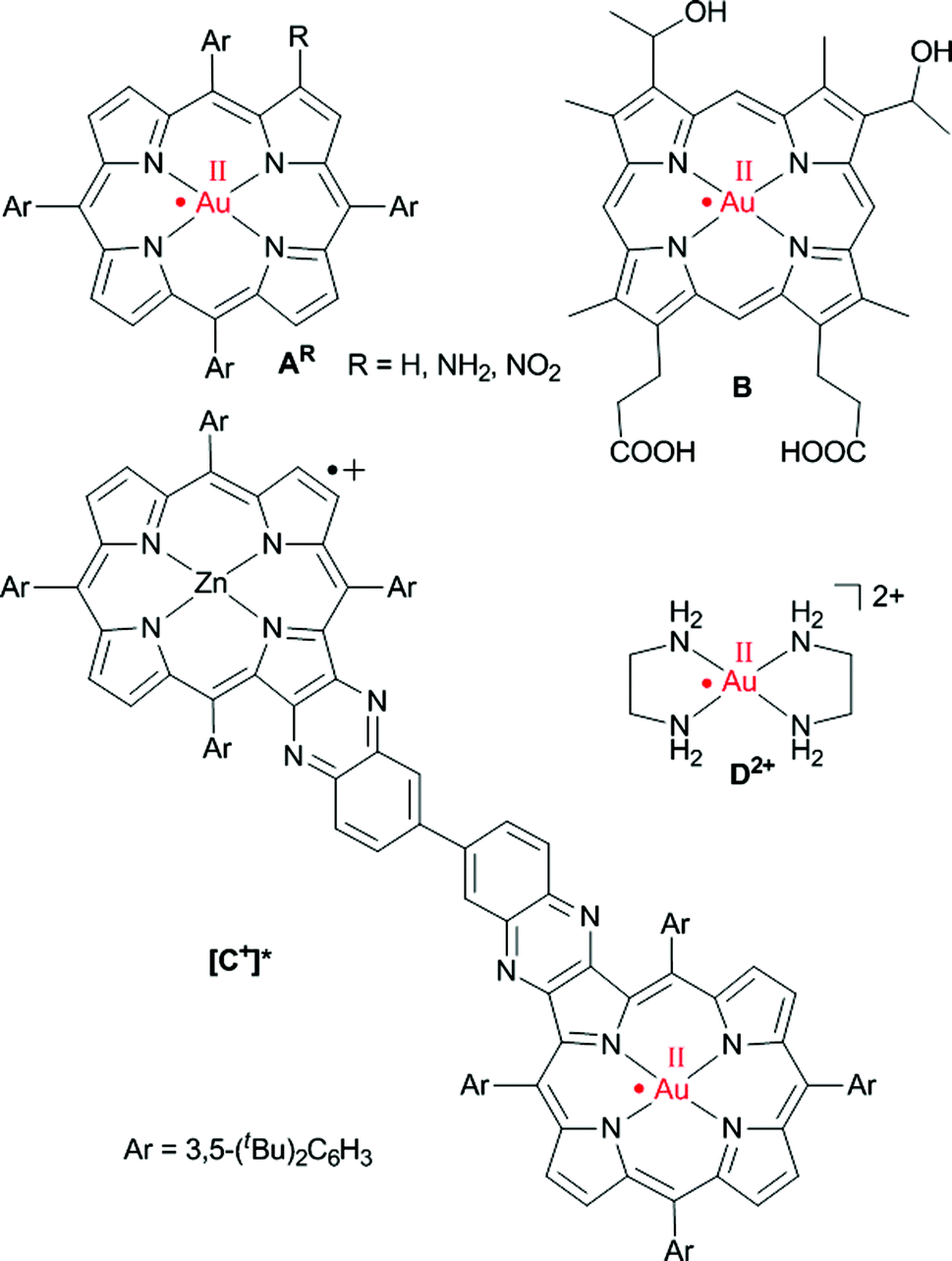 | ||
| Scheme 1 Genuine mononuclear gold(II) complexes with planar N4 coordination according to EPR spectroscopic results.9,13,15 | ||
The gold(II) porphyrin AH (Scheme 1) prepared by reduction of the corresponding gold(III) porphyrin cation [AH]+ with the strongly reducing naphthalene radical anion yielded a broad EPR resonance centred at gav = 2.06.9 Hyperfine coupling to 197Au has been reported for the central g line [A(197Au) = 27 G at 113 K; I(197Au) = ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) , natural abundance 100%].9 An EPR resonance with a significantly smaller peak-to-peak distance was observed for the gold(II) complex B of hematoporphyrin IX with g⊥ = 2.035, g∥ = 1.970 and A⊥(197Au) = A∥(197Au) = 15 G at 130 K suggesting a less pronounced metal character (Scheme 1).13b,e The charge-shifted state [C+]* of a ZnII–AuIII bis(tetraarylporphyrin) C+ yielded an EPR resonance with g = 2.182, 2.043, 1.979 and A(197Au) = 180, 14, n.r. G in frozen toluene solution at 143 K for the gold(II) centre (Scheme 1).13d The gold complex [Au(en)2]2+D2+ with the pure σ-donor ligand ethylenediamine trapped in a zeolite shows g∥ = 2.239, g⊥ = 2.051, A∥(197Au) = 188 G and A⊥(197Au) = 22 G at room temperature (Scheme 1).15 Gold(III)-centred reductions have been associated with a significantly higher reorganization energy (ca. 1.25 eV) than porphyrin-based reductions (ca. 0.6 eV).13c The large reorganization energy renders gold(III) porphyrins suitable electron acceptors in photoelectron transfer schemes.7 Moreover, gold(III)-associated counterions should dissociate upon AuIII to AuII reduction further retarding the back-electron transfer. For instance, chloride is associated to the AuIII centre in solid AuCl(TPP) by electrostatic forces with a gold chloride distance of 3.01(1) Å.16 Unfortunately, no solid structures of porphyrinato gold(II) complexes have been reported so far and further experimental or theoretical studies are lacking.
, natural abundance 100%].9 An EPR resonance with a significantly smaller peak-to-peak distance was observed for the gold(II) complex B of hematoporphyrin IX with g⊥ = 2.035, g∥ = 1.970 and A⊥(197Au) = A∥(197Au) = 15 G at 130 K suggesting a less pronounced metal character (Scheme 1).13b,e The charge-shifted state [C+]* of a ZnII–AuIII bis(tetraarylporphyrin) C+ yielded an EPR resonance with g = 2.182, 2.043, 1.979 and A(197Au) = 180, 14, n.r. G in frozen toluene solution at 143 K for the gold(II) centre (Scheme 1).13d The gold complex [Au(en)2]2+D2+ with the pure σ-donor ligand ethylenediamine trapped in a zeolite shows g∥ = 2.239, g⊥ = 2.051, A∥(197Au) = 188 G and A⊥(197Au) = 22 G at room temperature (Scheme 1).15 Gold(III)-centred reductions have been associated with a significantly higher reorganization energy (ca. 1.25 eV) than porphyrin-based reductions (ca. 0.6 eV).13c The large reorganization energy renders gold(III) porphyrins suitable electron acceptors in photoelectron transfer schemes.7 Moreover, gold(III)-associated counterions should dissociate upon AuIII to AuII reduction further retarding the back-electron transfer. For instance, chloride is associated to the AuIII centre in solid AuCl(TPP) by electrostatic forces with a gold chloride distance of 3.01(1) Å.16 Unfortunately, no solid structures of porphyrinato gold(II) complexes have been reported so far and further experimental or theoretical studies are lacking.
We had previously reported synthetically versatile meso-substituted tetraaryl porphyrins with trans-AB2C substitution pattern including A = nitrophenyl, aminophenyl or amidophenyl, C = phenyl carboxylic acid or ester and B = EWG or EDG substituted aryl groups.17 These porphyrins can be metallated17 and assembled to multiporphyrin amides,17b,17c electron donor substituted amide-linked dyads17a as well as electron donor (ferrocene) and electron acceptor (quinone) substituted amide-linked triads and tetrads17b with well defined sequences from the N-terminus to the C-terminus. The different meso substituents of the porphyrin amino acids at the B position can be used to modulate the solubilities and to fine-tune the redox potentials which allows to design redox gradients.17
With this family of porphyrins in hand, we disclose in this contribution the factors that control the relative stabilities of a gold(II) porphyrin and its valence isomeric gold(III) porphyrin radical anion. We report novel meso-substituted AuIII porphyrin amino acid derivatives with trans-AB2C substitution pattern for potential incorporation into electron transfer chains via amide bonds. These gold(III) porphyrins were inspected by cyclic voltammetry, UV/Vis spectroelectrochemistry and by EPR spectroscopy upon selective one-electron reduction with cobaltocene. We provide strong EPR and UV/Vis spectroscopic evidence that all singly reduced gold(III) porphyrins are well described as gold(II) porphyrins essentially irrespective of the meso-substituents A, B and C and that the porphyrin radical anions are higher energy valence tautomers of the ground state AuII valence isomers. Detailed EPR parameters of the gold(II) porphyrinato complexes were obtained by spectral simulations of the experimental spectra (g tensors, (super)hyperfine couplings, valence isomer ratios). The experimental data are corroborated and interpreted with the aid of density functional theory (DFT) calculations in the framework of electron transfer theory.
Results and discussion
Synthesis of free-base porphyrins and (porphyrinato)gold(III) complexes (series [1a]+–[3a]+ and series [4a]+–[4c]+)
The free-base porphyrins Ia–IVc were prepared according to literature procedures.17 Metallation of the free-base porphyrins was successful with potassium tetrachlorido aurate(III) in the presence of HOAc/NaOAc (Fleischer's route18), except for amino-substituted porphyrin IIa (Scheme 2). Best yields were obtained using four equivalents of KAuCl4 giving the cationic aurated porphyrins as poorly soluble tetrachlorido aurate salts.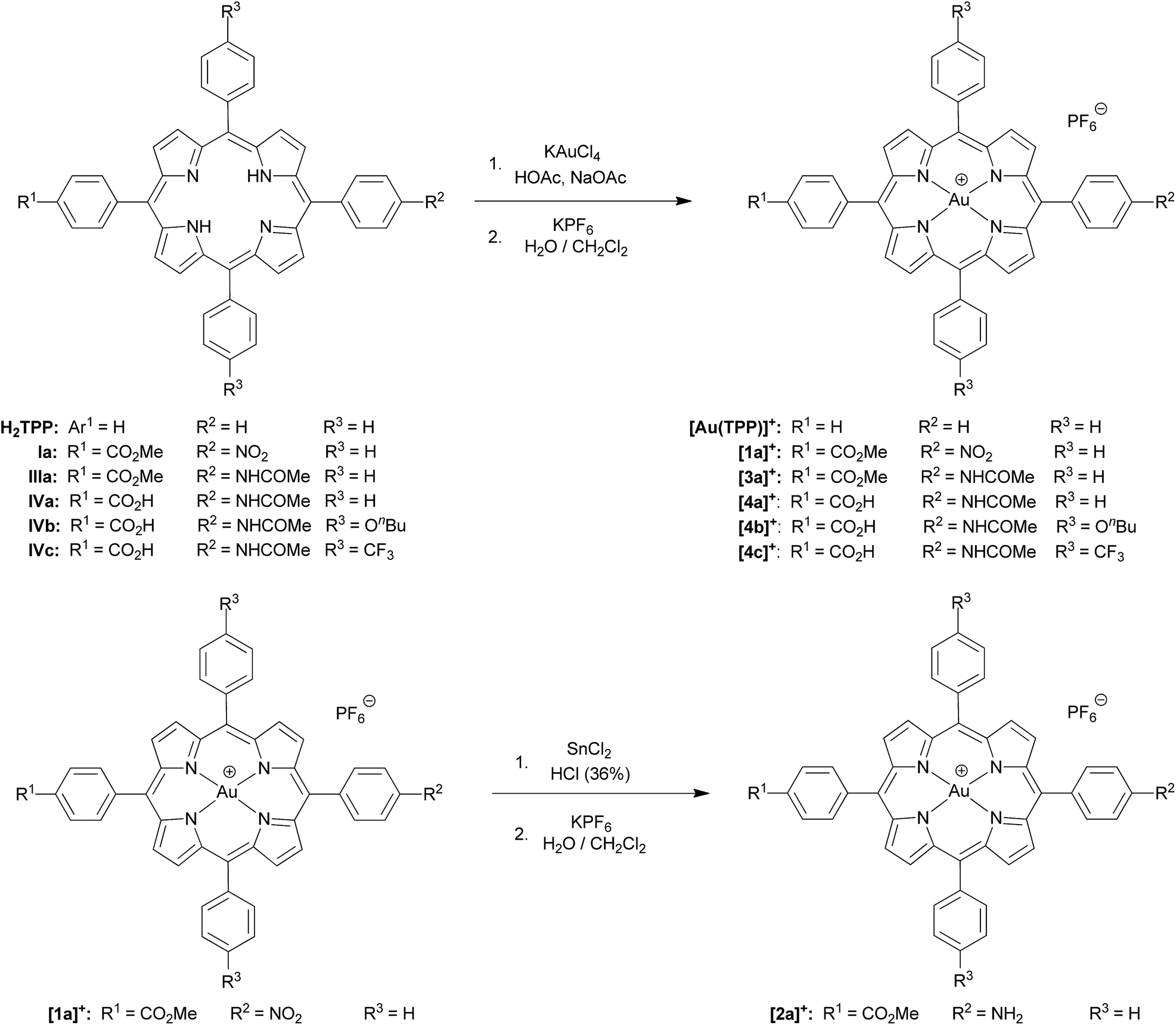 | ||
| Scheme 2 Synthesis of (porphyrinato)gold(III) complexes ([Au(TPP)]+, series [1a]+, [2a]+, [3a]+ and series [4a]+, [4b]+, [4c]+). | ||
Auration of the amino-substituted porphyrin IIa with KAuCl4/HOAc/NaOAc according to Fleischer's method resulted in acylation of the amino group. Auration of IIa according to Sauvage's protocol using [AuI(tht)2][BF4] followed by disproportionation to AuIII and Au0 was unsuccessful as well (tht = tetrahydrothiophene).19 Thus, [2a]+ was prepared via metallation and ion exchange of nitroporphyrin ester Ia to give [1a][PF6], followed by reduction of the nitro group of [1a]+ with SnCl2/HCl to give the aurated amino-substituted porphyrin [2a]Cl (Scheme 2). During this procedure, the gold(III) ion was neither reduced nor removed. Hence, the AuIII porphyrins are stable under protic conditions. Counterion exchange of [Au(porph)][AuCl4] or [2a]Cl with KPF6 yielded the corresponding soluble hexafluorophosphate salts which are conveniently purified by column chromatography.
