
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advanced 1,1-carboboration reactions with pentafluorophenylboranes
Gerald
Kehr
and
Gerhard
Erker
Organisch-Chemisches Institut, Universität Münster, Corrensstraße 40, D-48149 Münster, Germany. E-mail: erker@uni-muenster.de
First published on 8th October 2015
Abstract
The 1,1 carboboration reaction of a variety of metal-substituted alkynes with simple trialkylboranes R3B yields the respective alkenylboranes (Wrackmeyer reaction). The use of the strongly electrophilic R-B(C6F5)2 reagents allows for much milder reaction conditions and gives good yields of the respective bulky alkenylboranes from conventional terminal alkynes by means of 1,2-hydride migration. Even internal alkynes undergo 1,1-carboboration with the R-B(C6F5)2 reagents, in this case yielding alkenylboranes by means of C–C bond cleavage. Phosphorus, sulfur or even boron containing substituents can serve as the migrating alkynyl substituents in the advanced 1,1-carboboration reactions using the R-B(C6F5)2 reagents. Sequential 1,1-carboboration of geminal bis(alkynyl) derivatives of these elements with the R-B(C6F5)2 boranes yields boryl substituted phospholes, thiophenes or even boroles in quite a variety. Vicinal bis(alkynyl)arenes or heteroarene substrates undergo benzannulation reactions in this way. Many of the -B(C6F5)2 substituted 1,1-carboboration products can be used as reagents in cross coupling reactions. A recently disclosed organometallic analogue, namely a 1,1-carbozirconation reaction is described.
 Gerald Kehr | Gerald Kehr studied Chemistry at the University of Bayreuth and obtained his doctoral degree under the guidance of Prof. Bernd Wrackmeyer. After his postdoctoral stay with Prof. Jacques Livage in the group of Prof. Clement Sanchez at the University “Pierre et Marie Curie” (Paris, France) and Prof. Rudolph Willem at the Free University of Brussels (Belgium), he became a senior researcher at the Organisch-Chemisches Institut at Münster. His current research interests are frustrated Lewis pair and carboboration chemistry. Dr Gerald Kehr was fellow of the “Studienstiftung des deutschen Volkes” and is an author in more than 320 publications and patents. |
 Gerhard Erker | Gerhard Erker studied Chemistry in Cologne. He received his doctoral degree in 1973 from the Universität Bochum. After a postdoctoral stay at Princeton University, his habilitation at Bochum and a stay as a Heisenberg-fellow at the Max-Planck-Institut für Kohlenforschung in Mülheim he became a Professor at the Universität Würzburg in 1985. Since 1990 he was a full Professor at the WWU Münster. He served as the President of the German Chemical Society in 2000/1 and as a Member of the Senate of the Deutsche Forschungsgemeinschaft (2002–2008). He is a member of several Academies, and he has received a number of awards. He has a keen interest in organometallic chemistry and catalysis and recently an increasing interest in main group element chemistry, including 1,1-carboboration and frustrated Lewis pair chemistry. Since 2015 Gerhard Erker is a Senior Professor at the WWU Münster. |
Introduction: the Wrackmeyer reaction
In the mid-1960s Binger and Köster described the reaction of alkynylborate anions 1 with electrophiles, e.g. R2BCl, to give unusual alkenylboranes 2, that were formed by a reaction pathway involving 1,2-alkyl migration from the borate boron atom to the acetylenic α-carbon atom (see Scheme 1).1 The analogous reactions with R32PCl reagents led to phosphanyl substituted alkenylboranes 3.2 Since the alkynylborates had been prepared by alkynyl anion addition to the respective boranes, these reactions can be looked at as first, although quite specific examples of 1,1-carboboration reactions.3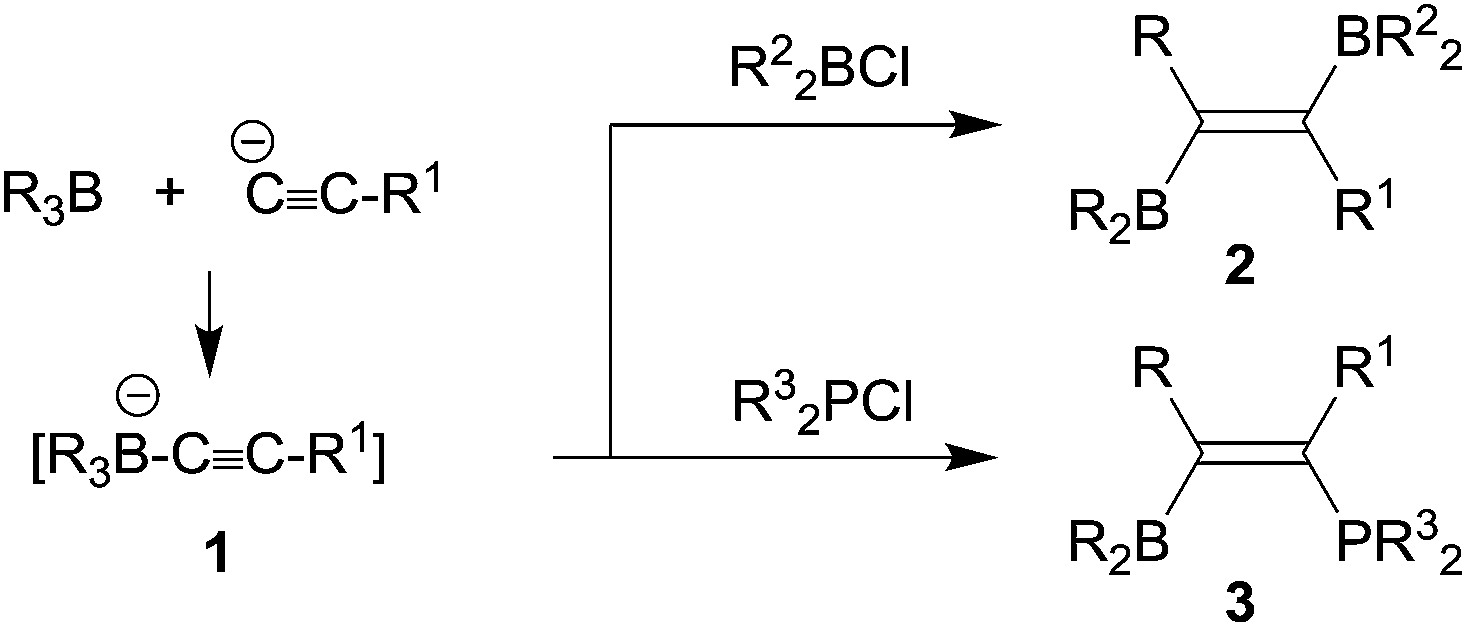 | ||
| Scheme 1 | ||
