DOI:
10.1039/C5SC03391H
(Edge Article)
Chem. Sci., 2016,
7, 650-654
Synthesis of open-shell ladder π-systems by catalytic C–H annulation of diarylacetylenes†
Received
9th September 2015
, Accepted 20th October 2015
First published on 22nd October 2015
Abstract
A new open-shell ladder-shaped π-system has been synthesized. Pentaleno[1,2-b:4,5-b′]difluorene derivatives, 8 fused ring systems bearing 5- and 6-membered rings, were constructed from alkynylfluorenone through a reaction sequence including Pd-catalyzed C–H/C–H annulation. X-ray crystallography and ESR spectroscopy revealed the open-shell character of these ladder-shaped molecules, which derives from their extended π-electron conjugation. Absorption peaks in the near IR region as well as narrow redox potentials observed by cyclic voltammetry indicated small optical and fundamental energy gaps of these fused ring systems.
Introduction
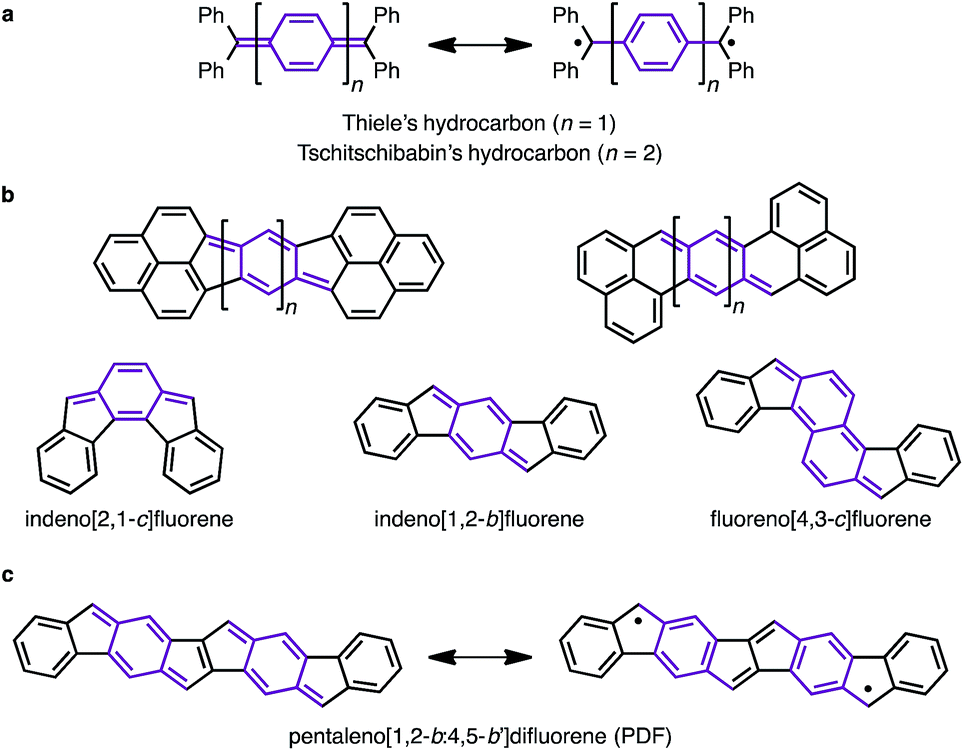
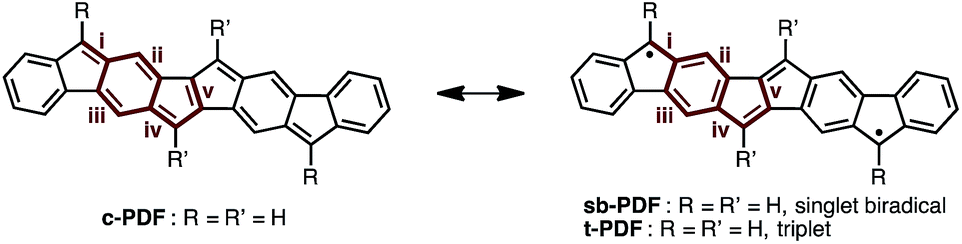
Since the discovery of Thiele's and Tschitschibabin's hydrocarbons (Fig. 1a),1 organic biradical compounds (open-shell π-systems) have attracted great interest from chemists, and more recently from materials scientists.2 Theoretical and experimental studies have revealed unique properties, such as a low energy gap, stimuli-responsive spin structure, and singlet fission.2–4 Despite their practical utility, however, high chemical reactivity and short lifetimes are common concerns in radical chemistry and in device applications. Among the various types of fused ring systems, linearly extended polycyclic structures bearing p-quinodimethane moieties (Fig. 1b) are found in either open-shell (phenalenyls) or closed-shell (indenyl) carbon-based π-systems depending upon the nature of the ring fusion.3,4 Although significant progress, such as C–H activation reactions,5 has been made toward the synthesis of novel polycyclic structures, the construction of extended carbon-based π-systems still represents a challenge.5b To push the chemistry of open-shell conjugated hydrocarbons forward, more powerful synthetic methods and a novel structural design are needed. Herein, we report the facile synthesis of a novel carbon-based π-system having 8 fused rings, pentaleno[1,2-b:4,5-b′]difluorene (PDF), which has possible resonance structures of closed shell and diradical as shown in Fig. 1c. Aryl-substituted PDFs were synthesized from simple alkynylfluorenone by applying our recently reported C–H activation reaction.6 The NMR, ESR, absorption, and CV measurements of PDF revealed their open-shell character with narrow HOMO–LUMO energy gaps.