Characterization of (porphyrinato)gold(III) complexes (series [1a]+–[3a]+ and series [4a]+–[4c]+)
Ester-substituted complexes [1a][PF6]–[3a][PF6] are sufficiently soluble in dichloromethane. However, THF is required for acids [4b][PF6] and [4c][PF6] and even methanol is necessary for [4a][PF6] in order to acquire NMR spectra with a satisfactory signal-to-noise ratio. This shows that the counterion and the meso substituents determine the solubility. All gold(III) complexes were characterised by 1H NMR, 13C NMR, 31P NMR and 2D NMR spectroscopy, IR spectroscopy and high-resolution mass spectrometry. The proton NMR spectra display the expected number and intensities of resonances. The chemical shifts vary according to the substitution patterns paralleling the shifts of the corresponding free-base porphyrins Ia–IIIa and IVa–IVc. The CH3-ester, NH2-amine and CH3-amide substituents display characteristic resonances at δ = 4.1, 4.7, 2.2 ppm, respectively. The [PF6]− counterions show the characteristic septet at δ = −144 ppm in the 31P NMR spectra. Upon auration the characteristic high-field pyrrol NH resonances of the free-base porphyrin disappear. Furthermore, auration of the free-base porphyrins consistently shifts the pyrrole CH proton resonances to lower field by 0.5 ppm, in accordance with the positive charge of the metal centre. In the IR spectra, characteristic absorptions for group vibrations are found for the ester, amine, amide, nitro, trifluoromethyl and acid substituents at around 1719, 1618, 1690, 1520/1346, 1324 and 1716 cm−1, respectively. The [PF6]− counterions display absorptions for the PF stretching and deformation modes at 835–843 and 556–558 cm−1, respectively. ESI mass spectra fully confirm the integrity and stability of the complex cations displaying peaks at m/z values corresponding to the intact complex cation (see Exp. section).With the exception of the electron-rich R3 = OnBu-substituted complex [4b]+ all gold(III) porphyrinato complexes [1a]+, [2a]+, [3a]+, [4a]+ and [4c]+ show hypsochromically shifted Soret bands as compared to their corresponding free-base porphyrins Ia, IIa, IIIa, IVa and IVc (hypso porphyrins8b). In all cases, the number of Q bands is reduced from four to two (or even to one) as expected for metalloporphyrins with local D4h symmetry of the porphyrin core. Expectedly, gold(III) porphyrinato complexes are non-emissive at room temperature in fluid solution as exemplarily checked for [1a][PF6], [3a][PF6] and [4c][PF6].8b,20
Redox chemistry of (porphyrinato)gold(III) complexes (series [1a]+–[3a]+ and series [4a]+–[4c]+)
Several reversible reductions are observed for cations [1a]+, [2a]+, [3a]+, [4b]+ and [4c]+ 10−3 M in 0.1 M [nBu4N][PF6]/THF solution (Fig. 1 and 2, Table 1). For solubility reasons complex [4a]+ was measured in MeOH and the solvent window of MeOH allows for only a single reversible reduction wave to be observed (Fig. 2, Table 1). All potentials are given relative to the ferrocene/ferrocenium couple. As expected from substituent effects, [1a]+ is more easily reduced to 1a (−0.92 V) than [Au(TPP)]+ to Au(TPP) (−0.97 V), while [2a]+ (−0.99 V) is more difficult to reduce. Similarly, the potentials shift to more negative values in the series [4c]+ (−1.00 V), [4a]+ (−1.02 V) and [4b]+ (−1.08 V), which is again explicable by the increasing electron donating nature of the substituents (CF3, H, OnBu). Similar to the corresponding free-base porphyrins the shifts are only small.17 The second reduction is especially facile with the electron withdrawing NO2 substituent (1a/[1a]−; −1.55 V). The nitro derivative [1a]+ shows even further reversible reductions. Hence, one of the [1a]+ reductions might be associated to the nitro substituent itself (vide infra).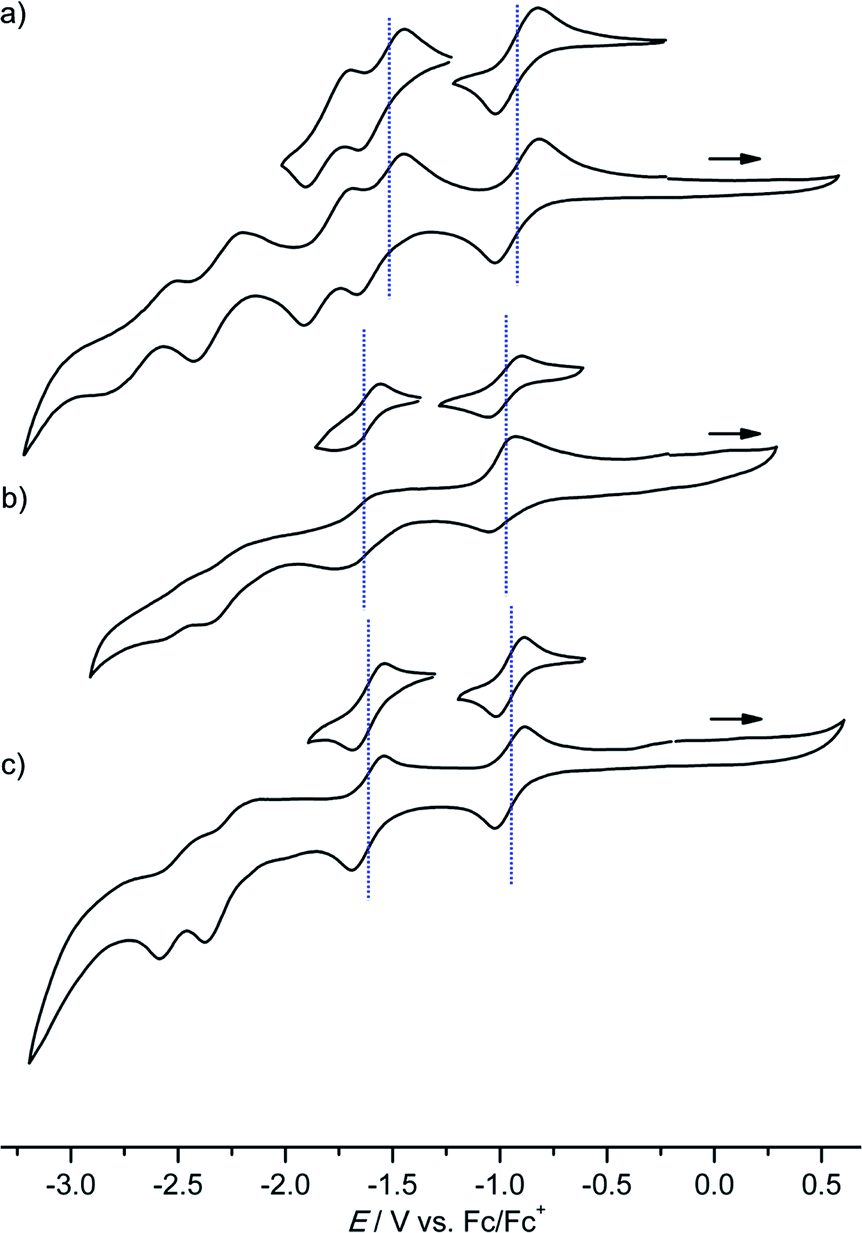 | ||
| Fig. 1 Cyclic voltammograms of (a) [1a]+, (b) [2a]+ and (c) [3a]+ 10−3 M in 0.1 M [nBu4N][PF6]/THF solution; potentials referenced against the ferrocene/ferrocenium couple. | ||
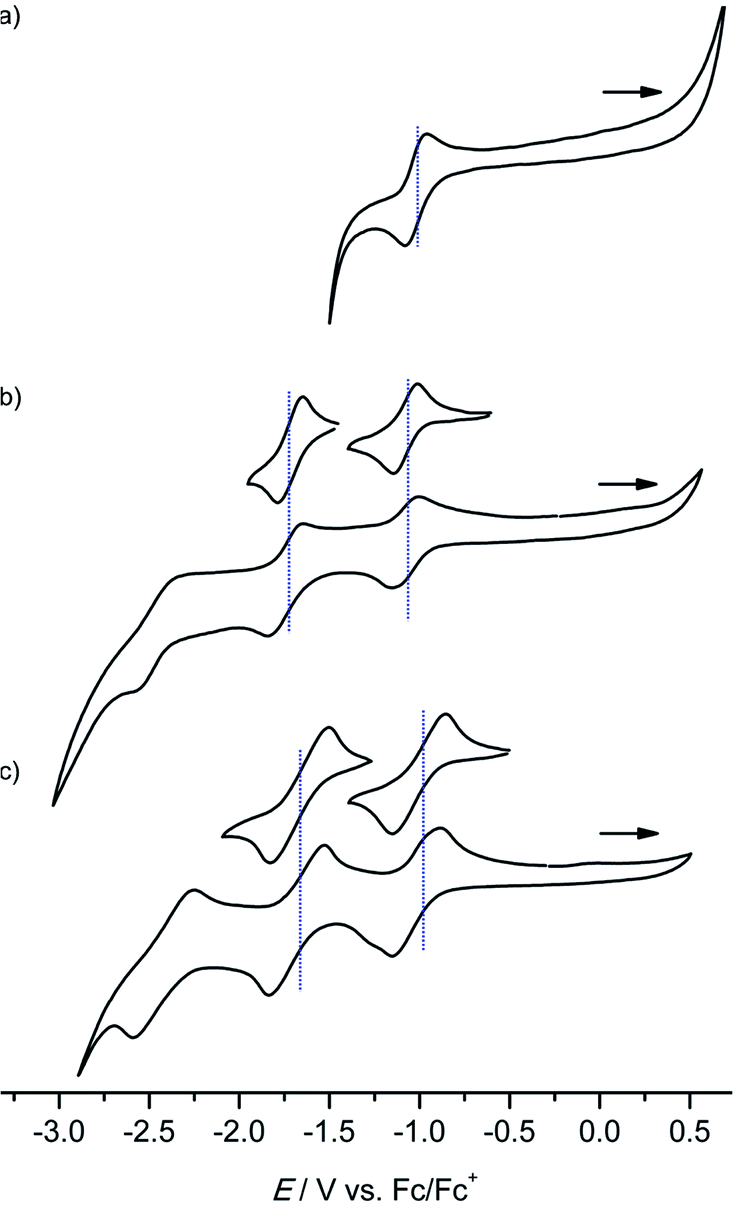 | ||
| Fig. 2 Cyclic voltammograms of (a) [4a]+ (MeOH), (b) [4b]+ and (c) [4c]+ 10−3 M in 0.1 M [nBu4N][PF6]/THF solution; potentials referenced against the ferrocene/ferrocenium couple. | ||
| E ½ ([Au(P)]+/Au(P)) | E ½ (Au(P)/[Au(P)]−) | E ½ ([Au(P)]−/[Au(P)]2−) | E ½ ([Au(P)]2−/[Au(P)]3−) | |
|---|---|---|---|---|
| a In MeOH. b Outside solvent window. | ||||
| [Au(TPP)][PF6] | −0.97 (−1.06/−0.88) | −1.65 (−1.73/−1.57) | −2.34 (−2.43/−2.25) | — |
| 1a[PF6] | −0.92 (−1.02/−0.82) | −1.55 (−1.66/−1.44) | −1.80 (−1.90/−1.70) | −2.31 (−2.42/−2.20) |
| 2a[PF6] | −0.99 (−1.06/−0.91) | −1.67 (−1.78/−1.56) | −2.27 (−2.38/−2.16) | −2.51 (−2.56/−2.45) |
| 3a[PF6] | −0.96 (−1.02/−0.89) | −1.63 (−1.70/−1.55) | −2.28 (−2.37/−2.19) | −2.50 (−2.58/−2.41) |
| 4a[PF6] | −1.02 (−1.08/−0.96) | |||
| 4b[PF6] | −1.08 (−1.15/−1.01) | −1.72 (−1.79/−1.65) | −2.46 (−2.57/−2.34) | |
| 4c[PF6] | −1.00 (−1.15/−0.87) | −1.67 (−1.83/−1.50) | −2.42 (−2.58/−2.25) | |
The differences between the first and second reduction potentials amount to 0.60–0.68 V which corresponds to very high comproportionation constants of KC > 1010 for the neutral complexes.21 Hence, disproportionation of the neutral complexes into the corresponding cations and anions can be safely neglected and spectral signatures after one-electron reduction will essentially be associated with the neutral complexes.