Wrackmeyer et al. developed this into a very useful neutral 1,1-carboboration variant (the “Wrackmeyer reaction”).4 They had found that e.g. trimethylstannyl-acetylenes 4 reacted rapidly with e.g. triethylborane to give the tetra-substituted alkenylboranes 5 (see Scheme 2).5 They found out that this reaction required a good metal containing migrating group at the alkyne substrate and showed that R3Ge- and R3Pb- (and some transition metal derived groups) were even better.4,6 Many examples using silyl substituents were used, although in these cases more forcing reaction conditions were often necessary.7 Intermediate metal/alkyne π-complexes were identified as reactive intermediates and even in a few cases characterized by X-ray diffraction.4,8 Subsequent developments carried out by the Wrackmeyer group involved silole9 and stannole syntheses8c,d,10,11 starting from the respective geminal bis(alkynyl) metal derivatives. A recent highlight of this development was the straightforward preparation of fused polycyclic silole systems such as compound 9 reported by Wrackmeyer et al.12
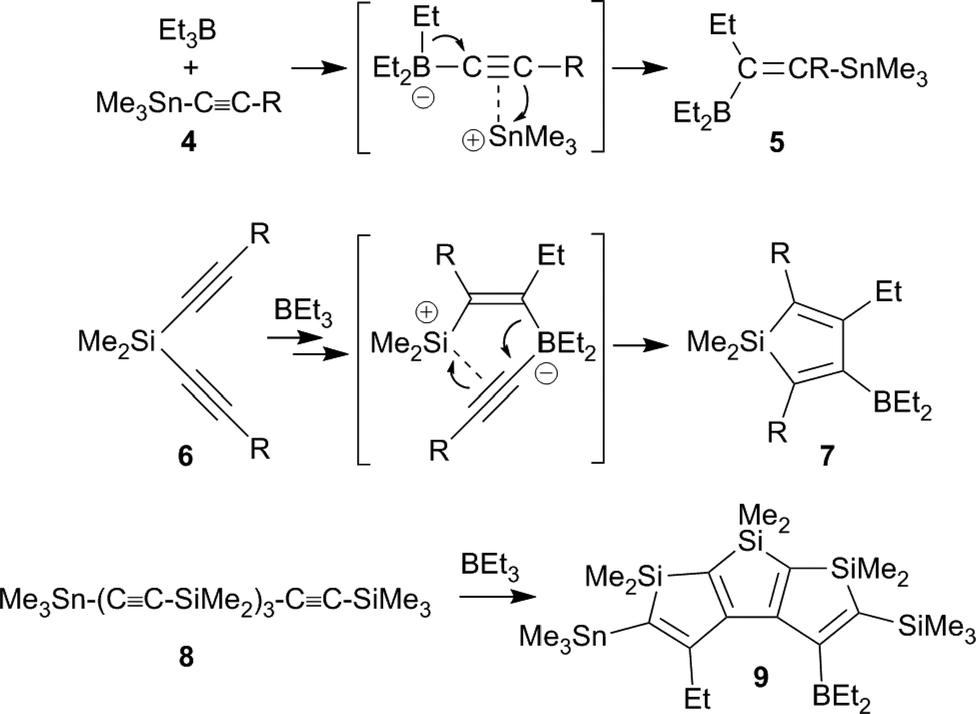 | ||
| Scheme 2 | ||
Paetzold et al. had described a few early experiments showing that 1,1-carboboration reactions could be accomplished in the absence of good metal containing migrating groups, although those reactions required high temperatures and the use of more strongly electrophilic dihalogenoboranes. A typical example is the formation of the alkenylborane 10 from benzyl-BBr2 with tert-butylacetylene (in this case H is migrating along the alkynyl framework) at 150 °C to give 10 (see Scheme 3). The products were actually identified by their oxidative deborylation products.13
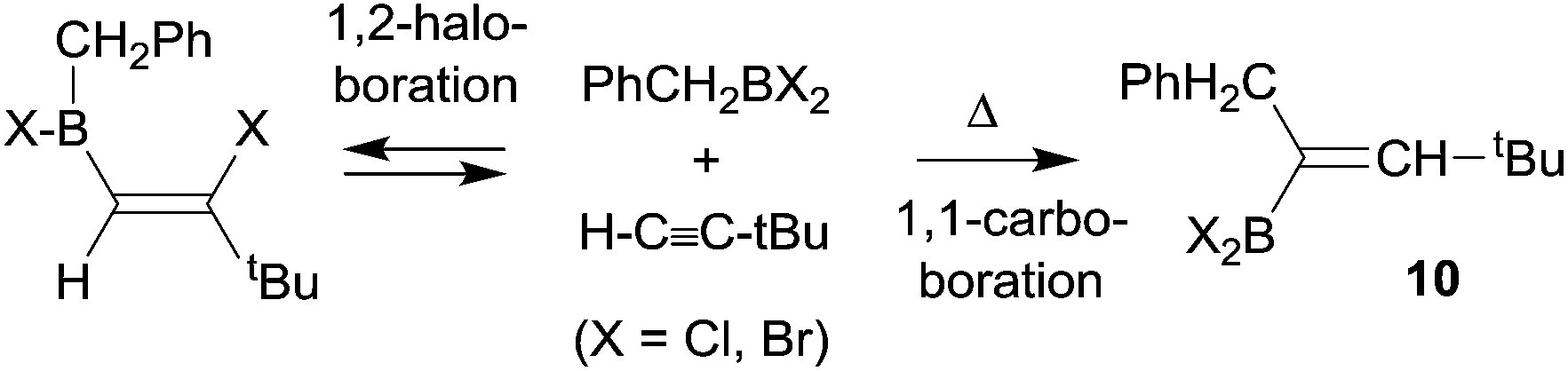 | ||
| Scheme 3 | ||
The advent of 1,1-carboboration with R-B(C6F5)2 reagents