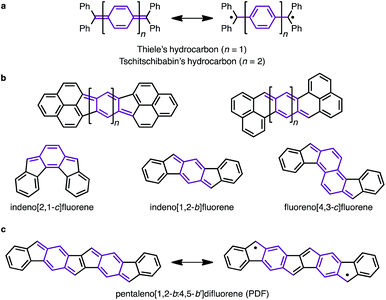 |
| | Fig. 1 (a) Thiele's and Tschitschibabin's hydrocarbons. (b) Representative polycyclic conjugated hydrocarbons containing p-quinodimethane moieties. (c) PDF (this work). p-Quinodimethane moieties are highlighted in purple. | |
Results and discussion
DFT study of PDF
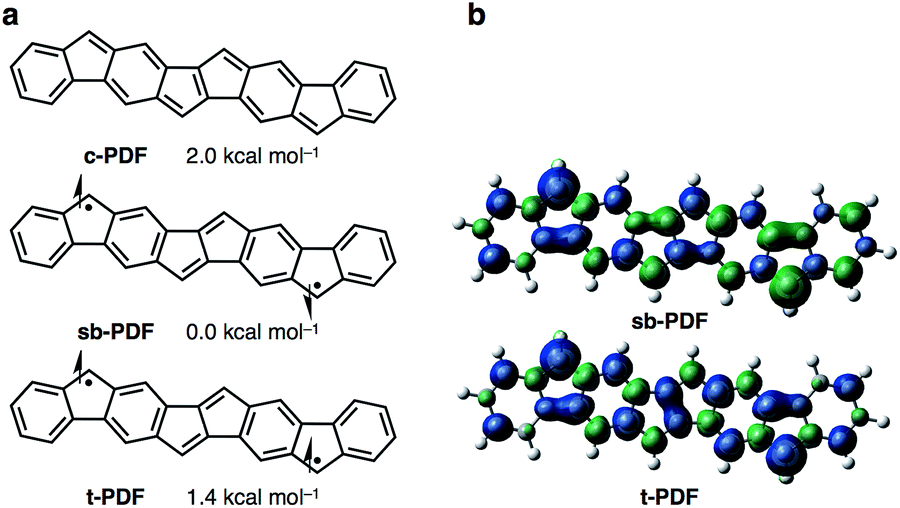
We began by performing a density functional theory (DFT) study on the electronic states of neutral PDF in order to estimate the physical properties of the target compounds. The structures of unsubstituted PDF was optimized at the (U)B3LYP/6-311+G(d,p) level. Fig. 2a shows the possible electronic states of PDF. Three electronic states, closed-shell (c-PDF), open-shell singlet biradical (sb-PDF), and triplet (t-PDF), were found. The relative energy values of the three electronic states indicated sb-PDF as the most favorable at the ground state, whereas c-PDF and t-PDF are slightly higher in energy (2.0 and 1.4 kcal mol−1, respectively).6 The biradical character (y) of sb-PDF estimated from the natural orbital occupancy number (NOON)7 of the lowest unoccupied natural orbital (LUNO) by B3LYP/6-311+G(d,p) method is 55%. In the open-shell structures, sb-PDF and t-PDF, spin densities are delocalized throughout the molecules and the highest densities were found at the C9-position of the fluorene moieties, as illustrated in Fig. 2b. Based on these theoretical studies, PDF is expected to exhibit open-shell character, and PDF may show singlet–triplet transition.