All gold(III) complexes were reduced electrochemically to the neutral species in an optically transparent thin layer electrochemical (OTTLE) cell using THF as solvent (MeOH for [4a]+). In all cases, isosbestic points were observed corroborating the reversible nature of the first reduction process (Fig. 3 and ESI†). The shifts of the Soret and Q bands as well as the observed isosbestic points closely resemble those found for the [Au(TPP)]+/Au(TPP) process in THF (ESI†) and in pyridine13c or in PhCN.9 In all cases, except for the [2a]+/2a and [4b]+/4b redox couples with the strongly electron-donating NH2 and OnBu substituents, the intensity of the Soret band decreases while for [2a]+/2a and [4b]+/4b the intensity increases (Fig. 3b and ESI†). A similar hyperchromic effect has been observed for the [ANH2]+/ANH2 couple with the amino group attached to a porphyrin beta position.13c
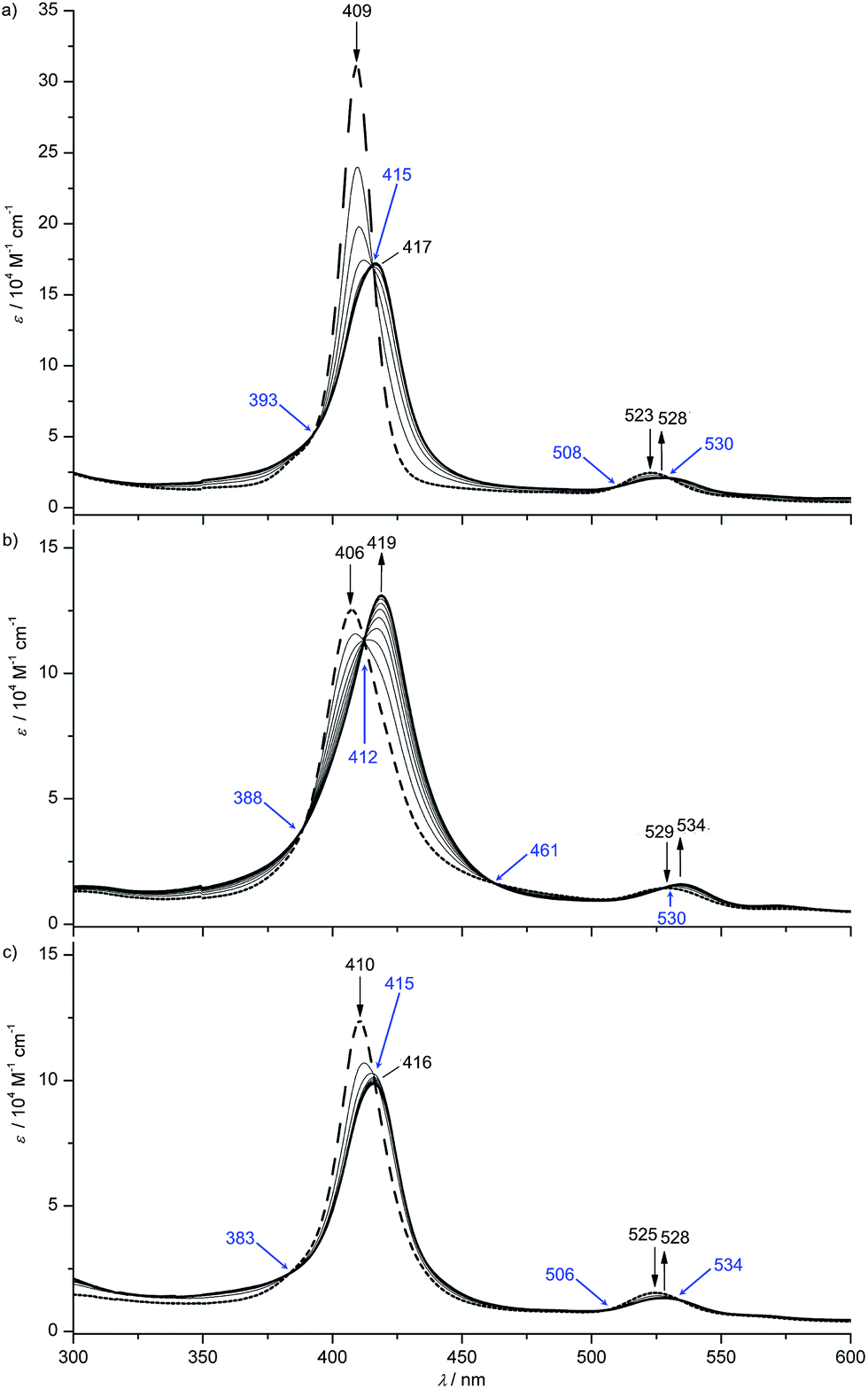 | ||
| Fig. 3 UV/Vis spectral changes upon the first electroreduction of (a) [1a]+, (b) [2a]+ and (c) [3a]+ in 0.1 M [nBu4N][PF6]/THF solution (isosbestic points indicated in blue). | ||
For chemical reductions, the gold(III) porphyrin complexes were dissolved in CH2Cl2 ([1a][PF6]–[1c][PF6]), THF ([4b][PF6]/[4c][PF6]) or MeOH ([4a][PF6]) (ca. 5 mM). In order to definitely prevent overreduction, these solutions were treated with slightly substoichiometric amounts of CoCp2 (E½ = −1.33 V in CH2Cl2vs. Fc/Fc+ (ref. 22)) in an EPR tube. The redox potential of CoCp2 is perfectly in-between the first and second reduction of the gold porphyrins (Fig. 1 and 2) further avoiding over-reduction. The reaction mixture in the tube was immediately frozen by immersing into liquid nitrogen and subjected to X-band EPR spectroscopy. Hence, we obtained significantly better resolved EPR spectra than previously reported for neutral porphyrinato gold complexes prepared by reduction of [AH]+ with the strongly reducing naphthalene radical anion in DMF (ca. −3 V vs. Fc/Fc+ (ref. 22)).9 In this case some over-reduction might have been occurred blurring the hyperfine couplings to gold and nitrogen nuclei.
Indeed, Au(TPP) as prepared by reduction of [Au(TPP)]+ by CoCp2 in CH2Cl2 shows a well-resolved EPR pattern which could be reasonably simulated by a rhombic g tensor with hyperfine interaction to a single 197Au nucleus (I = ; natural abundance 100%) and superhyperfine coupling to four 14N nuclei (I = 1, natural abundance 99.6%). The high resolution allows a very good estimation of the high-field parameters while the low-field parameters are less well-resolved (Table 2, Fig. 4). Compared to the isoelectronic Cu(TPP) complex (63/65Cu; I = ; combined natural abundance 100%; g1 = 2.197, g2 = g3 = 2.054)17d the metal coupling constant A1 [A1(197Au) = 43 G; A1(63/65Cu) = 197 G (ref. 17d)] is significantly reduced in Au(TPP). This suggests a more covalent character of the AuII–N bonds compared to the CuII–N bonds in their respective TPP2− complexes. For [C+]* with strongly electron donating meso substituents at the gold porphyrin a much larger hfc to 197Au has been reported [A1(197Au) = 180 G].13d In accordance with the stronger nephelauxetic effect of porphyrins, complex D2+ with the pure σ donor ligand ethylenediamine features a significantly larger hyperfine coupling to 197Au than Au(TPP) as well.15
| Major species | Minor species | |||||||
|---|---|---|---|---|---|---|---|---|
| g 1,2,3 | A(197Au)1,2,3/G | 4 × A(14N)1,2,3/G | Line width (Gauss/Lorentz) | Fraction/% | g 1,2,3 | 4 × A(14N)1,2,3/G | Line width (Gauss/Lorentz) | |
| a The nitro radical 1a′′ (23%) shows g1,2,3 = 2.031, 2.005, 1.948 and 4 × A(14N)1,2,3 = 2, 17, 2 G. | ||||||||
| Au(TPP) | 2.182, 2.056, 1.982 | 43, 20, 20 | 18, 22, 21 | 1.0/0.3 | 100 | — | — | — |
| 1a | 2.190, 2.056, 1.974 | 44, 29, 29 | 18, 22, 21 | 1.7/0.3 | 78 | 2.016, 2.005, 1.995a | 1, 12, 1 | 0.75/0.1 |
| 1a/TBACl | 2.190, 2.056, 1.974 | 44, 29, 29 | 18, 22, 21 | 1.7/0.3 | 65 | 2.016, 2.005, 1.994 | 1, 12, 1 | 0.9/0.3 |
| 2a | 2.192, 2.062, 1.963 | 46, 25, 25 | 18, 22, 22 | 1.7/0.3 | 94 | 2.018, 2.005, 1.994 | 1, 12, 1 | 0.9/0.2 |
| 3a | 2.192, 2.062, 1.968 | 44, 29, 29 | 18, 22, 21 | 1.7/0.3 | 95 | 2.016, 2.005, 1.994 | 1, 13, 1 | 0.9/0.3 |
| 4a | 2.175, 2.057, 1.973 | 43, 29, 29 | 18, 22, 21 | 1.7/0.3 | 100 | — | — | — |
| 4b | 2.175, 2.056, 1.972 | 42, 25, 25 | 18, 22, 21 | 3.0/0.3 | 100 | — | — | — |
| 4c | 2.175, 2.055, 1.974 | 44, 29, 29 | 18, 22, 21 | 1.7/0.3 | 100 | — | — | — |
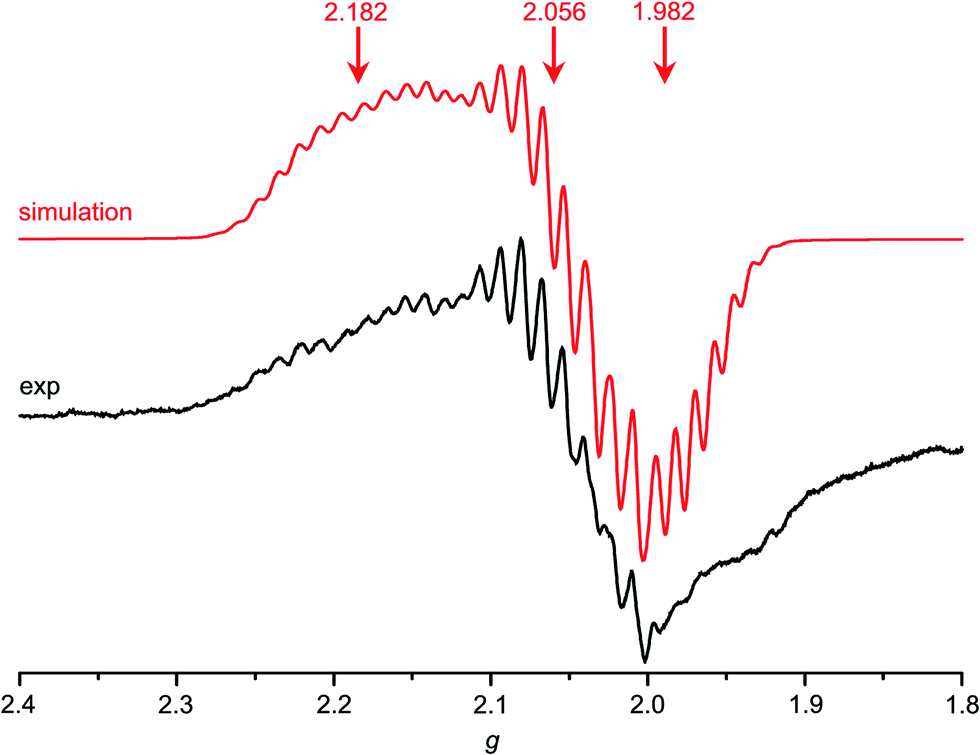 | ||
| Fig. 4 X-band EPR spectrum of Au(TPP) in frozen CH2Cl2 solution (77 K, 9.4 GHz) and corresponding simulation. | ||
For complexes 1a–3a the broad EPR resonance corresponding to the AuII valence isomer is less well resolved due to the lower symmetry and hence different superhyperfine interactions (Fig. 5). Furthermore, the broad AuII resonance is superimposed by a sharp slightly rhombic resonance around g = 2.0. For 2a and 3a, this sharp resonance accounts for approximately 5–6% of the total EPR intensity. The pattern can be satisfactorily simulated by g1,2,3 = 2.018, 2.005, 1.994 and hyperfine coupling to four nitrogen atoms (A1,2,3 = 1, 12, 1 G). These data fit to gold(III) porphyrin radical anions 2a′ and 3a′. For 4a–4c prepared in THF or MeOH, the corresponding gold(III) porphyrin radical anions 4a′, 4b′ and 4c′ are only present in negligible amounts (Fig. 6). Hence, in all these cases the equilibrium between the gold(II) valence isomers 2a–4c and their corresponding porphyrin radical anions 2a′–4c′ is in favour of the gold(II) isomers. The very strong preference of 4a–4c over 4a′–4c′ independent of the meso substituents might be due to a solvent effect overwhelming the substituent effects. Indeed, in THF or in MeOH solvent-separated ion-pairs [4a–4c]+//[PF6]− should be present while in CH2Cl2 solution contact ion pairs of [2a,3a][PF6] are formed. Indeed, reduction of [2a][PF6] or [3a][PF6] in THF resulted in EPR spectra mainly displaying the gold(II) valence isomers (ESI, Fig. S29 and S30†). The counterion location might affect the charge and spin distribution in the neutral species as well (vide infra).