Paetzold's examples had indicated that the use of more Lewis acidic boranes might be useful for the development of 1,1-carboboration chemistry. This was realized by turning to B(C6F5)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 and the related R-B(C6F5)2 reagents which in turn were often readily available by hydroboration routes using Piers' borane [HB(C6F5)2].15 We had found that the terminal alkyne 1-pentyne underwent selective 1,1-carboboration with the bis(pentafluorophenyl) substituted metallocene system 11 to give a E-/Z-mixture of the product 12 (see Scheme 4).16 The substituted alkyl group at boron migrated selectively in this process. We also reacted 1-pentyne with B(C6F5)3 under mild conditions and an E-/Z-mixture of the 1,1-carboboration products 13 was formed by C6F5 migration. In both examples subsequent E- to Z-alkenylborane isomerization was achieved by photolysis.
14 and the related R-B(C6F5)2 reagents which in turn were often readily available by hydroboration routes using Piers' borane [HB(C6F5)2].15 We had found that the terminal alkyne 1-pentyne underwent selective 1,1-carboboration with the bis(pentafluorophenyl) substituted metallocene system 11 to give a E-/Z-mixture of the product 12 (see Scheme 4).16 The substituted alkyl group at boron migrated selectively in this process. We also reacted 1-pentyne with B(C6F5)3 under mild conditions and an E-/Z-mixture of the 1,1-carboboration products 13 was formed by C6F5 migration. In both examples subsequent E- to Z-alkenylborane isomerization was achieved by photolysis.
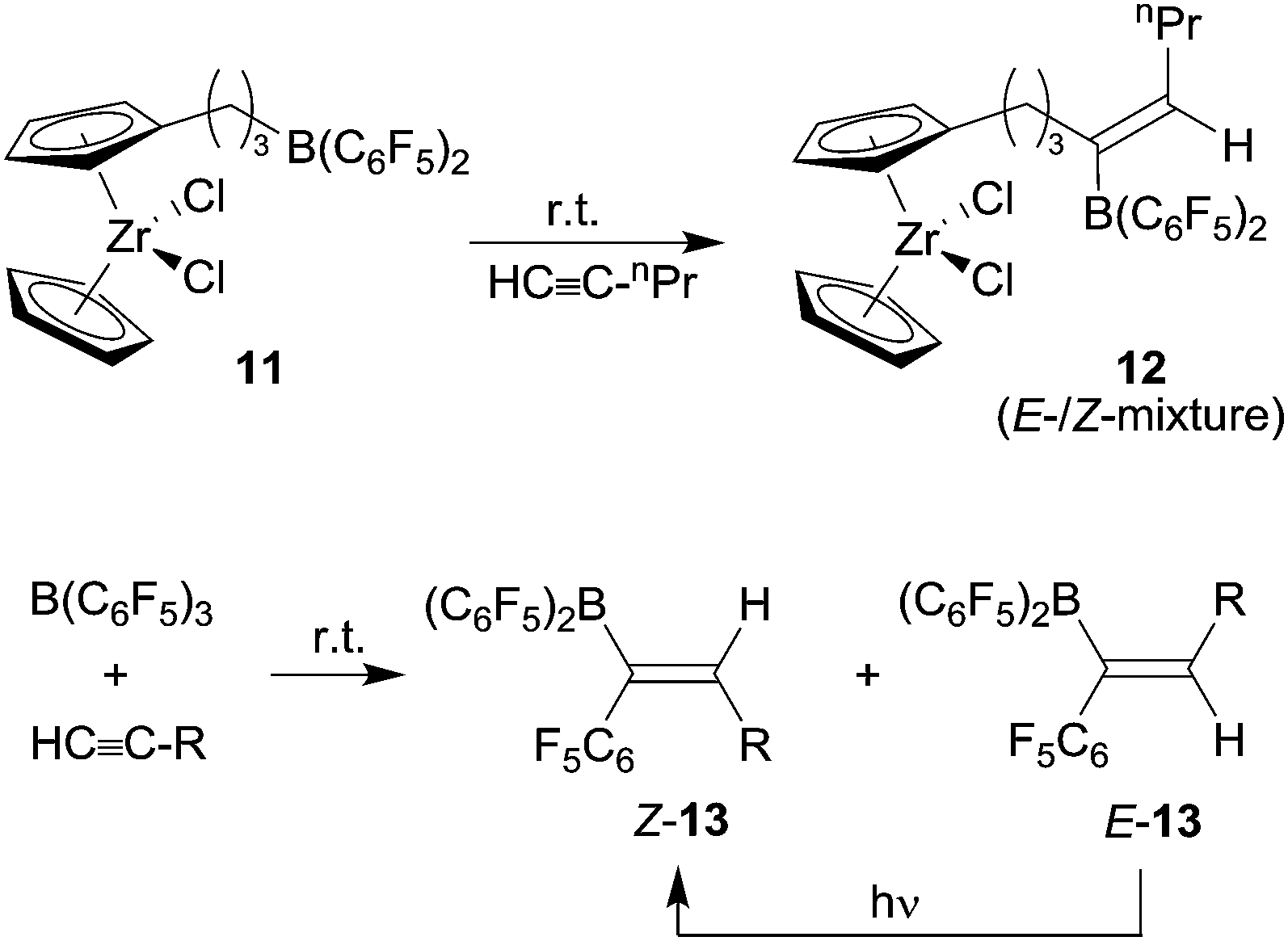 | ||
| Scheme 4 | ||
At about the same time Berke et al. reported the 1,1-carboboration reaction of phenylacetylene with B(C6F5)3 to give the E-/Z-mixture of the products 13 (R: Ph) (see Scheme 4).17 From these early examples we subsequently developed the 1,1-carboboration reaction of alkynes with R-B(C6F5)2 reagents into a viable alternative to the ubiquitous hydroboration route to alkenylboranes.18 We used the products as reagents in cross-coupling reactions18 as well as borane Lewis acid components in catalytic metal-free frustrated Lewis pair hydrogenation reactions.19 Most of this and related work has been reviewed by us,20 so this will not be repeated here.