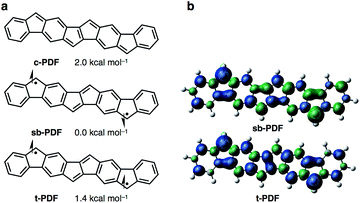 |
| | Fig. 2 (a) Electronic states of PDF with energy values relative to the most stable states. (b) Spin density distributions. Blue and green surfaces represent α and β spin density, respectively. Isovalue is 0.003. Calculated at the (U)B3LYP/6-311+G(d,p) level. | |
Synthesis of Ar4PDF (1a, b)
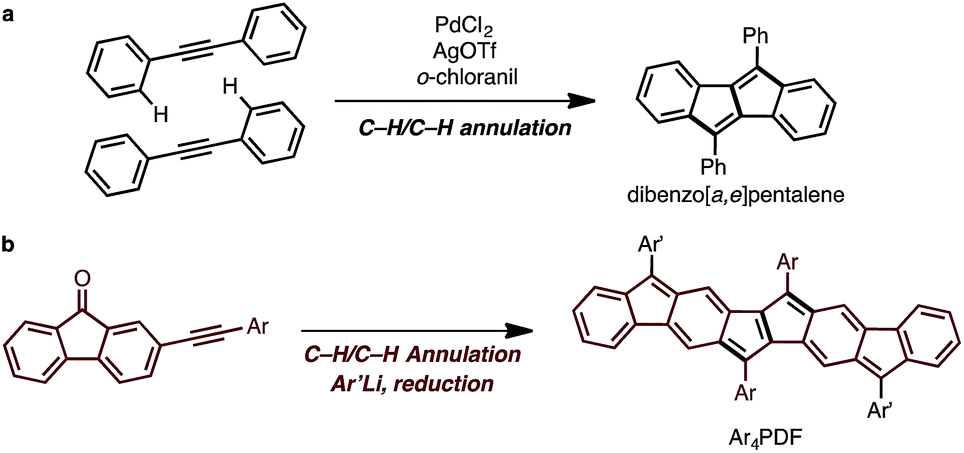
The synthetic strategy for Ar4PDF is shown in Fig. 3. We have previously developed a Pd-catalyzed oxidative C–H/C–H annulation of diarylacetylenes8 to form dibenzo[a,e]pentalene frameworks in the presence of PdCl2/AgOTf/o-chloranil9 (Fig. 3a). We thus envisaged that elongated ladder-shaped frameworks could be easily constructed from alkynylfluorenones using the C–H/C–H annulation reaction (Fig. 3b). The thus-formed pentalene-fused fluorenones could then be converted into Ar4PDF by sequential nucleophilic addition of an aryllithium and reductive dehydroxylation, a commonly used method for the synthesis of quinoidal hydrocarbons.4
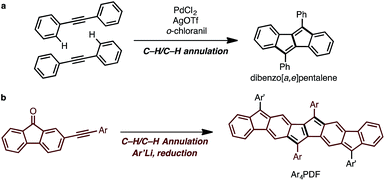 |
| | Fig. 3 (a) Pd-catalyzed oxidative C–H/C–H annulation for the synthesis of dibenzo[a,e]pentalene. (b) Strategy for the synthesis of Ar4PDF. | |
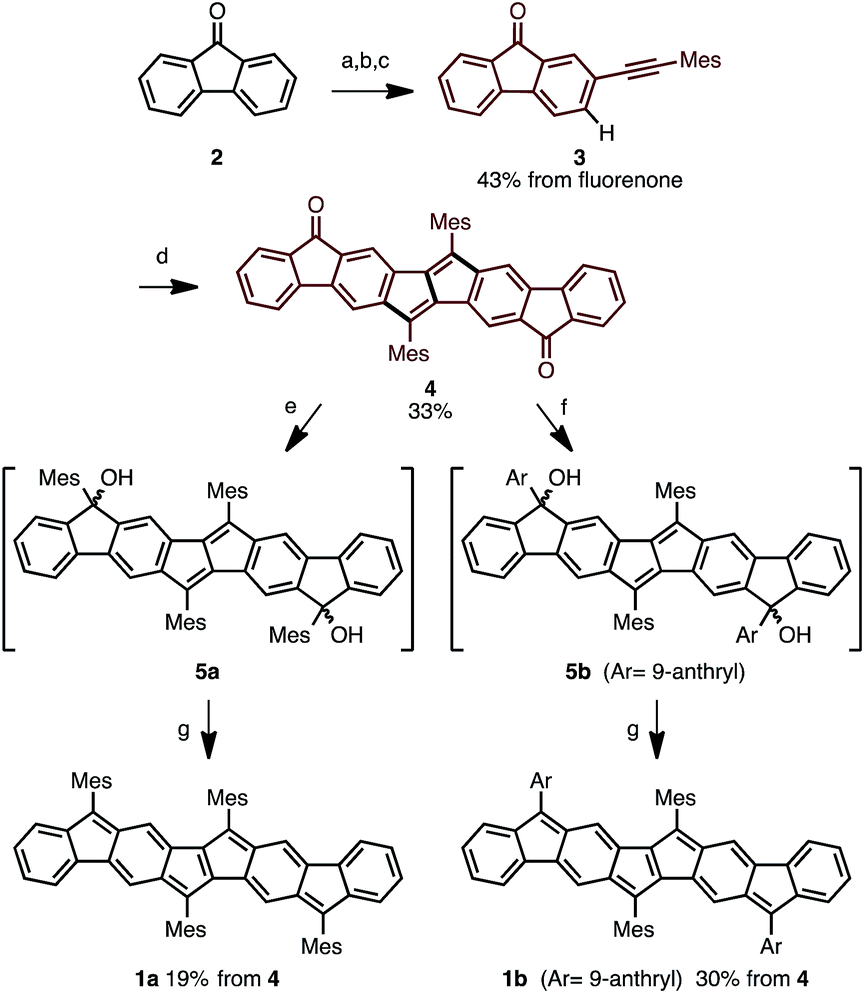
Following this strategy, Ar4PDF (1a and 1b) were prepared in six steps from fluorenone (2), as shown in Scheme 1. To begin with, 2-(mesitylethynyl)fluorenone (3), which can be obtained by mono-bromination of fluorenone10 and subsequent Sonogashira cross-coupling,11 was subjected to Pd-catalyzed C–H/C–H annulation. The mesityl (2,4,6-trimethylphenyl) group was chosen for two reasons: (i) an aryl group with no ortho-C–H bond is required for the selective cleavage of fluorenone C–H bond; and (ii) the bulky mesityl group can stabilize the reactive pentalene moiety. Gratifyingly, the C–H cleavage proceeded