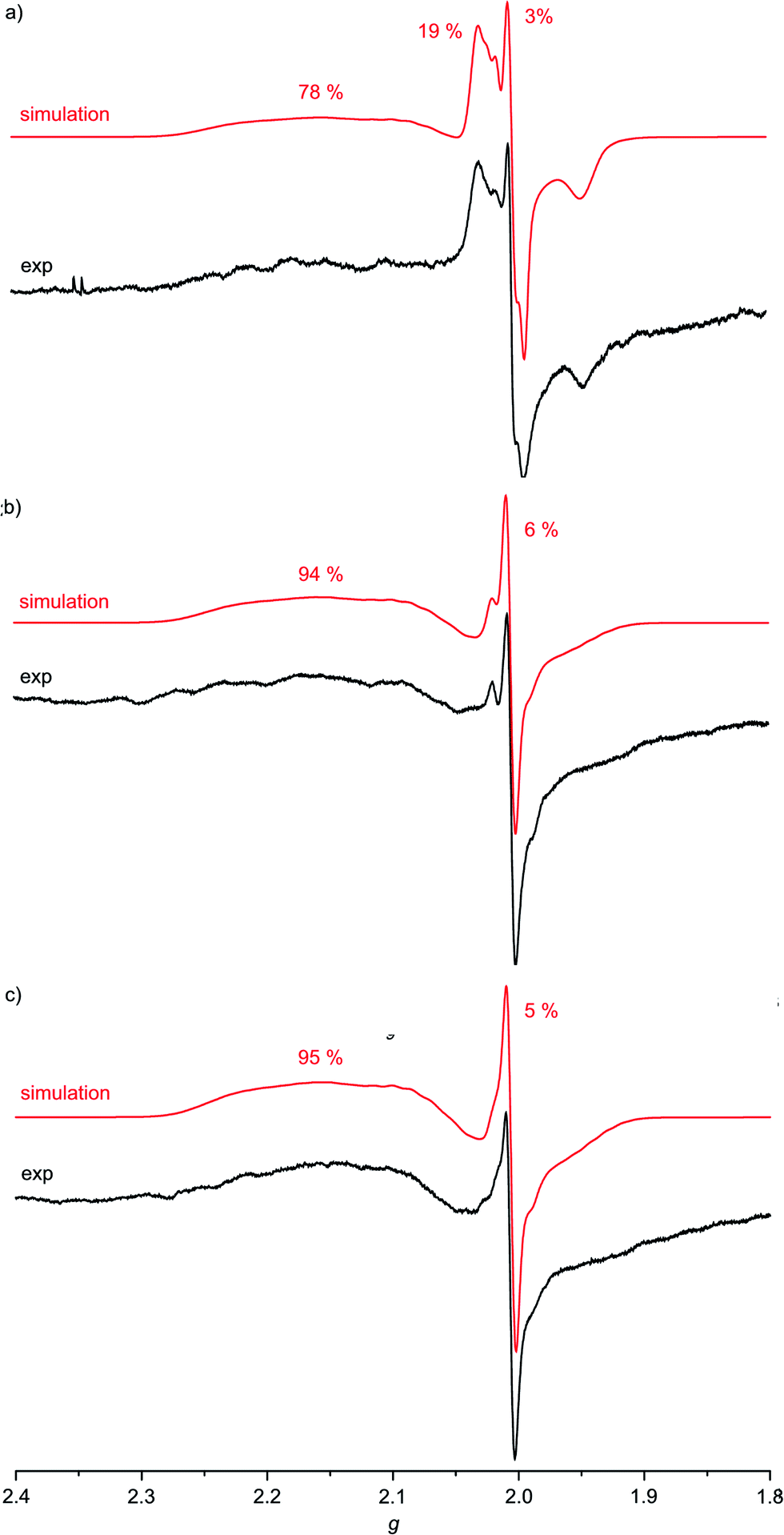 | ||
Fig. 5 X-band EPR spectra of (a) 1a/1a′/1a′′ (78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:19), (b) 2a/2a′ (94:6) and (c) 3a/3a′ (95:5) in frozen CH2Cl2 solution (77 K, 9.4 GHz) and corresponding simulations. :3:19), (b) 2a/2a′ (94:6) and (c) 3a/3a′ (95:5) in frozen CH2Cl2 solution (77 K, 9.4 GHz) and corresponding simulations. | ||
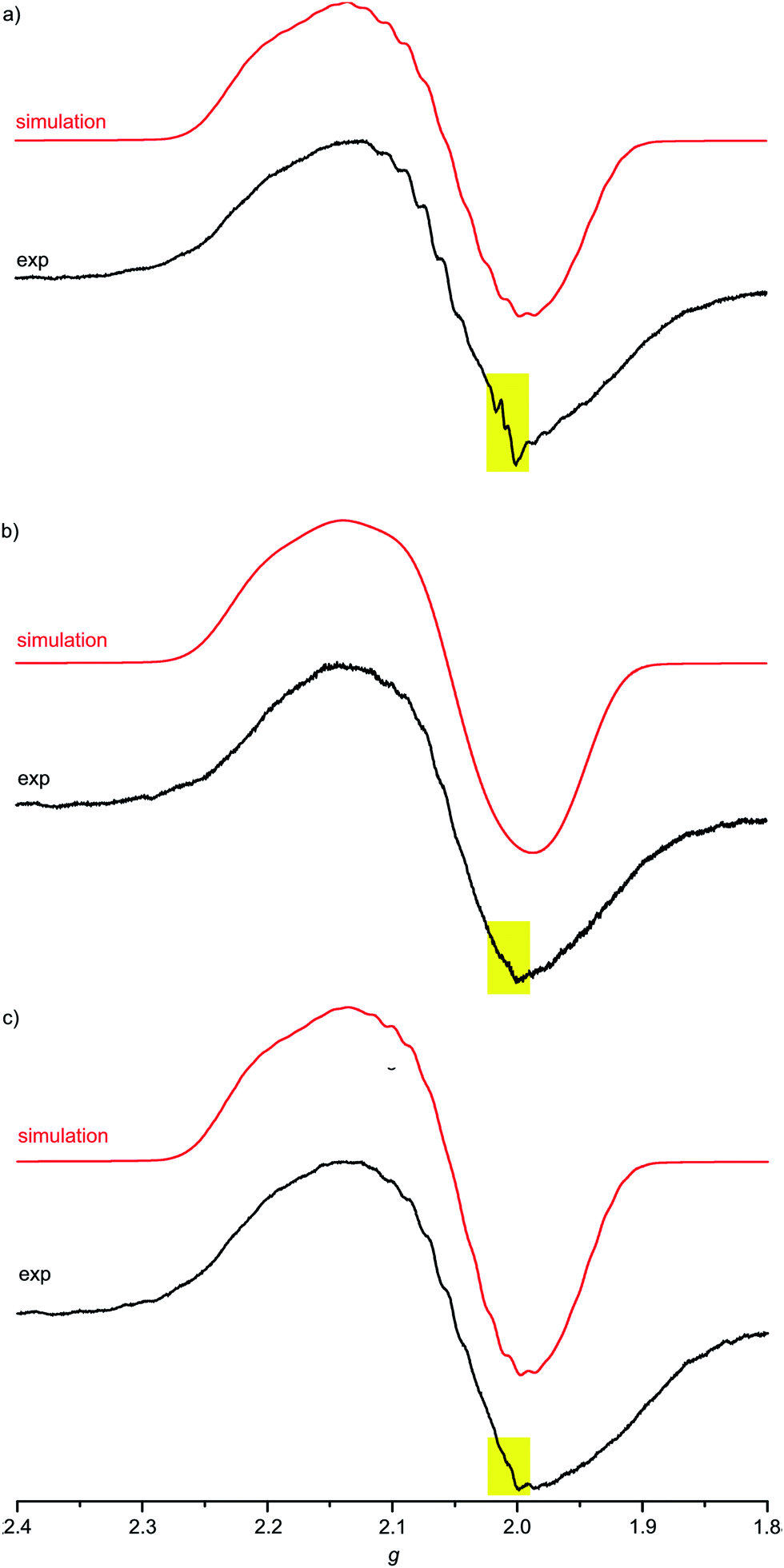 | ||
| Fig. 6 X-band EPR spectra of (a) 4a (MeOH), (b) 4b (THF) and (c) 4c (THF) in frozen solution (77 K, 9.4 GHz) and corresponding simulations; yellow squares highlight the presence of traces of porphyrin radical anions 4a′, 4b′ and 4c′, respectively. | ||
The much more intense sharp EPR resonance present in the EPR spectrum of 1a obtained from [1a][PF6] in CH2Cl2 (Fig. 5a) differs from the sharp resonances assigned to the porphyrin π radical anions 2a′ and 3a′. Indeed, simulations of the resonance suggests the presence of a further radical species with g1,2,3 = 2.031, 2.005, 1.948 and hyperfine coupling to a single nitrogen atom (A1,2,3 = 2, 17, 2 G). This is in good accordance with a nitroarene radical anion.23 Hence, this distinct EPR resonance is assigned to a nitrophenyl radical anion valence isomer 1a′′. The radical distribution 1a:1a′:1a′′ is estimated as 78:3:19. The decomposition into the component spectra is displayed in the ESI.† The effect of the type of counterions was probed by adding two equivalents of [nBu4N]Cl to the solution prior to reduction of the gold(III) porphyrin with CoCp2. No significant changes are observed for Au(TPP), 3a (CH2Cl2) or 4c (THF) in the presence of chloride. However, the presence of chloride transforms the 1a:1a′:1a′′ radical mixture almost completely into a 1a:1a′ mixture (65:35) as only the gold(II) resonance and the porphyrin radical anion resonance are observed under these conditions (ESI,†Table 2), similar to the 2a′ and 3a′ cases. Hence, for the nitro derivative 1a, three possible valence isomers are possible: the gold(II) radical (1a), the porphyrin based π radical (1a′) and a further nitro group based π radical (1a′′). Assuming, that rapid freezing does not strongly affect the equilibria of valence isomers, we can conclude that the environment, namely anions and the solvent, appears to influence these valence isomeric equilibria significantly. The substituents influence the equilibria as well, especially, when a strongly electron accepting nitro group is present. A conceivable intervalence transition between 1a and 1a′/1a′′ is not detected in the UV/Vis spectrum by comparison with the spectra of 2a and 3a (Fig. 3). This might be associated with the different orbital symmetries of 1a and 1a′/1a′′.
The electronic structure of the gold(II) radicals 1a–4c, the valence isomeric equilibrium 1a/1a′′ and the effect of counterions will be addressed by theoretical methods in the next section.
DFT studies of (porphyrinato)gold(III) complexes (series [1a]+–[3a]+ and series [4a]+–[4c]+) and the corresponding reduced species (series 1a–3a and series 4a–4c)
Both the geometries of the cationic gold(III) porphyrins [Au(TPP)]+, [1a]+–[4c]+ and the structures of all corresponding neutral species Au(TPP), 1a–3a and 4a–4c were optimised by DFT methods (B3LYP, LANL2DZ, IEFPCM CH2Cl2; Table 3, Fig. 7 and ESI†). The most significant differences between the cationic gold(III) complexes and their neutral congeners are found in the Au–N distances which increase by ca. 4% from 2.051 to 2.124 Å in all cases (Table 3). The large changes of the Au–N distances (Table 3) contribute to the reorganisation energy of the reduction process.13c The gold ions are located nearly perfectly in the centre of the four pyrrolic nitrogen atoms in all complexes. The macrocycle itself displays only minor distortions both in the cations as well as in the neutral complexes. A minor increase of the saddling (B2u) distortion is noted in the neutral complexes (Table 3). These metrical data of 1a–4c strongly suggest that the reduction of the metal centre to AuII is favoured in the electronic ground state. A reduction of the porphyrin to its radical anion 1a′–4c′ should result in pronounced macrocycle distortions as well as in small AuIII–N (radical anion) bond distances which is not observed. The calculated Mulliken spin densities are in full accordance with these structural parameters. In all neutral complexes the majority of the spin density is located at the metal centre (Mulliken spin density at Au: 0.44), especially in the 5dx2−y2 orbital (Fig. 7 and ESI†). The remainder is distributed over the pyrrolic nitrogen atoms in the σ-orbitals pointing towards the metal centre (Mulliken spin density at N: 0.14). This clearly advocates a gold-centred radical localised in the σ-system of the almost planar molecule rather than a porphyrin radical anion with the spin delocalised in the π-system of the porphyrin. The DFT determined AuII electronic ground states of 1a–4c perfectly match the experimentally derived ground states. The spin densities are also in full agreement with experimentally determined EPR parameters (g values, 197Au hyperfine coupling and 14N superhyperfine coupling). Compared with the isoelectronic Cu(TPP) [Mulliken spin density at Cu: 0.58; Mulliken spin density at N: 0.105] the spin densities are more delocalised onto the nitrogen atoms which is in agreement with the EPR results as well (ESI†).17d| Au–N/Å | Centre–N/Åa | Cα–N–N′–C′α (ruffling, B1u)/° | N–centre–N′ (saddling, B2u)a/° | N–O/Å | Torsion angle with respect to porphyrin plane C5–C12–C38–C43 | Au⋯F/Å | |
|---|---|---|---|---|---|---|---|
| a Centre denotes the geometric centre of the four pyrrole nitrogen atoms. b Constrained distances. | |||||||
| [Au(TPP)]+ | 2.051/2.051/2.051/2.051 | 2.051/2.051/2.051/2.051 | −0.49 to +0.50 | 179.51/179.51 | |||
| Au(TPP) | 2.124/2.124/2.124/2.124 | 2.124/2.124/2.124/2.124 | −0.05 to +0.04 | 178.46/178.46 | |||
| [1a]+ | 2.050/2.050/2.050/2.050 | 2.050/2.050/2.050/2.050 | −2.23 to +1.95 | 179.57/179.57 | 1.281/1.281 | 66.1 | |
| 1a | 2.124/2.124/2.124/2.124 | 2.124/2.124/2.124/2.124 | −0.27 to −0.05 | 178.38/178.38 | 1.283/1.283 | 62.4 | |