Under slightly more forcing conditions the 1,1-carboboration reaction could even be used for C–C bond activation reactions. Cleavage of unactivated carbon–carbon bonds is still relatively rare, it is mostly achieved using metal complex initiation or catalysis.21 1,1-Carboboration in some cases allows to achieve a metal-free cleavage of non-activated sp-carbon-sp2 (or sp3)-carbon-σ-bonds. The reaction of 4-octyne with B(C6F5)3 is a typical example: at 110 °C the reaction proceeds cleanly by C–C bond cleavage and 1,2-migration of the n-propyl group along the alkynyl framework to give 14 (see Scheme 5). Compound 14 was subsequently converted by a Pd-catalyzed cross coupling reaction to the boron free product 15.22 In a way this 1,1-carboboration reaction resembles the reverse of a Fritsch–Buttenberg–Wiechel-rearrangement, although the reaction mechanisms of these two reactions are not related.23 Piers et al. provided nice examples of 1,1-carboboration reactions at reactive C6F5-substituted borole frameworks with tolane that also proceeded with C–C bond cleavage (see Scheme 5).24
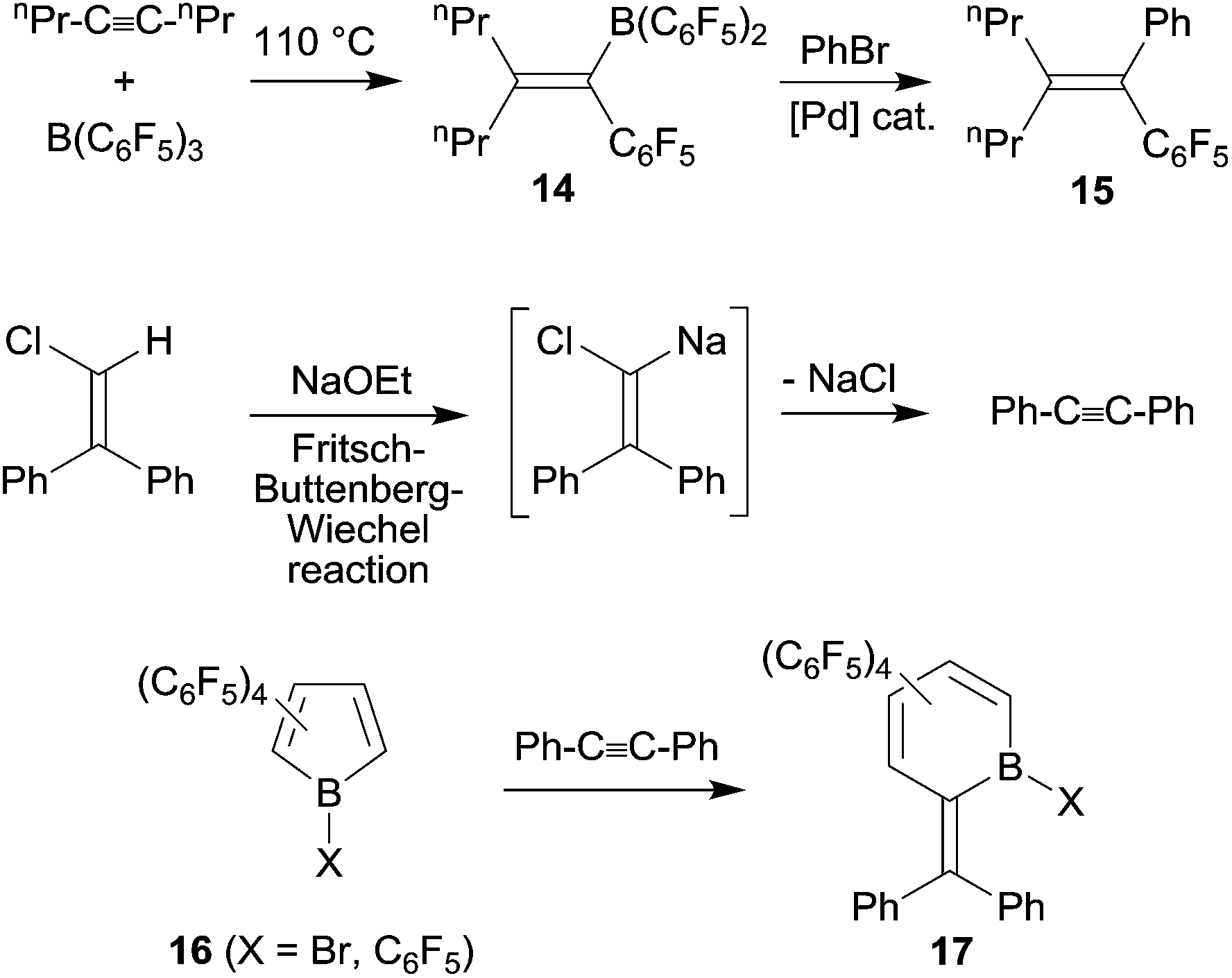 | ||
| Scheme 5 | ||
A strength of the original Wrackmeyer reaction had been the formation of unsaturated cyclic compounds (e.g. siloles, see Scheme 2) from di- and oligoacetylene precursors. This has been introduced into the reaction schemes of the B(C6F5)2 based “advanced 1,1-carboboration” reaction protocols, which has resulted in a variety of remarkable developments. Some of these will be briefly highlighted in the following chapters of this perspective.
Benzannulation