selectively at the C3-position of 3 to afford dione 4 in 33% yield as a brown solid. Treatment of 4 with mesityllithium or 9-anthryllithium successfully gave diols 5a and 5b, respectively, as a mixture of diastereomers. After removal of unreacted starting materials, the diols 5 were mixed with excess SnCl2 at room temperature to produce Ar4PDFs 1a and 1b bearing the [6-5-6-5-5-6-5-6] fused ring systems. The products were obtained as deep green solids regardless of the aryllithium reagent used. It should be noted that 1a showed high durability against air exposure for several months in solid state, whereas 1b decomposed slowly under air. The quinoidal molecules 1a and 1b could even be isolated by silica-gel column chromatography in air. The m/z values and isotope patterns in the high-resolution mass spectra of these products were fully consistent with the calculated values.
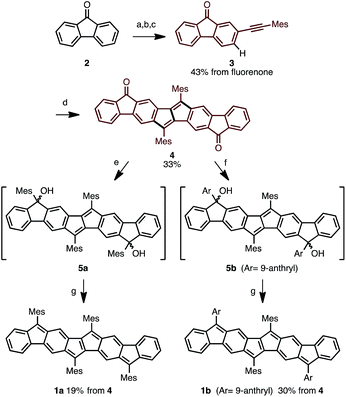 |
| | Scheme 1 Synthesis of ladder-shaped open-shell molecules 1a, b. Reaction conditions: (a) Br2, H2O, 80 °C, 4 h; (b) Me3SiCCH, PdCl2(PPh3)2, CuI, THF, NEt3, reflux, 9 h; (c) MesI, PdCl2(PPh3)2, CuI, H2O, DBU, benzene, 80 °C, 6 h; (d) PdCl2, o-chloranil, AgOTf, DMAc, 80 °C, 18 h; (e) MesLi, THF, −78 °C, 1.5 h; (f) 9-anthrylLi, THF, −78 °C, 1.5 h; (g) SnCl2, toluene, r.t., 4 h. Mes = 2,4,6-trimethylphenyl, DMAc = N,N-dimethylacetamide. | |
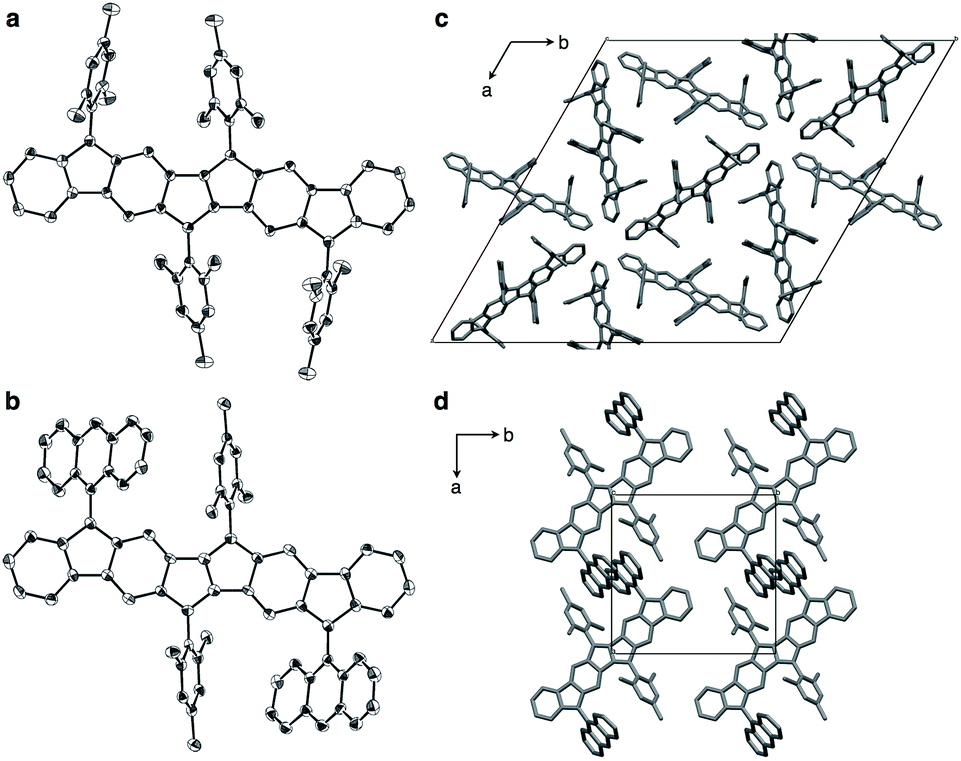
X-ray structures of 1a and 1b
Compounds 1a and 1b could be crystallized from CS2/Et2O and their exact molecular structures were unambiguously confirmed by X-ray diffraction (Fig. 4). Dihedral angles between PDF planes and the outer aryl groups on the fluorene moieties were 74.8° (1a) and 57.3° (1b), respectively, reflecting the bulkiness of the aryl groups. In the packing structures of 1a and 1b, no intermolecular π–π stacking of the PDF core was found. The 9-anthryl moieties of 1b were stacked intermolecularly as shown in Fig. 4d.