| 1a′′ | 2.057/2.058/2.058/2.057 | 2.057/2.058/2.058/2.057 | −1.39 to +1.09 | 179.25/179.25 | 1.349/1.349b | 50.8 | |
| [1a⋯PF6] | 2.047/2.050/2.052/2.051 | 2.048/2.050/2.050/2.050 | −2.71 to +1.47 | 179.82/179.83 | 1.282/1.282 | 68.3 | 3.146 |
| [1a⋯ PF6]− | 2.127/2.119/2.127/2.124 | 2.126/2.121/2.126/2.122 | −1.54 to +1.78 | 178.75/178.75 | 1.284/1.285 | 61.4 | 4.076 |
| [1a′′⋯PF6]− | 2.057/2.057/2.056/2.055 | 2.056/2.056/2.057/2.056 | −6.18 to +6.03 | 179.35/179.35 | 1.315/1.316 | 50.2 | 3.249 |
| [2a]+ | 2.051/2.052/2.051/2.052 | 2.052/2.052/2.052/2.051 | −3.54 to +3.44 | 179.60/179.60 | |||
| 2a | 2.124/2.125/2.125/2.125 | 2.125/2.125/2.125/2.125 | −2.26 to +2.35 | 178.28/178.28 | |||
| [3a]+ | 2.051/2.051/2.051/2.051 | 2.051/2.051/2.051/2.051 | −0.39 to +0.63 | 179.55/179.55 | |||
| 3a | 2.124/2.124/2.124/2.124 | 2.124/2.124/2.124/2.124 | −0.85 to +0.69 | 178.43/178.43 | |||
| [4a]+ | 2.051/2.051/2.051/2.051 | 2.051/2.051/2.051/2.051 | −1.05 to +1.19 | 179.58/179.58 | |||
| 4a | 2.124/2.124/2.124/2.124 | 2.124/2.124/2.124/2.124 | −0.76 to +0.74 | 178.42/178.42 | |||
| [4b]+ | 2.052/2.052/2.052/2.052 | 2.052/2.052/2.052/2.052 | −1.03 to +0.90 | 179.56/179.56 | |||
| 4b | 2.125/2.125/2.125/2.125 | 2.125/2.125/2.125/2.125 | −0.77 to +0.69 | 178.35/178.35 | |||
| [4c]+ | 2.051/2.051/2.051/2.051 | 2.051/2.051/2.051/2.051 | −0.49 to +0.89 | 179.56/179.56 | |||
| 4c | 2.124/2.124/2.124/2.124 | 2.124/2.124/2.124/2.124 | −0.92 to +1.10 | 178.53/178.53 | |||
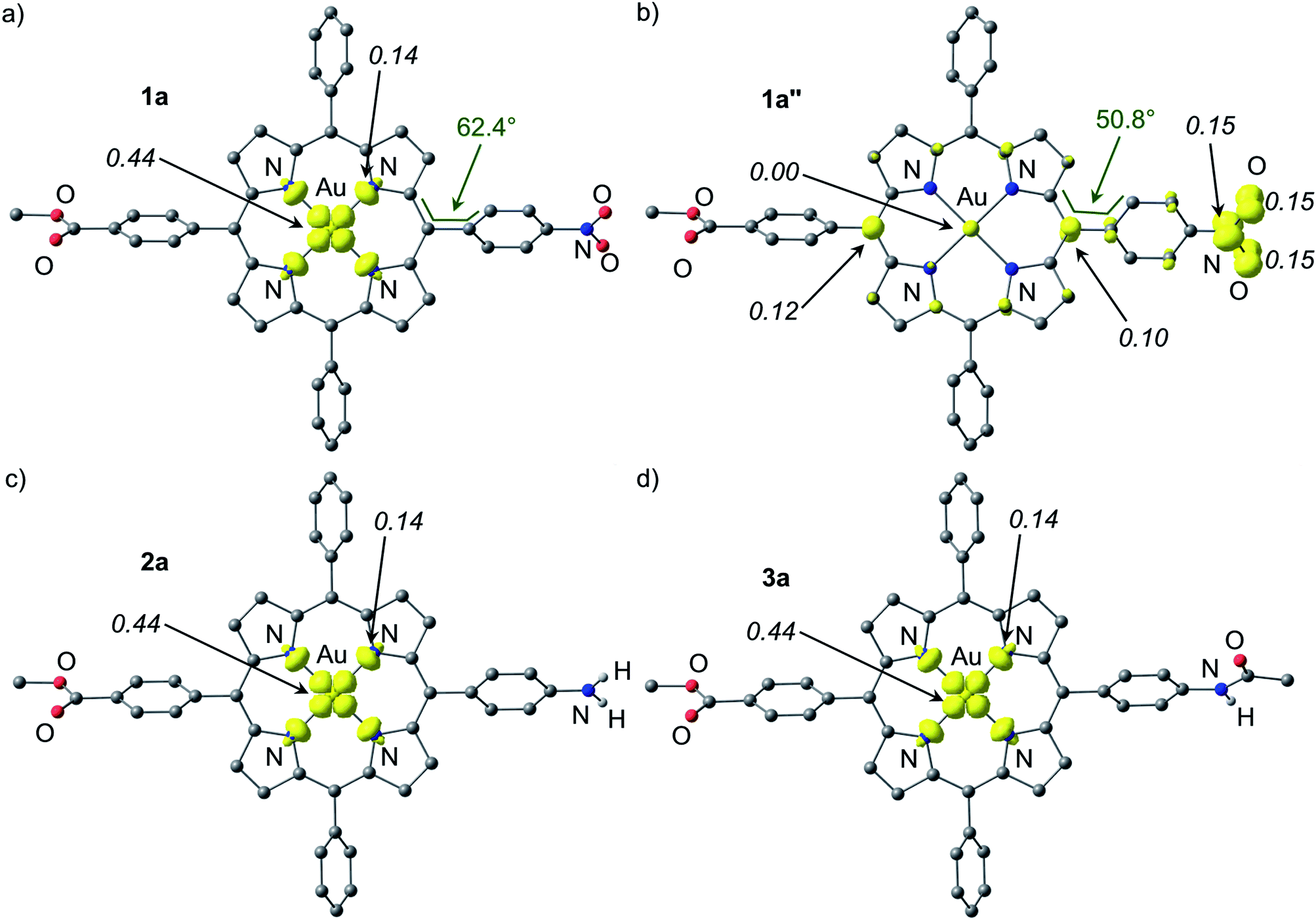 | ||
| Fig. 7 Spin densities of DFT optimised geometries of (a) 1a, (b) 1a′′ (constrained), (c) 2a and (d) 3a (isosurface value at 0.01 a.u. in yellow; 0.006 a.u. for 1a′′; UB3LYP, LANL2DZ, IEFPCM CH2Cl2; CH hydrogen atoms omitted; Mulliken spin densities in italics). | ||
The special case of the nitro derivative 1a which displays significant amounts of the nitrobenzene π radical anion valence isomer 1a′′ in the EPR spectrum (Fig. 5a) was treated by DFT methods as well. However, all geometry optimisation attempts (with the employed functional, basis set and tight convergence criteria) converged to the stable AuII valence isomer 1a. In order to get an impression on the spin density distribution in valence isomer 1a′′, the nitrobenzene radical anion [C6H5NO2]˙− was separately optimised by DFT methods giving NO distances of 1.349 Å. These NO distances were then constrained to 1.349 Å in geometry optimizations of 1a′′ giving the (constrained) optimised structure of 1a′′ as shown in Fig. 7. The Au–N bond lengths of 1a′′ are fully consistent with a gold(III) oxidation state (Table 3). Compared to [1a]+ and 1a the C6H4NO2 torsion angle with respect to the porphyrin plane C5–C12–C38–C43 is significantly reduced from 66.1° and 62.4° to 50.8° suggesting a conjugative electron withdrawing effect of the gold(III) porphyrin as expected for a π-centred radical. The spin density is mainly located at the NO2 substituent and partially delocalised over the π-system of the porphyrin. The Mulliken spin density at the gold atom in 1a′′ is essentially zero (Fig. 7).
As an unconstrained optimization of 1a′′ was unsuccessful, we investigated the effect of the counterion [PF6]− on the charge and spin distribution in [1a⋯PF6]− and [1a′′⋯PF6]−, respectively. Indeed, we succeeded in optimising both valence isomers [1a⋯PF6]− and [1a′′⋯PF6]− without any constraints (Fig. 8). The AuII valence isomer [1a⋯PF6]− is preferred by 12 kJ mol−1. In this AuII isomer [1a⋯PF6]− the [PF6]− ion is not coordinated to the metal (Au⋯F 4.076 Å) but only hydrogen-bonded to two CH groups of the aryl substituents (Fig. 8a). The spin density is again localised at the metal centre (Mulliken spin density at Au: 0.44) and the pyrrolic nitrogen atoms (Mulliken spin density at N: 0.14). In the nitro-based π radical [1a′′⋯PF6]− the [PF6]− ion is much closer to the gold centre (Au⋯F 3.249 Å, Fig. 8b). The presence of the negative charge close to the metal centre stabilises the AuIII oxidation state and indeed the gold ion carries no spin density. Au–N distances (2.057 Å; [1a′′⋯PF6]−) fully agree with a gold(III) porphyrin but not with a gold(II) porphyrin (2.127 Å; [1a⋯PF6]−). The N–O distances have increased from 1.284 Å in [1a⋯PF6]− to 1.315 Å in [1a′′⋯PF6]− as expected for population of N–O antibonding orbitals. The spin density is largely confined to the NO2 substituent and partially delocalized to the π-system of the porphyrin. The C5–C12–C38–C43 torsion angle of the nitrophenyl substituent decreases from 61.4° ([1a⋯PF6]−) to 50.2° ([1a′′⋯PF6]−) similar to the 1a/1a′′ (constrained) pair. In essence, the intramolecular electron transfer pathway between [1a⋯PF6]− and [1a′′⋯PF6]− encompasses the Au–N and Au⋯F distances (totally symmetric stretching vibration of the gold coordination sphere), the symmetric NO2 stretching mode and a phenyl torsional motion (Fig. 8).
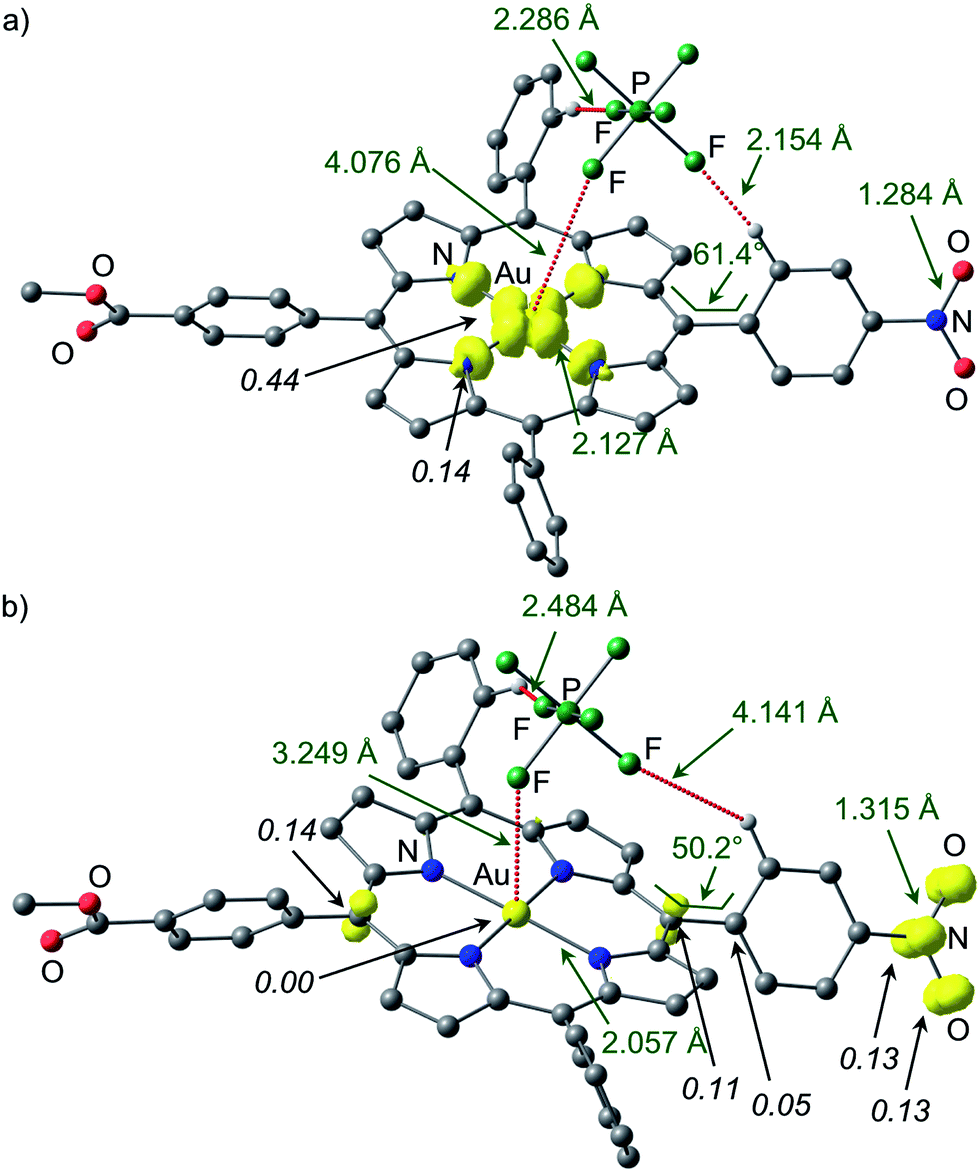 | ||
| Fig. 8 Spin densities of DFT optimised geometries of (a) [1a⋯PF6]− and (b) [1a′′⋯PF6]− (isosurface value at 0.01 a.u. in yellow; UB3LYP, LANL2DZ, IEFPCM CH2Cl2; hydrogen atoms which are not involved in hydrogen bonds are omitted; Mulliken spin densities in italics). | ||
With respect to photoinduced electron transfer reactions using porphyrinato gold(III) complexes as electron acceptors we suggest that the initial kinetic reduction product of a porphyrinato gold(III) complex should be a gold(III) porphyrin π radical anion (such as 1a′–4c′) due to the smaller activation barrier and the better electronic coupling to electron donors (Scheme 3). In a following intramolecular valence isomerisation the electron shifts to the central gold ion (σ-system) with concomitant dissociation of the counterion giving the thermodynamic AuII product (such as 1a–4c) (Scheme 3). The latter chemical reaction will render the whole photoinduced ET process irreversible, which is advantageous for further reactivity of the redox sites. In the case of nitro substituted porphyrins a further valence isomer [1a′′⋯PF6]− with a nitrophenyl π radical anion is existent as well. Both the solvent, the present ions and the substituents determine the final charge and spin distribution.