o-Bis(alkynyl)benzenes have been used as the starting materials for the synthesis of naphthalene derivates by a sequence of consecutive 1,1-carboboration reactions. This application of the advanced carboboration reaction contributes an interesting addition to the existing repertoire of benzannulation reactions of 1,2-bis(alkynyl)benzenes.25 We have shown that the readily available systems 18 reacted smoothly with a small series of R–B(C6F5)2 reagents to form the naphthalenes 20, probably in a two-step reaction sequence proceeding via the intermediate 19 (see Scheme 6). The B(C6F5)2 substituent of product 20 was subsequently utilized for cross-coupling reactions and the trimethylsilyl-substituents could conveniently be removed.26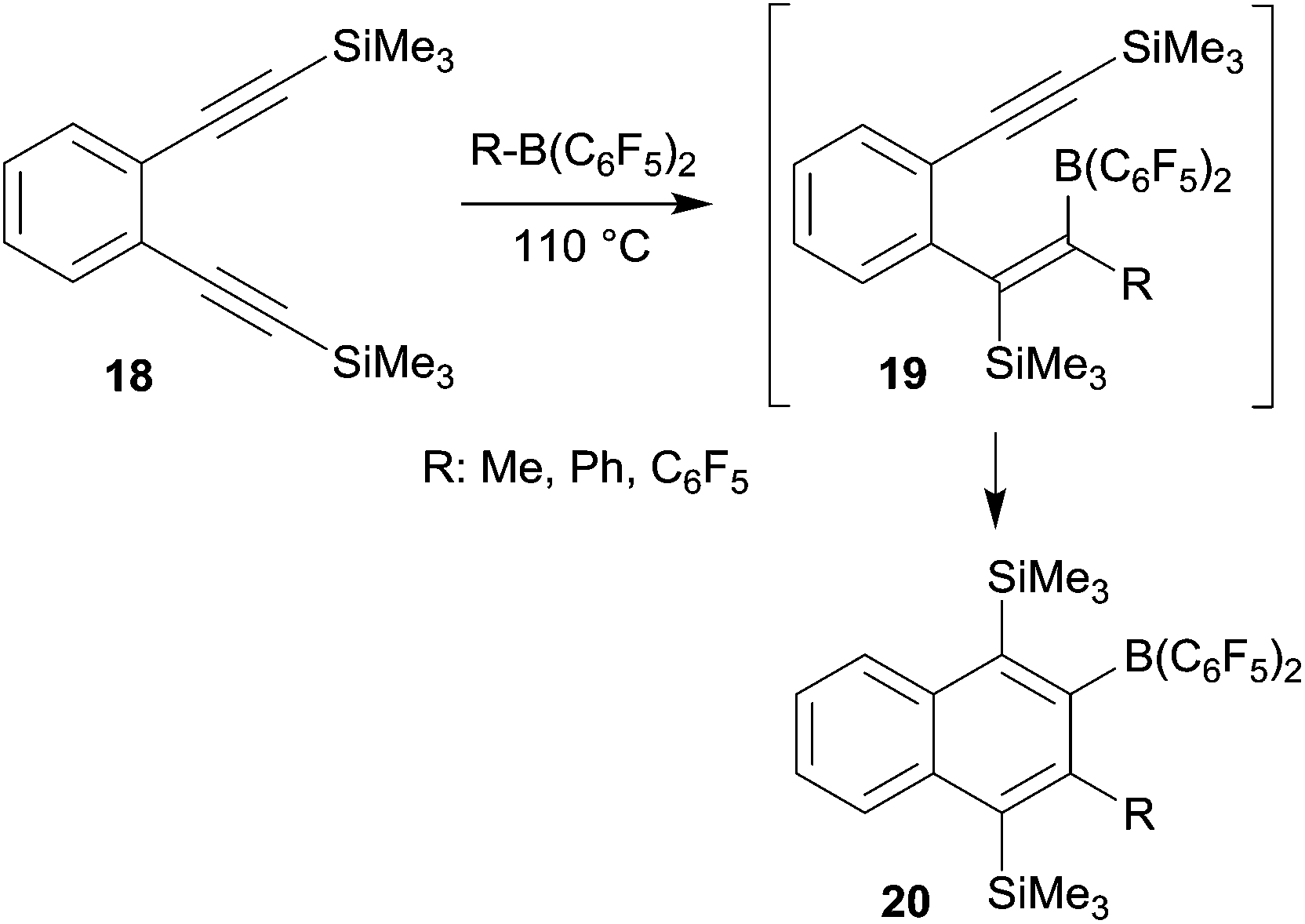 | ||
| Scheme 6 | ||
The reaction of o-bis(Mes2P-ethynyl)benzene (21) with B(C6F5)3 gave a different product type 22. In this case we assumed that a competing pathway was favoured which represents the seldomly observed carbocation alternative to the 1,1-carboboration reaction (see Scheme 7).27 It may be that the ensuing formation of a B–P interaction favours this alternative pathway.
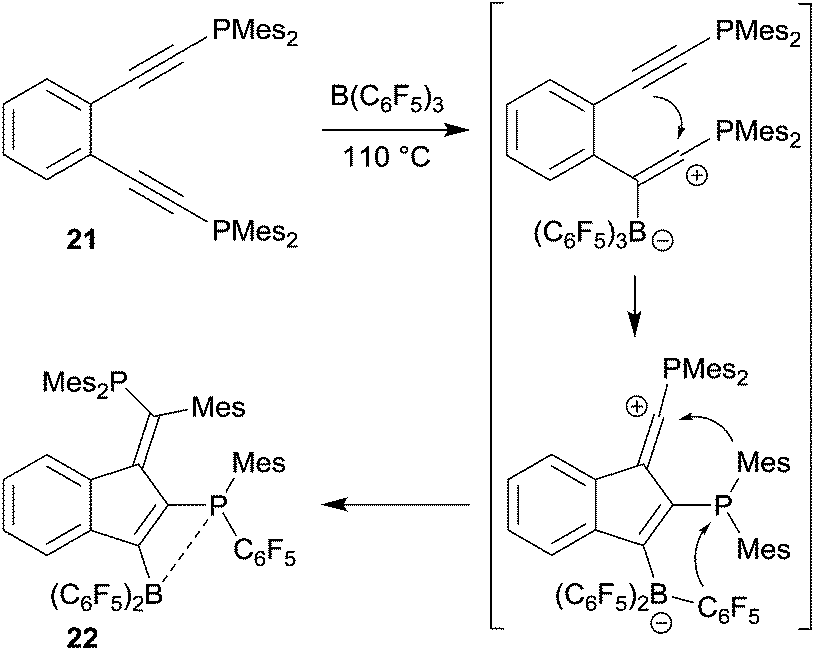 | ||
| Scheme 7 | ||
Benzannulated heterocycles are often prepared by forming the heterocyclic ring at the already present benzenoid aromatic ring. 1,1-Carboboration reactions allow for an alternative strategy. This could be shown in a variety of examples using the B(C6F5)3 borane reagent (see Scheme 8).28 Its reaction with the indole derivate 23 initially gave only a conventional single 1,1-carboboration product 24 at the distal alkynyl moiety. But this was apparently reversible to eventually cleanly yield the respective carbazole derivative 25 under thermodynamic control. In a similar way, the benzothiophenes 27a,b and a functionalized quinoline were prepared by 1,1-carboboration of the respective 2,3-bis(alkynyl) heteroarenes.