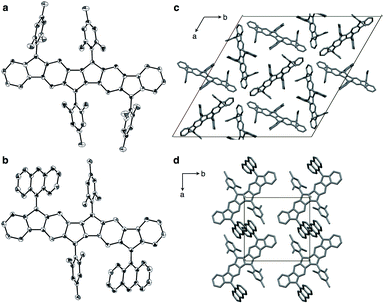 |
| | Fig. 4 ORTEP drawings of 1a (a) and 1b (b) with 50% probability (all hydrogen atoms and solvent molecules are omitted for clarity; half of the entire structure constitutes an asymmetric unit), and packing structure of 1a (c) and 1b (d) along c axis. | |
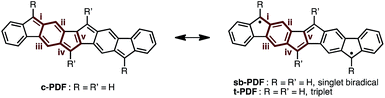
Table 1 summarizes the bond lengths of 1a and 1b, and unsubstituted PDFs optimized at the B3LYP/6-311+G(d,p) level of theory. Despite different aryl groups (mesityl or 9-anthryl) being attached to the fluorene moieties, no significant difference was found in the bond lengths and angles of the PDF moiety in 1a and 1b, besides a small difference in bond i. Bonds i–v are double bonds in the closed-shell structure (c-PDF), whereas, in the open-shell structures (sb-PDF and t-PDF), three bonds (i, iv, and v) are single bonds and two bonds (ii and iii) are conjugated bonds (bond order ∼ 1.5). The small difference in the bond lengths between 1a and 1b indicated that the effect of the packing modes were negligible in this case. Clearly, the five bonds of sb-PDF and t-PDF are longer than those of c-PDF, especially i, iv, and v. In all bonds, little differences are observed between sb-PDF and t-PDF. The bond lengths of 1a and 1b observed by X-ray crystallography at 103 K were between those of open-shell (sb-PDF and t-PDF) and closed-shell (c-PDF), and rather close to the open-shell structures (see also Table S2 in ESI† for detail). We concluded that the PDF core structure of 1a and 1b is a hybrid of the closed shell and open shell forms in the solid state.
Table 1 Selected bond lengths of PDFs 1a, b, and calculated analogues (Å)
|
|
|
|
1a
|
1b
|
c-PDF
|
sb-PDF
|
t-PDF
|
|
Optimized at the (U)B3LYP/6-311+G(d,p) level.