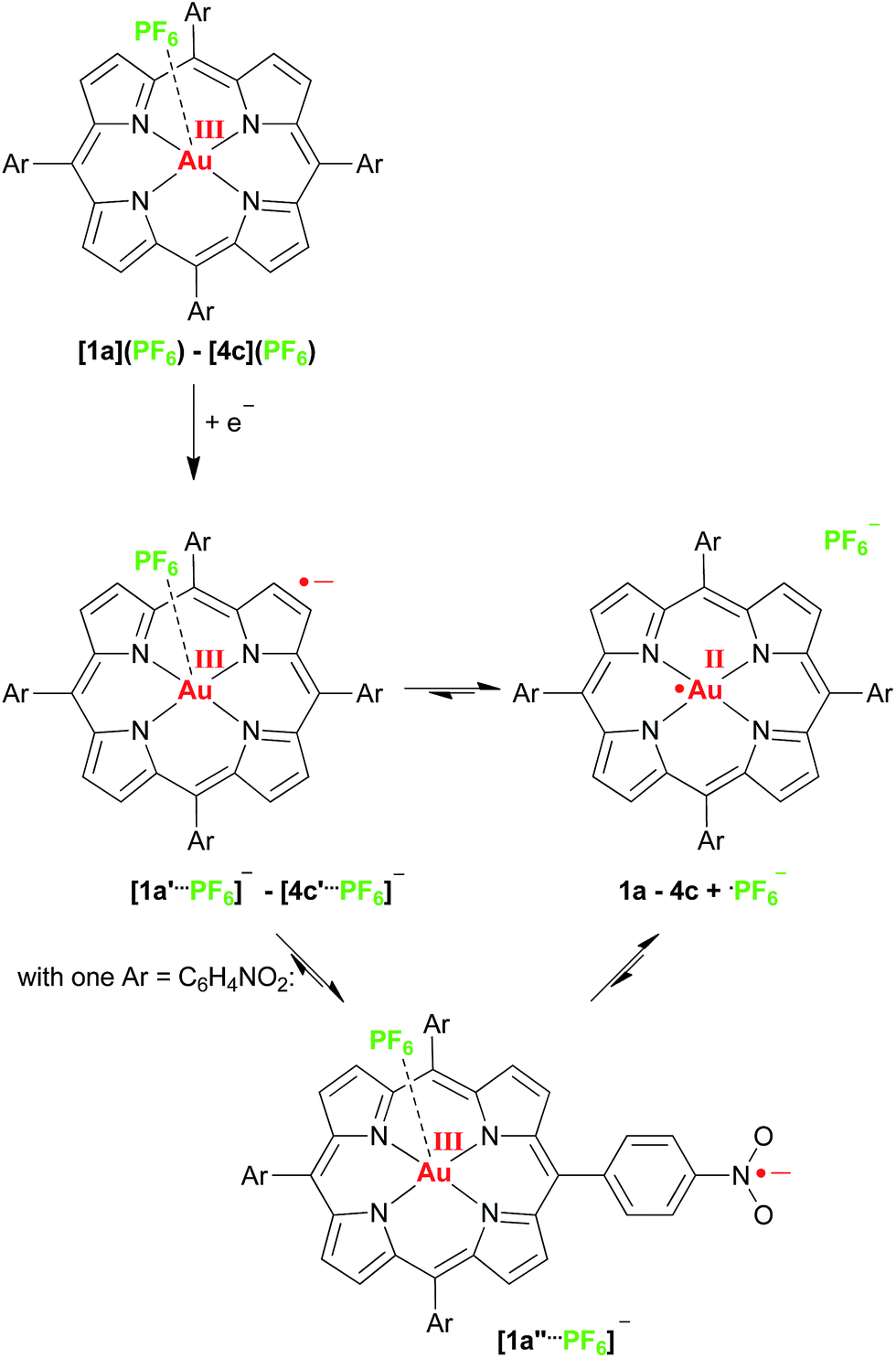 | ||
| Scheme 3 Valence tautomeric equilibria of gold(II) porphyrins and gold(III) porphyrin radical anions. | ||
Experimental
Porphyrins H2TPP, Ia, IIa, IIIa, IVa, IVb and IVc were prepared according to published procedures.17 Solvents were dried by standard methods. Other reagents were used as received from commercial suppliers (Acros, Sigma-Aldrich). NMR spectra were recorded on a Bruker Avance DRX 400 spectrometer at 400.31 MHz (1H), 100.05 MHz (13C{1H}), 162.05 MHz (31P{1H}). Resonances are reported in ppm versus the solvent signal as an internal standard. CD2Cl2 (1H: δ = 5.32 ppm; 13C: δ = 53.8 ppm), d8-THF (1H: δ = 1.73, 3.58 ppm; 13C: δ = 25.5, 67.7 ppm) MeOD (1H: δ = 3.31, 4.87 ppm; 13C: δ = 49.0 ppm) and versus external H3PO4 (85%) (31P: δ = 0 ppm); (s) = singlet, (pt) = pseudo triplet (unresolved doublet of doublets), (br, s) = broad singlet, septet (sept). IR spectra were recorded with a BioRad Excalibur FTS 3100 spectrometer as KBr disks; (vs) = very strong, (s) = strong, (m) = medium, (w) = weak. ESI mass spectra were recorded on a Micromass Q-TOF-Ultima spectrometer. Electrochemical experiments were carried out on a BioLogic SP-50 voltammetric analyzer using a glassy carbon working electrode, a platinum wire as the counter electrode and a 0.01 M Ag/AgNO3 electrode as the reference electrode. The measurements were carried out at a scan rate of 100 mV s−1 for cyclic voltammetry experiments and for square wave voltammetry experiments using a concentration of 10−3 M in 0.1 M [nBu4N][PF6] as the supporting electrolyte in THF (MeOH). Potentials are given relative to the ferrocene/ferrocenium couple. Spectroelectrochemical experiments were performed using a thin layer quartz glass (path length 1 mm) cell kit (GAMEC Analysentechnik, Illingen, Germany) equipped with a Pt gauze working electrode, a Pt counter electrode and a Ag/AgNO3 reference electrode (10−5 M solutions in THF or in MeOH, containing 0.1 M [nBu4N][PF6]). X-band CW EPR spectra were measured on a Miniscope MS 300 (Magnettech GmbH, Germany). g-Values are referenced to external Mn2+ in ZnS (g = 2.118, 2.066, 2.027, 1.986, 1.946, 1.906). Simulations were performed with the program package EasySpin.24 UV/Vis/NIR spectra were recorded on a Varian Cary 5000 spectrometer using 1.0 cm cells (Hellma, suprasil).DFT calculations
Density functional calculations were carried out with the Gaussian09/DFT series25 of programs. The B3LYP formulation of density functional theory was used employing the LANL2DZ basis set. To include solvent effects the integral equation formalism polarisable continuum model (IEFPCM CH2Cl2) was employed. No (symmetry) constraints were imposed on the molecules, except for the NO distance constraint for 1a′. The presence of energy minima of the ground states was checked by analytical frequency calculations.EPR measurements of radical anions
Under an inert atmosphere a solution of the respective gold(III) porphyrin complex (c = 5 × 10−3 M) in CH2Cl2 ([Au(TPP)][PF6], [1a][PF6], [2a][PF6], [3a][PF6]), MeOH ([4a][PF6]) or THF ([4b][PF6], [4c][PF6]) was treated with 0.95 equivalents of cobaltocene CoCp2. The X-band EPR spectrum of the sample was measured immediately after freezing the solution to 77 K. The effect of chloride was measured by addition of 2.0 equivalents of [nBu4N]Cl prior to the reduction.[5,10,15,20-Tetraphenylporphyrinato]gold(III) hexafluorophosphate [Au(TPP)][PF6]
5,10,15,20-Tetraphenylporphyrin (50 mg, 0.08 mmol), potassium tetrachloridoaurate(III) (121 mg, 0.32 mmol) and sodium acetate (302 mg, 3.68 mmol) were dissolved in glacial acetic acid (20 mL). The reaction mixture was heated to reflux for 20 h, allowed to cool to room temperature, and diluted with dichloromethane (100 mL). The mixture was washed with water (2 × 50 mL), saturated sodium carbonate solution (2 × 50 mL) and water (1 × 50 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was evaporated to dryness and the residue dissolved in dichloromethane (50 mL). The organic phase was stirred with a saturated aqueous solution of potassium hexafluorophosphate (10 mL) for 72 h. The mixture was diluted with dichloromethane (100 mL) and washed with water (2 × 50 mL), dried over anhydrous magnesium sulfate, and filtered. The filtrate was removed under reduced pressure and the residue purified by chromatography over silica [dichloromethane:methanol (20:1); Rf = 0.40] to yield [Au(TPP)][PF6] (66 mg, 0.09 mmol, 88%) as a purple crystalline solid. C44H28AuF6N4P (954.7). UV/Vis: λmax(THF)/nm 409 (ε/M−1 cm−1 258000), 523 (12400). IR: νmax/cm−1 1638 (m), 1617 (s), 839 (vs, PF), 556 (m, PF6,def). NMR: δH (400 MHz, CD2Cl2) 7.93 (12H, m, H5/5/10/10/15/15/20/203/4/3/4/3/4/3/4), 8.24 (8H, m, H5/10/15/202/2/2/2), 9.36 (8H, s, H2,3,7,8,12,13,17,18); δC (100 MHz, CD2Cl2) 124.1 (s, C5/10/15/20), 128.1 (s, C5/10/15/203/3/3/3), 129.8 (s, C5/10/15/204/4/4/4), 132.7 (s, C2/3/7/8/12/13/17/18), 134.5 (s, C5/10/15/202/2/2/2), 137.3 (s, C1/4/6/9/11/14/16/19), 138.7 (s, C5/10/15/201/1/1/1). δP (162 MHz, CD2Cl2) −143.8 (sept, 1JPF = 710 Hz). MS (ESI): m/z 809.99 (100%) [M]+. HR-MS (ESI): m/z 809.1993 (calcd for C44H28AuN4: 809.1980). CV (Fc/Fc+, THF): E½/V −2.350, −1.650, −0.975.
[10,20-Di(phenyl)-15-(4-(methoxycarbonylphenyl))-5-(4-nitrophenyl)porphyrinato]gold(III) hexafluorophosphate [1a][PF6]
10,20-Di(phenyl)-15-(4-(methoxycarbonylphenyl))-5-(4-nitrophenyl)porphyrin Ia (100 mg, 0.14 mmol), potassium tetrachloridoaurate(III) (212 mg, 0.56 mmol) and sodium acetate (529 mg, 6.44 mmol) were dissolved in glacial acetic acid (40 mL). The reaction mixture was heated to reflux for 22 h, allowed to cool to room temperature, and diluted with dichloromethane (200 mL). The mixture was washed with water (2 × 100 mL), saturated aqueous sodium carbonate solution (2 × 100 mL) and water (1 × 100 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was evaporated to dryness and the residue dissolved in dichloromethane (100 mL). The organic phase was stirred with a saturated aqueous solution of potassium hexafluorophosphate (20 mL) for 72 h. The mixture was diluted with dichloromethane (100 mL) and washed with water (2 × 50 mL), dried over anhydrous magnesium sulfate, and filtered. The filtrate was removed under reduced pressure and the residue purified by chromatography over silica [dichloromethane:methanol (10:1); Rf = 0.35] to yield [1a][PF6] (64 mg, 0.06 mmol, 43%) as a purple crystalline solid. C46H29AuF6N5O4P (1057.7). UV/Vis: λmax(THF)/nm 410 (ε/M−1 cm−1 329000), 523 (17600). IR: νmax/cm−1 1717 (s, COester), 1597 (m), 1520 (s, NOasym), 1439 (m), 1346 (s, NOsym), 1277 (s, OCOdef), 1106 (m), 1034 (s), 1018 (s), 837 (vs, PF), 556 (s, PF6,def). NMR: δH (400 MHz, CD2Cl2) 4.12 (3H, s, H156), 7.95 (6H, m, H10/10/20/203/3/4/4), 8.29 (4H, d, H2/210/20), 8.39 (2H, d, 3JHH = 8.2 Hz, H152), 8.50 (2H, d, 3JHH = 8.5 Hz, H52), 8.57 (2H, d, 3JHH = 8.1 Hz, H153), 8.75 (2H, d, 3JHH = 8.5 Hz, H53), 9.35 (8H, m, H2,3,7,8,12,13,17,18); δC (100 MHz, CD2Cl2) 52.5 (s, C156), 121.4 (s, C15), 123.4 (s, C53), 124.8 (s, C10/20), 128.4 (s, C10/203/3), 129.4 (s, C153), 130.0 (s, C10/204/4), 132.2 (s, C154), 132.8–133.7 (multiple s, C2/3/7/8/12/13/17/18), 134.8 (s, C10/20/152/2/2), 136.5 (s, C52), 136.9–138.0 (multiple s, C1/4/6/9/11/14/16/19), 138.9 (s, C10/201/1), 143.3 (s, C151), 145.4 (s, C51), 149.4 (s, C54), 167.1 (s, C155). δP (162 MHz, CD2Cl2) −144.1 (sept, 1JPF = 711 Hz). MS (ESI): m/z 912.11 (100%) [M]+. HR-MS (ESI): m/z 912.1905 (calcd for C46H29AuN5O4: 912.1885). CV (Fc/Fc+, THF): E½/V −2.300, −1.795, −1.560, −0.920.
[5-(4-Aminophenyl)-10,20-di(phenyl)-15-(4-(methoxycarbonylphenyl))porphyrinato]gold(III) hexafluorophosphate [2a][PF6]
[10,20-Di(phenyl)-15-(4-(methoxycarbonylphenyl))-5-(4-nitrophenyl)porphyrinato]gold(III) hexafluorophosphate [1a][PF6] (100 mg, 0.09 mmol) and tin(II) chloride dihydrate were suspended in concentrated hydrochloric acid (36%, 50 mL). The reaction mixture was stirred in the dark under argon for 20 h. The mixture was diluted with dichloromethane (200 mL), washed with water (2 × 100 mL), saturated sodium carbonate solution (2 × 100 mL), and water (1 × 100 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was evaporated to dryness and the residue dissolved in dichloromethane (100 mL). The organic phase was stirred with a saturated aqueous solution of potassium hexafluorophosphate (20 mL) for 72 h. The mixture was diluted with dichloromethane (100 mL) and washed with water (2 × 50 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was removed under reduced pressure and the residue purified by chromatography over silica [dichloromethane:methanol (25:1); Rf = 0.22] to yield [2a][PF6] (31 mg, 0.03 mmol, 34%) as a reddish-brown solid. C46H31AuF6N5O2P (1027.7). UV/Vis: λmax(THF)/nm 406 (ε/M−1 cm−1 126000), 529 (10800), 589 (4800). IR: νmax/cm−1 1723 (s, COester), 1638 (s), 1618 (vs, NH2,def), 1277 (s, OCOdef), 966 (s), 835 (vs, PF), 567 (vs), 557 (s, PF6,def). NMR: δH (400 MHz, CD2Cl2) 4.14 (3H, s, H156), 4.7 (2H, br s, NH2), 7.25 (2H, d,3JHH = 8.3 Hz, H53), 7.95 (6H, m, H10/10/20/203/4/3/4), 8.03 (2H, d, 3JHH = 8.4 Hz, H52), 8.27 (4H, m, H10/202/2), 8.37 (2H, d, 3JHH = 8.3 Hz, H152), 8.57 (2H, d, 3JHH = 8.3 Hz, H153), 9.40 (8H, m, H2,3,7,8,12,13,17,18); δC (100 MHz, CD2Cl2) 52.2 (s, C156), 114.6 (s, C53), 122.6 (s, C15), 124.4 (s, C10/20), 127.5 (s, C5), 128.4 (s, C10/203/3), 129.4 (s, C153), 130.2 (s, C10/204/4), 132.3 (s, C154), 132.8–133.6 (multiple s, C2/3/7/8/12/13/17/18), 134.8 (s, C10/20/152/2/2), 135.6 (s, C51), 136.5 (s, C52), 137.1–137.7 (multiple s, C1/4/6/9/11/14/16/19), 139.0 (s, C10/201/1), 143.5 (s, C151), 149.5 (s, C54), 167.2 (s, C155). δP (162 MHz, CD2Cl2) −143.5 (sept, 1JPF = 710 Hz). MS (ESI): m/z 882.09 (100%) [M]+. HR-MS (ESI): m/z 882.2163 (calcd for C46H31AuN5O2: 882.2143). CV (Fc/Fc+, THF): E½/V −2.500 (irrev.), −2.280, −1.645, −0.990.