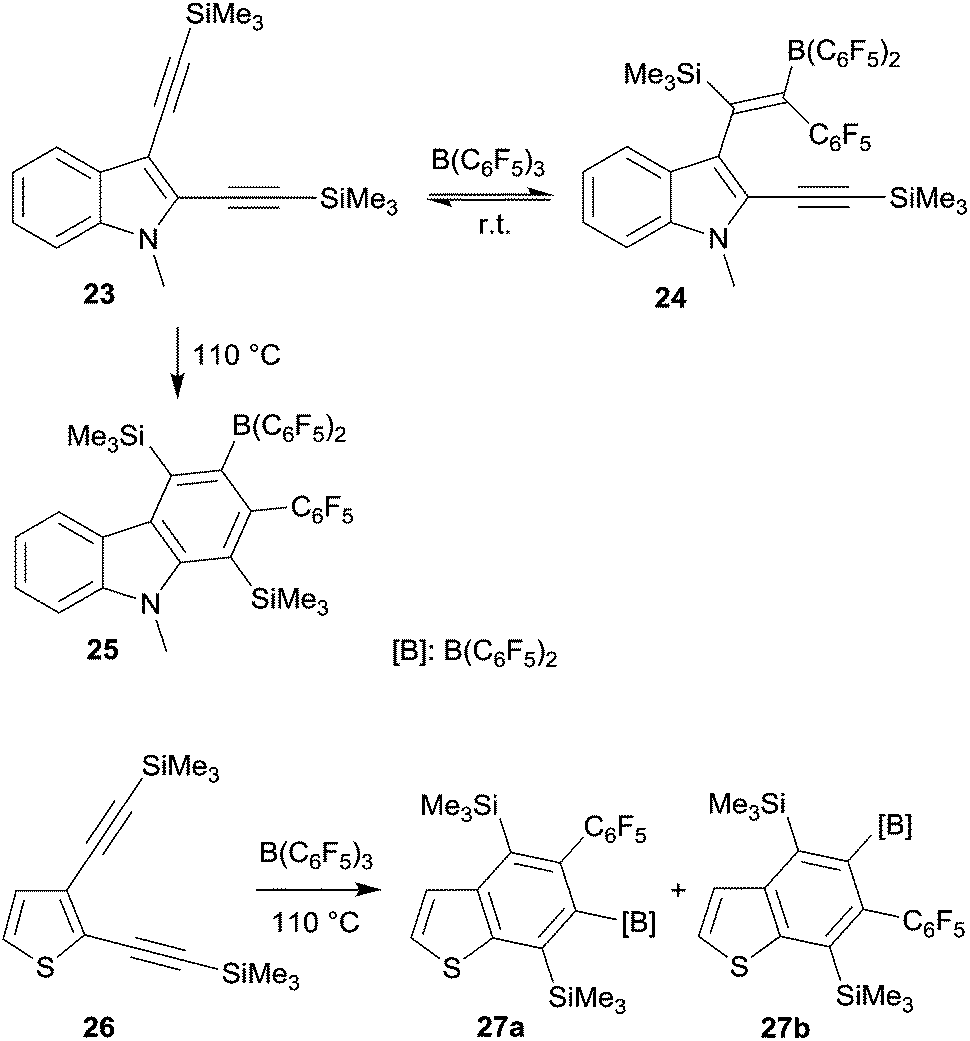 | ||
| Scheme 8 | ||
Five-membered heterocycles by 1,1-carboboration
We have shown that thiolate substituents can serve as suitable migrating groups in 1,1-carboboration reactions (see Scheme 9 for typical examples).29 This effect was used to develop a sequential 1,1-carboboration route to a boryl substituted thiophene derivative. The product 30 was deborylated by treatment with acetic acid. It also served as a cross-coupling reagent for the synthesis