|
| i |
1.401(3) |
1.416(6) |
1.388 |
1.410 |
1.410 |
| ii |
1.363(3) |
1.364(6) |
1.370 |
1.378 |
1.381 |
| iii |
1.372(3) |
1.374(7) |
1.374 |
1.388 |
1.390 |
| iv |
1.424(3) |
1.420(6) |
1.407 |
1.445 |
1.457 |
| v |
1.407(5) |
1.415(9) |
1.417 |
1.453 |
1.476 |
NMR, ESR, and SQUID measurement of 1a
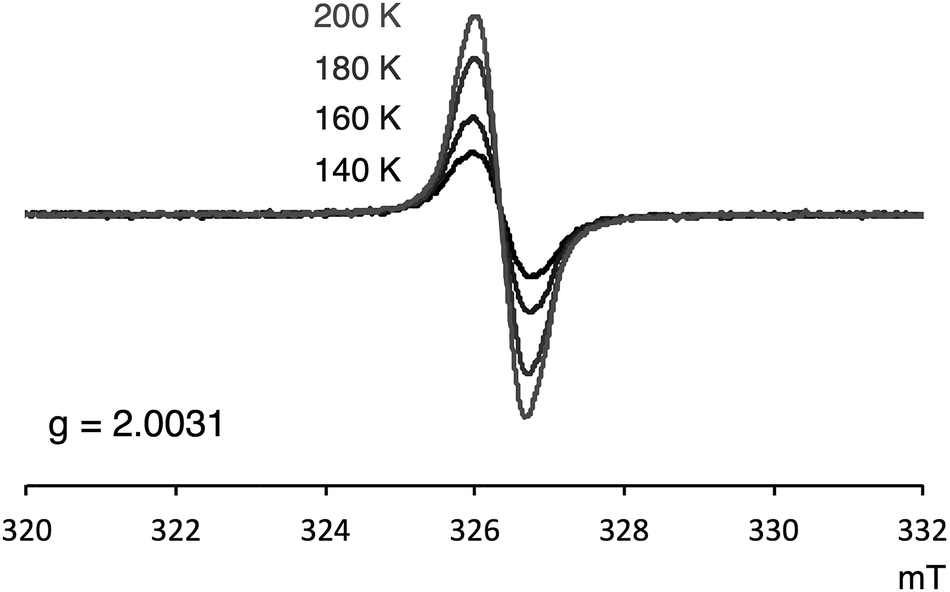
Several experimental measurements of 1a and 1b strongly implicated their open-shell character. For example, broad signals in the 1H NMR spectra were observed, which clearly suggests the paramagnetic character of these compounds. Only three singlets, which can be assigned to the mesityl group,12 were observed in the 1H NMR spectra of both 1a and 1b. Even at −90 °C, the 1H NMR signals of 1a were still broad (slightly sharper) thus indicating appreciable triplet character (see Fig. S4 in ESI†). ESR was also measured to confirm the open-shell properties of PDF. As shown in Fig. 5, the powder solid of 1a exhibited ESR signals with no obvious spin–spin coupling probably due to the long spin–spin distances and no intermolecular stacking. The g values of 1a was 2.0031, which are in the range of typical carboradical species.2 The singlet ground state of 1a was indicated by VT ESR and preliminary SQUID measurements. The ESR signal of 1a decreased as temperature decreased (Fig. 5), and the temperature dependence of magnetization obtained from SQUID measurement also supported singlet–triplet transition of 1a. From the result of SQUID measurement, singlet–triplet energy gap (ΔES–T) could be estimated to 3.4 kcal mol−1 (see ESI† for detail).
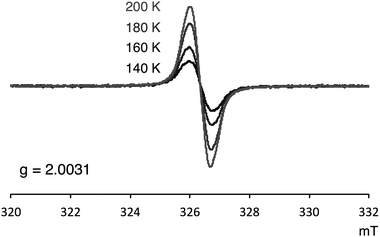 |
| | Fig. 5 VT ESR spectra of 1a in solid state. | |
UV-vis-near-IR absorption spectra and CV measurement
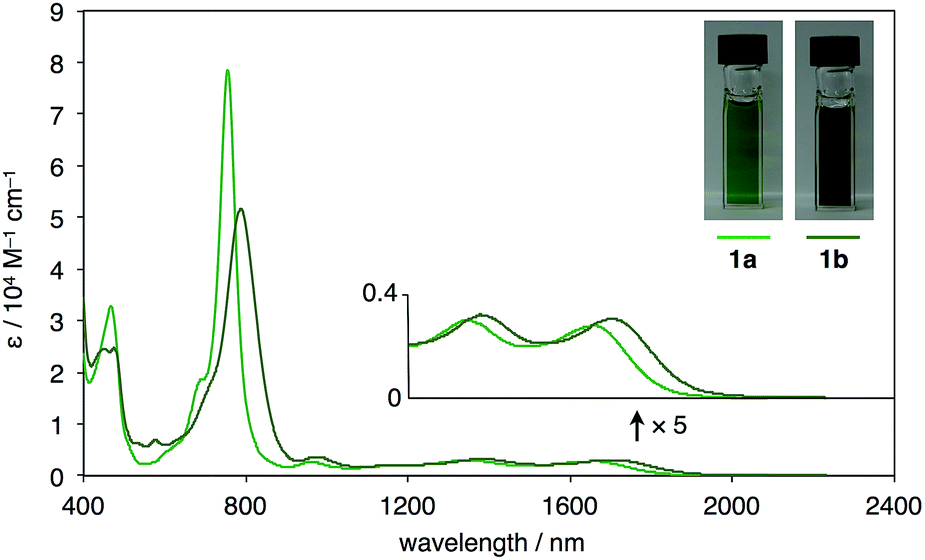
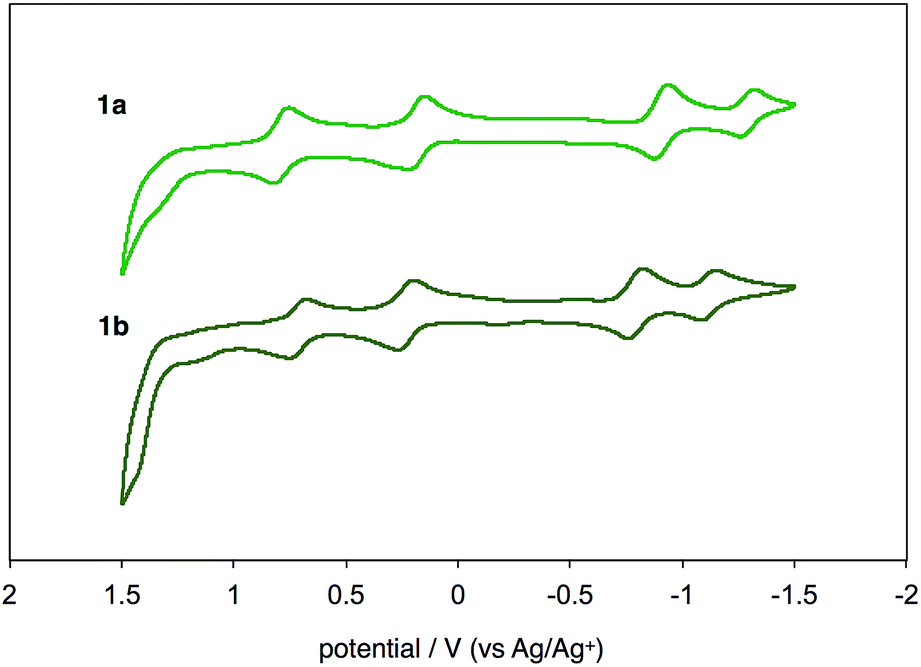