[5-(4-(N-Acetylaminophenyl))-10,20-di(phenyl)-15-(4-(methoxycarbonylphenyl))porphyrinato]gold(III) hexafluorophosphate [3a][PF6]
5-(4-(N-Acetylaminophenyl))-10,20-di(phenyl)-15-(4-(methoxycarbonylphenyl))porphyrin IIIa (50 mg, 0.07 mmol), potassium tetrachloridoaurate(III) (104 mg, 0.28 mmol) and sodium acetate (258 mg, 3.15 mmol) were dissolved in glacial acetic acid (20 mL). The reaction mixture was heated to reflux for 24 h, allowed to cool to room temperature, and diluted with dichloromethane (100 mL). The mixture was washed with water (2 × 50 mL), saturated aqueous sodium carbonate solution (2 × 50 mL) and water (1 × 50 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was evaporated to dryness and the residue dissolved in dichloromethane (50 mL). The organic phase was stirred with a saturated aqueous solution of potassium hexafluorophosphate (10 mL) for 72 h. The mixture was diluted with dichloromethane (50 mL) and washed with water (2 × 50 mL), dried over anhydrous magnesium sulfate, and filtered. The filtrate was removed under reduced pressure and the residue purified by chromatography over silica [dichloromethane:methanol (10:1); Rf = 0.43] to yield [3a][PF6] (32 mg, 0.03 mmol, 49%) as a purple crystalline solid. C48H33AuF6N5O3P (1069.7). UV/Vis: λmax(THF)/nm 410 (ε/M−1 cm−1 124000), 525 (10500). IR: νmax/cm−1 2964 (m, NH), 1717 (s, COester), 1677 (s, COamide), 1616 (m), 1262 (s, OCOdef), 1096 (s), 1020 (s), 839 (s), 803 (vs, PF), 708 (m), 557 (m, PF6,def). NMR: δH (400 MHz, CD2Cl2) 2.33 (3H, s, H56), 4.11 (3H, s, H156), 7.93 (6H, m, H10/10/20/203/4/3/4), 8.04 (1H, s, NH), 8.12 (2H, d,3JHH = 8.3 Hz, H53), 8.19 (2H, d, 3JHH = 8.4 Hz, H52), 8.24 (4H, m, H10/202/2), 8.34 (2H, d, 3JHH = 8.3 Hz, H152), 8.55 (2H, d, 3JHH = 8.3 Hz, H153), 9.37 (8H, m, H2,3,7,8,12,13,17,18); δC (100 MHz, CD2Cl2) 24.9 (s, C56), 53.2 (s, C156), 119.2 (s, C53), 122.6 (s, C15), 124.5 (s, C10/20), 124.8 (s, C5), 128.4 (s, C10/203/3), 129.3 (s, C153), 130.2 (s, C10/204/4), 132.0 (s, C154), 132.5–133.3 (multiple s, C2/3/7/8/12/13/17/18), 134.8 (s, C10/20/152/2/2), 135.4 (s, C52), 137.0–137.9 (multiple s, C1/4/6/9/11/14/16/19), 139.0 (s, C10/201/1), 140.4 (s, C51), 141.5 (s, C54), 143.4 (s, C151), 169.9 (s, C155). δP (162 MHz, CD2Cl2) −143.5 (sept, 1JPF = 711 Hz). MS (ESI): m/z 924.01 (100%) [M]+. HR-MS (ESI): m/z 924.2229 (calcd for C48H33AuN5O3: 924.2249). CV (Fc/Fc+, THF): E½/V −2.490 (irrev.), −2.300, −1.630, −0.990.
[5-(4-(N-Acetylaminophenyl))-10,20-di(phenyl)-15-(4-(carboxyphenyl))porphyrinato]gold(III) hexafluorophosphate [4a][PF6]
5-(4-(N-Acetylaminophenyl))-10,20-di(phenyl)-15-(4-(carboxyphenyl))porphyrin IVa (50 mg, 0.07 mmol), potassium tetrachloridoaurate(III) (104 mg, 0.28 mmol) and sodium acetate (258 mg, 3.15 mmol) were dissolved in glacial acetic acid (20 mL). The reaction mixture was heated to reflux for 24 h, allowed to cool to room temperature and diluted with dichloromethane (100 mL). The mixture was washed with water (2 × 50 mL), saturated aqueous sodium carbonate solution (2 × 50 mL) and water (1 × 50 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was evaporated to dryness and the residue dissolved in dichloromethane (50 mL). The organic phase was stirred with a saturated aqueous solution of potassium hexafluorophosphate (10 mL) for 72 h. The mixture was diluted with dichloromethane (50 mL) and washed with water (2 × 50 mL), dried over anhydrous magnesium sulfate, and filtered. The filtrate was removed under reduced pressure and the residue purified by chromatography over silica [dichloromethane:methanol (10:1); Rf = 0.41] to yield [4a][PF6] (24 mg, 0.02 mmol, 28%) as a reddish-brown solid. C47H31AuF6N5O3P (1055.7). UV/Vis: λmax(MeOH)/nm 408 (ε/M−1 cm−1 204000), 522 (12300). IR: νmax/cm−1 2955, 2914, 2872 (m, OH), 1712 (m, COacid), 1695 (m, COamide), 1638 (s), 1618 (s), 1432 (s), 1385 (s), 1363 (s), 1232 (s, COCdef), 1155 (s), 1121 (m), 839 (vs, PF), 775 (s), 770 (s), 558 (s, PF6,def). NMR: δH (400 MHz, CD3OD) 2.33 (3H, s, H56), 7.95 (6H, m, H10/10/20/203/4/3/4), 8.12 (2H, d,3JHH = 8.3 Hz, H53), 8.20 (2H, d, 3JHH = 8.4 Hz, H52), 8.24 (2H, d, 3JHH = 8.3 Hz, H152), 8.26 (4H, m, H10/202/2), 8.45 (2H, d, 3JHH = 8.3 Hz, H153), 9.36 (8H, m, H2,3,7,8,12,13,17,18); δC (100 MHz, CD3OD) 24.0 (s, C56), 120.1 (s, C53), 124.8 (s, C15), 125.0 (s, C10/20), 127.1 (s, C5), 127.4 (s, C10/203/3), 128.1 (s, C153), 129.1 (s, C10/204/4), 131.4 (s, C154), 131.9 (br s, C2/3/7/8/12/13/17/18), 133.5 (s, C52), 134.8 (s, C10/20/152/2/2), 136.9 (br s, C1/4/6/9/11/14/16/19), 138.7 (s, C10/201/1), 140.0 (s, C51), 140.2 (s, C151), 141.4 (s, C54), 165.9 (s, C155), 170.9 (s, C55). δP (162 MHz, CD3OD) −143.5 (sept, 1JPF = 710 Hz). MS (ESI): m/z 910.18 (100%) [M]+. HR-MS (ESI): m/z 910.2115 (calcd for C47H31AuN5O3: 910.2093). CV (Fc/Fc+, MeOH): E½/V −1.030.
[5-(4-(N-Acetylaminophenyl))-10,20-di((4-butoxy)phenyl)-15-(4-(carboxyphenyl))porphyrinato]gold(III) hexafluorophosphate [4b][PF6]
5-(4-(N-Acetylaminophenyl))-10,20-di((4-butoxy)phenyl)-15-(4-(carboxyphenyl))porphyrin IVb (75 mg, 0.09 mmol), potassium tetrachloridoaurate(III) (133 mg, 0.36 mmol) and sodium acetate (328 mg, 4.00 mmol) were dissolved in glacial acetic acid (40 mL). The reaction mixture was heated to reflux for 24 h, allowed to cool to room temperature, and diluted with dichloromethane (200 mL). The mixture was washed with water (2 × 100 mL), saturated aqueous sodium carbonate solution (2 × 100 mL) and water (1 × 100 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was evaporated to dryness and the residue dissolved in dichloromethane (100 mL). The organic phase was stirred with a saturated aqueous solution of potassium hexafluorophosphate (20 mL) for 72 h. The mixture was diluted with dichloromethane (100 mL) and washed with water (2 × 50 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was removed under reduced pressure and the residue purified by chromatography over silica [dichloromethane:methanol (10:1); Rf = 0.40] to yield [4b][PF6] (72 mg, 0.06 mmol, 66%) as a purple solid. C55H47AuF6N5O5P (1199.9). UV/Vis: λmax(THF)/nm 422 (ε/M−1 cm−1 86000), 527 (9100), 571 (3200). IR: νmax/cm−1 2957, 2924, 2870, 2855 (m, OH), 1716 (sh, COacid), 1699 (s, COamide), 1605 (s), 1505 (m), 1247 (s, COCdef), 843 (vs, PF), 804 (s), 558 (s, PF6,def). NMR: δH (400 MHz, d8-THF) 1.10 (6H, t, 3JHH = 7.3 Hz, H10/208/8), 1.66 (4H, m, H10/207/7), 1.96 (4H, m, H10/206/6), 2.14 (3H, s, H56), 4.30 (2H, t, 3JHH = 6.4 Hz, H10/205/5), 7.44 (2H, d, 3JHH = 7.4 Hz, H10/203/3), 8.12 (4H, m, H5/52/3), 8.17 (2H, d, 3JHH = 7.3 Hz, H10/202/2), 8.37 (2H, d, 3JHH = 7.1 Hz, H152), 8.53 (2H, d, 3JHH = 7.0 Hz, H153), 9.36 (8H, m, H2,3,7,8,12,13,17,18), 9.48 (1H, s, NH); δC (100 MHz, d8-THF) 14.3 (s, C10/208/8), 20.4 (s, C10/207/7), 24.4 (s, C56), 32.5 (s, C10/206/6), 67.6 (s, C10/205/5), 114.7 (s, C10/203/3), 118.8 (s, C53), 122.9 (s, C15), 124.7 (s, C10/20), 125.1 (s, C5), 129.8 (s, C153), 131.7 (s, C10/201/1), 132.5 (s, C154), 133.1–133.8 (br s, C2/3/7/8/12/13/17/18), 135.2 (s, C152), 135.7 (s, C52), 136.6 (s, C10/202/2), 137.6 (s, C51), 138.5 (br s, C1/4/6/9/11/14/16/19), 141.3 (s, C54), 143.9 (s, C151), 161.5 (s, C10/204/4), 167.5 (s, C155), 169.3 (s, C55). δP (162 MHz, d8-THF) −143.5 (sept, 1JPF = 710 Hz). MS (ESI): m/z 1054.26 (100%) [M]+. HR-MS (ESI): m/z 1054.3218 (calcd for C55H47AuN5O5: 1054.3243). CV (Fc/Fc+, THF): E½/V −2.450, −1.745, −1.070.
[5-(4-(N-Acetylaminophenyl))-10,20-bis(4-(trifluoromethylphenyl))-15-(4-(carboxyphenyl))porphyrinato]gold(III) hexafluorophosphate [4c][PF6]
5-(4-(N-Acetylaminophenyl))-10,20-bis(4-(trifluoromethylphenyl))-15-(4-(carboxyphenyl))porphyrin IVc (63 mg, 0.07 mmol), potassium tetrachlorido aurate(III) (104 mg, 0.28 mmol), and sodium acetate (258 mg, 3.15 mmol) were dissolved in glacial acetic acid (20 mL). The reaction mixture was heated to reflux for 24 h, allowed to cool to room temperature and diluted with dichloromethane (100 mL). The mixture was washed with water (2 × 50 mL), saturated aqueous sodium carbonate solution (2 × 50 mL) and water (1 × 50 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was evaporated to dryness and the residue dissolved in dichloromethane (50 mL). The organic phase was stirred with a saturated aqueous solution of potassium hexafluorophosphate (10 mL) for 72 h. The mixture was diluted with dichloromethane (50 mL) and washed with water (2 × 50 mL), dried over anhydrous magnesium sulfate and filtered. The filtrate was removed under reduced pressure and the residue purified by chromatography over silica [dichloromethane:methanol (10:1); Rf = 0.30] to yield [4c][PF6] (71 mg, 0.06 mmol, 85%) as a purple, crystalline solid. C49H29AuF12N5O3P (1191.7). UV/Vis: λmax(THF)/nm 409 (ε/M−1 cm−1 170000), 525 (15100), 571 (3200). IR: νmax/cm−1 2959, 2922, 2851 (w, OH), 1721 (s, COacid), 1689 (s, COamide), 1616 (m), 1591 (m), 1515 (m), 1406 (m), 1324 (vs, CF), 1168 (m), 1126 (m), 1109 (m), 1069 (s), 1034 (m), 1017 (s), 842 (vs, PF), 820 (s), 800 (s), 706 (m), 556 (s, PF6,def). NMR: δH (400 MHz, d8-THF) 2.21 (3H, s, H56), 8.08 (2H, d, 3JHH = 8.2 Hz, H53), 8.14 (2H, d, 3JHH = 8.2 Hz, H52), 8.25 (2H, d, 3JHH = 7.5 Hz, H10/202/2), 8.39 (2H, d, 3JHH = 7.1 Hz, H152), 8.53 (2H, d, 3JHH = 7.5 Hz, H10/203/3), 8.53 (2H, d, 3JHH = 7.5 Hz, H153), 9.40 (8H, m, H2,3,7,8,12,13,17,18), 9.41 (1H, s, NH); δC (100 MHz, d8-THF) 24.3 (s, C56), 118.8 (s, C53), 122.9 (s, C5), 123.5 (s, C15), 124.3 (s, C10/20), 125.6 (s, C10/202/2), 127.0 (s, C10/204/4), 129.8 (s, C153), 132.4 (s, C154), 133.1–133.8 (multiple s, C2/3/7/8/12/13/17/18), 135.3 (s, C152), 135.7 (s, C5/5/10/102/3/2/3), 137.7 (s, C51), 137.9–138.7 (multiple s, C1/4/6/9/11/14/16/19), 142.2 (s, C54), 143.9 (s, C10/15/201/1/1), 161.5 (s, C10/204/4), 167.5 (s, C155), 169.2 (s, C55). δP (162 MHz, d8-THF) −143.3 (sept, 1JPF = 711 Hz). MS (ESI): m/z 1045.97 (100%) [M]+. HR-MS (ESI): m/z 1046.1863 (calcd for C49H29AuF6N5O3: 1046.1840). CV (Fc/Fc+, THF): E½/V −2.300, −1.590, −0.990.
Conclusions
Auration of meso-tetraaryl substituted AB2C porphyrins with KAuCl4 in the presence of HOAc/NaOAc cleanly gives the corresponding gold(III) porphyrinato complex cations. Amino-substituted porphyrins are N-acetylated under these conditions and have to be prepared from the corresponding nitro-substituted gold(III) porphyrins by reduction with SnCl2/HCl. The gold(III) complexes can be reduced at least three times. The potentials slightly depend on the electron withdrawing and donating nature of the substituents. The first reduction is addressed by UV/Vis spectroelectrochemistry and by EPR spectroscopy. Upon one-electron reduction, the Soret band experiences a small bathochromic shift. The intensity of the Soret band of the electron rich complexes [2a]+ (R2 = NH2) and [4b]+ (R3 = OnBu) slightly increases upon reduction while all other neutral complexes feature less intense Soret bands as compared to their parent AuIII complexes. These spectral data clearly suggest the presence of an unreduced porphyrinato ligand in all cases under these conditions. Chemical one-electron reduction of the porphyrinato gold(III) hexafluorophosphate salts by cobaltocene yields the corresponding AuII porphyrin complexes with a characteristic EPR pattern displaying hyperfine coupling to 197Au and 14N. The degree of 197Au hfc and g anisotropy places the gold contribution to the spin density in (tetraphenylporphyrinato)gold(II) complexes in between that of [Au(en)2]2+ (ref. 15) and the neutral gold hematoporphyrin IX complex.13e DFT calculations fully support the metal centred reduction in all cases, essentially irrespective of the substituent at the meso aryl groups. Only, the nitro substituent reduction competes significantly with the AuIII reduction and a valence isomeric equilibrium between the AuII valence isomer and the nitro π radical anion valence isomer is established. DFT calculations suggest that the position of the counterion triggers the position of the equilibria between the different valence isomers that interconvert by an intramolecular electron transfer process. These findings allows the usage of meso-substituted AuIII porphyrins as electron acceptors and electron storage materials in photo-induced redox processes, almost irrespective of the substitution pattern. Hence, the substituents fine-tune the redox potential or other properties such as solubility without compromising the thermodynamically preferred metal site of one-electron reduction. Combination of electron-accepting gold(III) porphyrins bearing carboxylic acid, amine and amide substituents, as introduced in this report, with light-harvesting porphyrins and electron donating porphyrins via amide connectivity17 are currently explored in our laboratory and will be reported in due course.Acknowledgements
The authors S. P., A. W. v. L. and K. H. thank the Center of Innovative and Emerging Materials (CINEMA) for financial support. We thank the students Katrin Limberg and Heike Pfaff for preparative assistance.Notes and references
- (a) D. Gust and T. A. Moore, in The Porphyrin Handbook, ed. K. M. Kadish, K. M.Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 8, p. 153 Search PubMed; (b) D. Gust, T. A. Moore and A. L. Moore, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, vol. 3, p. 272 Search PubMed; (c) S. Fukuzumi and H. Imahori, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, vol. 2, p. 927 Search PubMed; (d) S. Fukuzumi and D. M. Guldi, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, vol. 2, p. 270 Search PubMed.
- (a) S. Ishihara, J. Labuta, W. Van Rossom, D. Ishikawa, K. Minami, J. P. Hill and K. Ariga, Phys. Chem. Chem. Phys., 2014, 16, 9713 RSC; (b) N. A. Rakow and K. S. Suslick, Nature, 2000, 406, 710 CrossRef CAS PubMed; (c) N. A. Rakow, A. Sen, M. C. Janzen, J. B. Ponder and K. S. Suslick, Angew. Chem., Int. Ed., 2005, 44, 4528 CrossRef CAS PubMed; (d) S. H. Lim, L. Feng, J. W. Kemling, C. J. Musto and K. S. Suslick, Nat. Chem., 2009, 1, 562 CrossRef CAS PubMed; (e) P. Heier, N. D. Boscher, P. Choquet and K. Heinze, Inorg. Chem., 2014, 53, 11086 CrossRef CAS PubMed; (f) P. Heier, T. Bohn, P. Choquet, N. D. Boscher and K. Heinze, J. Mater. Chem. A, 2014, 2, 1560 RSC; (g) N. D. Boscher, T. Bohn, P. Heier, F. Moisy, B. Untereiner, K. Heinze and P. Choquet, Sens. Actuators, B, 2014, 191, 553 CrossRef CAS PubMed; (h) P. Heier, C. Förster, D. Schollmeyer, N. Boscher, P. Choquet and K. Heinze, Dalton Trans., 2013, 42, 906 RSC.
- (a) M. Ethirajan, Y. Chen, P. Joshia and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340 RSC; (b) E. D. Sternberg, D. Dolphin and C. Brücker, Tetrahedron, 1998, 54, 4151 CrossRef CAS.
- (a) L. He, T. Chen, Y. You, H. Hu, W. Zheng, W.-L. Kwong, T. Zou and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 12532 CAS; (b) C. T. Lum, R. W.-Y. Sun, T. Zou and C.-M. Che, Chem. Sci., 2014, 5, 1579 RSC; (c) C.-M. Che and R. W.-Y. Sun, Chem. Commun., 2011, 47, 9554 RSC; (d) R. W.-Y. Sun, C. K.-L. Li, D.-L. Ma, J. J. Yan, C.-N. Lok, C.-H. Leung, N. Zhu and C.-M. Che, Chem.–Eur. J., 2010, 16, 3097 CrossRef CAS PubMed; (e) R. W.-Y. Sun and C.-M. Che, Coord. Chem. Rev., 2009, 253, 1682 CrossRef CAS PubMed; (f) C.-M. Che, R. W.-Y. Sun, W.-Y. Yu, C.-B. Ko, N. Zhu and H. Sun, Chem. Commun., 2003, 1718 RSC.
- (a) C.-Y. Zhou, P. W. H. Chan and C.-M. Che, Org. Lett., 2006, 8, 325 CrossRef CAS PubMed; (b) A. Nijamudheen, D. Jose and A. Datta, J. Phys. Chem. C, 2011, 115, 2187 CrossRef CAS.
- D. Tanaka, Y. Inuta, M. Sakamoto, A. Furube, M. Haruta, Y.-G. So, K. Kimoto, I. Hamada and T. Teranishi, Chem. Sci., 2014, 5, 2007 RSC.
- (a) E. Göransson, J. Boixel, J. Fortage, D. Jacquemin, H.-C. Becker, E. Blart, L. Hammarström and F. Odobel, Inorg. Chem., 2012, 51, 11500 CrossRef PubMed; (b) K. Ohkubo, R. Garcia, P. J. Sintic, T. Khoury, M. J. Crossley, K. M. Kadish and S. Fukuzumi, Chem.–Eur. J., 2009, 15, 10493 CrossRef CAS PubMed; (c) J. Fortage, A. Scarpaci, L. Viau, Y. Pellegrin, E. Blart, M. Falkenström, L. Hammarström, I. Asselberghs, R. Kellens, W. Libaers, K. Clays, M. P. Eng and F. Odobel, Chem.–Eur. J., 2009, 15, 9058 CrossRef CAS PubMed; (d) J. Fortage, J. Boixel, E. Blart, H. C. Becker and F. Odobel, Inorg. Chem., 2009, 48, 518 CrossRef CAS PubMed; (e) J. Fortage, J. Boixel, E. Blart, L. Hammarström, H. C. Becker and F. Odobel, Chem.–Eur. J., 2008, 14, 3467 CrossRef CAS PubMed; (f) K. Ohkubo, P. J. Sintic, N. V. Tkachenko, H. Lemmetyinen, W. E. Z. Ou, J. Shao, K. M. Kadish, M. J. Crossley and S. Fukuzumi, Chem. Phys., 2006, 326, 3 CrossRef CAS PubMed; (g) S. Fukuzumi, K. Ohkubo, W. E. Z. Ou, J. Shao, K. M. Kadish, J. A. Hutchison, K. P. Ghiggino, P. J. Sintic and M. J. Crossley, J. Am. Chem. Soc., 2003, 125, 14984 CrossRef CAS PubMed; (h) K. Kilså, J. Kajanus, A. N. Macpherson, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2001, 123, 3069 CrossRef PubMed; (i) E. K. L. Yeow, P. J. Sintic, N. M. Cabral, J. N. H. Reek, M. J. Crossley and K. P. Ghiggino, Phys. Chem. Chem. Phys., 2000, 2, 4281 RSC; (j) I. M. Dixon, J.-P. Collin, J.-P. Sauvage, F. Barigelletti and L. Flamigni, Angew. Chem., Int. Ed., 2000, 39, 1292 CrossRef CAS; (k) D. B. Amabilino and J.-P. Sauvage, New J. Chem., 1998, 395 RSC; (l) A. Harriman, F. Odobel and J.-P. Sauvage, J. Am. Chem. Soc., 1995, 117, 9461 CrossRef CAS; (m) V. Heitz, S. Chardon-Noblat and J.-P. Sauvage, Tetrahedron Lett., 1991, 32, 197 CrossRef CAS; (n) A. M. Brun, A. Harriman, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1991, 113, 8657 CrossRef CAS.
- (a) Z. Abou-Gamra, A. Harriman and P. Neta, J. Chem. Soc., Faraday Trans. 2, 1986, 2337 RSC; (b) A. Antipas, D. Dolphin, M. Gouterman and E. C. Johnson, J. Am. Chem. Soc., 1978, 100, 7705 CrossRef CAS; (c) M. E. Jamin and R. T. Iwamoto, Inorg. Chim. Acta, 1978, 27, 135 CrossRef CAS.
- K. M. Kadish, W. E. Z. Ou, J. Shao, P. J. Sintic, K. Ohkubo, S. Fukuzumi and M. J. Crossley, Chem. Commun., 2002, 356 RSC.
- (a) J. H. Waters and H. B. Gray, J. Am. Chem. Soc., 1965, 87, 3534 CrossRef CAS; (b) R. L. Schlupp and A. H. Maki, Inorg. Chem., 1974, 13, 44 CrossRef CAS; (c) A. J. Blake, J. A. Greig, A. J. Holder, T. I. Hyde, A. Taylor and M. Schröder, Angew. Chem., Int. Ed. Engl., 1990, 29, 197 CrossRef PubMed; (d) A. P. Koley, S. Purohit, S. Ghosh, L. S. Prasad and P. T. Manoharan, J. Chem. Soc., Dalton Trans., 1988, 2607 RSC; (e) A. P. Koley, S. Purohit, L. S. Prasad, S. Ghosh and P. T. Manoharan, Inorg. Chem., 1992, 31, 305 CrossRef CAS; (f) A. P. Koley, L. S. Prasad, P. T. Manoharan and S. Ghosh, Inorg. Chim. Acta, 1992, 194, 219 CrossRef CAS; (g) L. Ihlo, M. Kampf, R. Böttcher and R. Kirmse, Z. Naturforsch., B: J. Chem. Sci., 2002, 57, 171 CAS; (h) M. Kampf, J. Griebel and R. Kirmse, Z. Anorg. Allg. Chem., 2004, 630, 2669 CrossRef CAS PubMed; (i) M. Kampf, R.-M. Olk and R. Kirmse, Z. Anorg. Allg. Chem., 2002, 628, 34 CrossRef CAS.
- F. G. Herring, G. Hwang, K. C. Lee, F. Mistry, P. S. Phillips, H. Willner and F. Aubke, J. Am. Chem. Soc., 1992, 114, 1271 CrossRef CAS.
- S. Seidel and K. Seppelt, Science, 2000, 290, 117 CrossRef CAS.
- (a) Z. Ou, W. Zhu, Y. Fang, P. J. Sintic, T. Khoury, M. J. Crossley and K. M. Kadish, Inorg. Chem., 2011, 50, 12802 CrossRef CAS PubMed; (b) A. Klein, in Spectroelectrochemistry, ed. W. Kaim and A. Klein, RSC publishing, 2008, p. 91 Search PubMed; (c) Z. Ou, K. M. Kadish, W. E. J. Shao, P. J. Sintic, K. Ohkubo, S. Fukuzumi and M. J. Crossley, Inorg. Chem., 2004, 43, 2078 CrossRef CAS PubMed; (d) S. Fukuzumi, K. Ohkubo, W. E. Z. Ou, J. Shao, K. M. Kadish, J. A. Hutchison, K. P. Ghiggino, P. J. Sintic and M. J. Crossley, J. Am. Chem. Soc., 2003, 125, 14984 CrossRef CAS PubMed; (e) G. Gencheva, D. Tsekova, G. Gochev, D. Mehandjiev and P. R. Bontchev, Inorg. Chem. Commun., 2003, 6, 325 CrossRef CAS.
- (a) F. Mohr, S. Sanz, E. Vergara, E. Cerrada and M. Laguna, Gold Bull., 2006, 39, 212 CrossRef CAS; (b) F. Mohr, S. Sanz, E. R. T. Tiekink and M. Laguna, Organometallics, 2006, 25, 3084 CrossRef CAS; (c) E. Cerrada, M. Laguna, L. A. Mendez and F. Mohr, J. Am. Chem. Soc., 2005, 127, 852 CrossRef PubMed.
- Z. Qu, L. Giurgiub and E. Roduner, Chem. Commun., 2006, 2507 RSC.
- R. Timkovich and A. Tulinsky, Inorg. Chem., 1977, 4, 962 CrossRef.
- (a) J. Melomedov, J. R. Ochsmann, M. Meister, F. Laquai and K. Heinze, Eur. J. Inorg. Chem., 2014, 2902 CrossRef CAS PubMed; (b) J. Melomedov, J. R. Ochsmann, M. Meister, F. Laquai and K. Heinze, Eur. J. Inorg. Chem., 2014, 1984 CrossRef CAS PubMed; (c) J. Melomedov, A. Wünsche von Leupoldt, M. Meister, F. Laquai and K. Heinze, Dalton Trans., 2013, 42, 9727 RSC; (d) K. Heinze and A. Reinhart, Dalton Trans., 2008, 469 RSC.
- E. B. Fleischer and A. Laszlo, Inorg. Nucl. Chem. Lett., 1969, 5, 373 CrossRef CAS.
- (a) J.-C. Chambron, V. Heitz and J.-P. Sauvage, New J. Chem., 1997, 21, 237 CAS; (b) N. Solladié, J.-C. Chambron and J.-P. Sauvage, J. Am. Chem. Soc., 1999, 121, 3684 CrossRef.
- (a) V. W.-W. Yam and E. C.-C. Cheng, Top. Curr. Chem., 2007, 281, 269–309 CrossRef CAS; (b) M. P. Eng, T. Ljungdahl, J. Andréasson, J. Mårtensson and B. Albinsson, J. Phys. Chem. A, 2005, 109, 1776–1784 CrossRef CAS PubMed.
- J. Heinze, Angew. Chem., Int. Ed., 1984, 23, 831 CrossRef PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- (a) D. H. Geske and A. H. Maki, J. Am. Chem. Soc., 1969, 82, 2671 CrossRef; (b) D. H. Geske, J. L. Ragle, M. A. Bambenek and A. L. Balch, J. Am. Chem. Soc., 1964, 86, 987 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision A.02), Gaussian, Inc., Wallingford CT, 2009.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and IR spectra, spectral changes upon reduction of [Au(TPP)][PF6], [4a][PF6], [4b][PF6], [4c][PF6], EPR spectra of 1a in the absence and presence of chloride, DFT calculations of Au(TPP), Cu(TPP), 4a, 4b and 4c, Cartesian coordinates of all optimised structures. See DOI: 10.1039/c5sc03429a |
| This journal is © The Royal Society of Chemistry 2016 |