UV-vis-near-IR absorption spectra as well as cyclic voltammogram (CV) proved small energy level gaps derived from open-shell states. Compounds 1a and 1b exhibit their highest absorption peaks centered at 756 and 786 nm, respectively, along with three bands in the near-IR region (Fig. 6). The lowest energy absorption bands at 1652 nm (1a) and 1702 nm (1b) can be seen. Cyclic voltammetry of 1a and 1b were also measured (Fig. 7). Two oxidation steps and two reduction steps were clearly observed in the CV chart of 1a and 1b. From CV results, fundamental energy gaps can be roughly estimated to 0.75 eV (1a) and 0.73 eV (1b), which are very close to the values obtained from the near-IR absorption peaks.
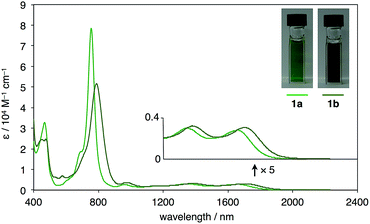 |
| | Fig. 6 UV-vis-NIR absorption spectra and photographs of CS2 solution (ca. 1 × 10−4 M) of 1a and 1b. | |
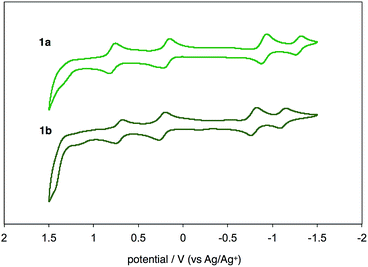 |
| | Fig. 7 Cyclic voltammograms of 1a and 1b in CH2Cl2 with n-Bu4N+(CF3SO2)2N− as supporting electrolyte, and scan rate of 0.1 V s−1. | |
Conclusions
We have achieved the rapid synthesis of linearly elongated, hexagon/pentagon-fused π-systems (pentaleno[1,2-b:4,5-b′]difluorene derivatives) by Pd-catalyzed C–H/C–H annulation. The synthesized molecules exhibited open-shell properties with small energy gap, which were confirmed by ESR spectra, NIR absorption, and CV. Although there is still room for improvement in the efficiency of key reactions, the successful rapid access to previously untapped molecular structures speaks well for the significant opportunities of C–H activation reactions in the chemistry of organic radical materials. Theoretical studies as well as further experimental investigations of the obtained open-shell π-systems are ongoing in our laboratory.
Acknowledgements
This work was supported by the ERATO program from JST (K. I.), the Funding Program for Next Generation World-Leading Researchers from JSPS (K. I.) and the National Science Foundation (CHE-1301485 to M. M. H.). T. M. is a recipient of JSPS fellowship for young scientists. We thank Prof. Kunio Awaga and Prof. Michio M. Matsushita (Nagoya Univ.) for the measurement of SQUID, and Dr Bradley D. Rose (Univ. of Oregon) for fruitful discussions. ITbM is supported by the World Premier International Research Center (WPI) Initiative, Japan.
Notes and references
-
(a) J. Thiele and H. Balhorn, Chem. Ber., 1904, 37, 1463 CrossRef;
(b) A. E. Tschitschibabin, Chem. Ber., 1907, 40, 1810 CrossRef CAS.
-
(a) Z. Sun and J. Wu, J. Mater. Chem., 2012, 22, 4151 RSC;
(b) Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857 RSC;
(c) M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed;
(d) Z. Sun, Z. Zeng and J. Wu, Chem.–Asian J., 2013, 8, 2894 CrossRef CAS PubMed;
(e) T. Kubo, Chem. Lett., 2015, 44, 111 CrossRef.
-
(a) K. Ohashi, T. Kubo, K. Masui, K. Yamamoto, K. Nakatsuji, T. Takui, Y. Kai and I. Murata, J. Am. Chem. Soc., 1998, 120, 2018 CrossRef CAS;
(b) T. Kubo, A. Shimizu, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Morita and K. Nakatsuji, Org. Lett., 2007, 9, 81 CrossRef CAS PubMed;
(c) Y. Li, W.-K. Heng, B. S. Lee, N. Aratani, J. L. Zafra, N. Bao, R. Lee, Y. M. Sung, Z. Sun, K.-W. Huang, R. D. Webster, J. T. López Navarrete, D.-H. Kim, A. Osuka, J. Casado, J. Ding and J. Wu, J. Am. Chem. Soc., 2012, 134, 14913 CrossRef CAS PubMed;
(d) S. Das, S. Lee, M. Son, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, Z. Sun, W. Zeng, R. Li, K. Huang, J. Ding, D. Kim and J. Wu, Chem.–Eur. J., 2014, 20, 11410 CrossRef CAS PubMed.
-
(a) D. T. Chase, A. G. Fix, B. D. Rose, C. D. Weber, S. Nobusue, C. E. Stockwell, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 11103 CrossRef CAS PubMed;
(b) D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127 CrossRef CAS PubMed;
(c) A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906 CrossRef CAS PubMed;
(d) D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349 CrossRef CAS PubMed;
(e) B. D. Rose, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Org. Lett., 2012, 14, 2426 CrossRef CAS PubMed;
(f) A. G. Fix, P. E. Deal, C. L. Vonnegut, B. D. Rose, L. N. Zakharov and M. M. Haley, Org. Lett., 2013, 15, 1362 CrossRef CAS PubMed.
-
(a) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed;
(b) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS PubMed.
- We also calculated by (U)CAM-B3LYP/6-311+G(d,p) and (U)M06-2X/6-311+G(d,p). See ESI† for detail.
- D. Döhnert and J. Koutecký, J. Am. Chem. Soc., 1980, 102, 1789 CrossRef.
- T. Maekawa, Y. Segawa and K. Itami, Chem. Sci., 2013, 4, 2369 RSC.
-
(a) K. Mochida, K. Kawasumi, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2011, 133, 10716 CrossRef CAS PubMed;
(b) K. Kawasumi, K. Mochida, T. Kajino, Y. Segawa and K. Itami, Org. Lett., 2012, 14, 418 CrossRef CAS PubMed;
(c) Q. Zhang, K. Kawasumi, Y. Segawa, K. Itami and L. T. Scott, J. Am. Chem. Soc., 2012, 134, 15664 CrossRef CAS PubMed;
(d) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739 CrossRef CAS PubMed;
(e) K. Kawasumi, K. Mochida, Y. Segawa and K. Itami, Tetrahedron, 2013, 69, 4371 CrossRef CAS;
(f) K. Ozaki, K. Kawasumi, M. Shibata, H. Ito and K. Itami, Nat. Commun., 2015, 6, 6251 CrossRef CAS PubMed.
- X. Zhang, J. Han, P.-F. Li, X. Ji and Z. Zhang, Synth. Commun., 2009, 3804 CrossRef CAS.
-
(a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef;
(b) M. J. Mio, L. C. Kopel, J. B. Braun, T. L. Gadzikwa, K. L. Hull, R. G. Brisbois, C. J. Markworth and P. A. Grieco, Org. Lett., 2002, 4, 3199 CrossRef CAS PubMed.
- Peak assignment was supported by calculation of the 1H NMR chemical shift of 1a by using GIAO B3LYP/6-311+G(2d,p)//B3LYP/6-31G(d) level of theory. See ESI† for detail.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses, NMR, UV-vis-nearIR absorption, CV and crystallographic table. CCDC 1421865 (1a), 1421866 (1b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03391h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a