e.g. of a substituted bithiophene derivative 31. Stephan et al. showed that the closely related bis(alkynyl) tellurium compound 32 underwent a consecutive series of 1,1-carboboration reactions with e.g. B(C6F5)3 to form an example of less often observed six-membered heterocyclic ring systems (33) (see Scheme 10).10,30 A dimeric product was formed in a reversible competitive pathway.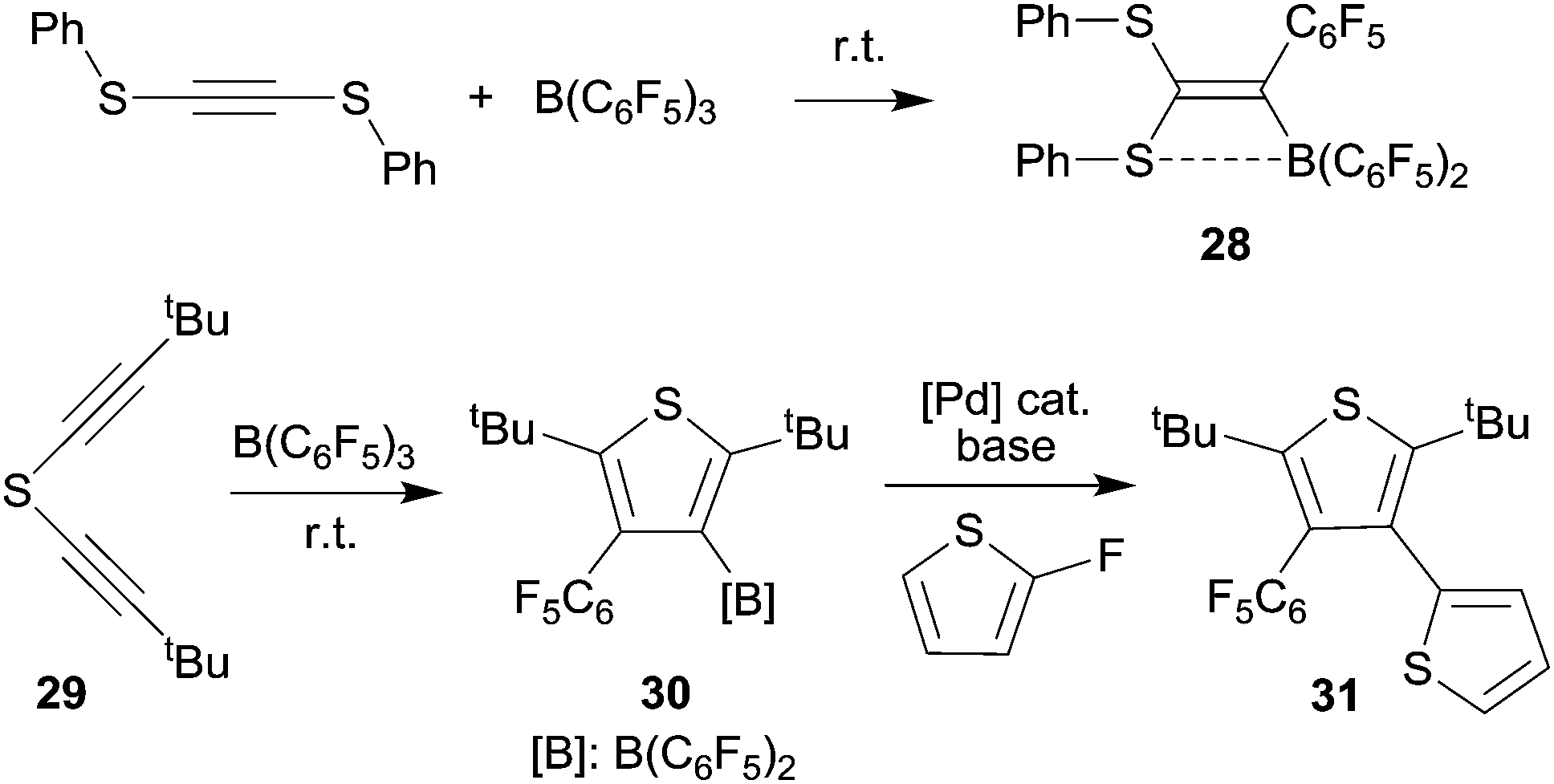 | ||
| Scheme 9 | ||
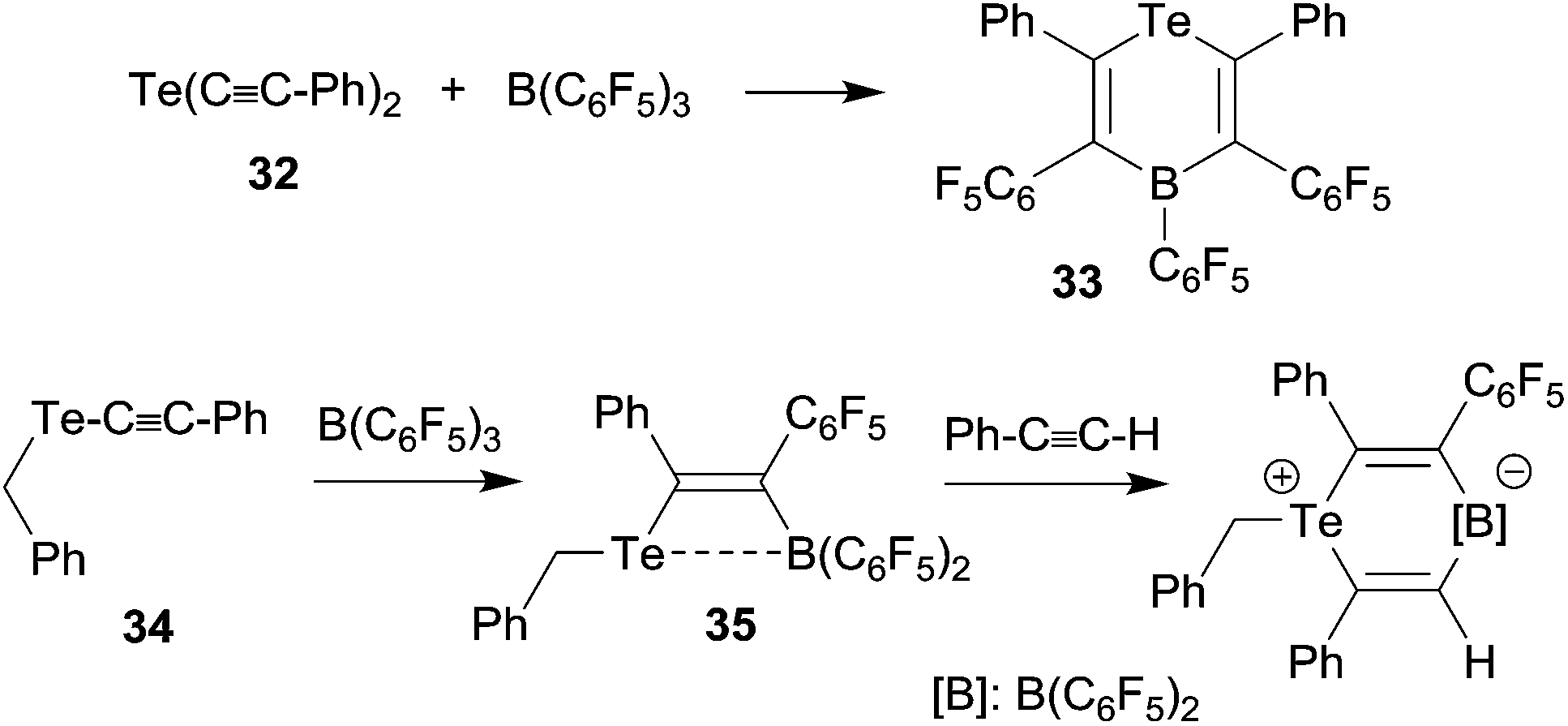 | ||
| Scheme 10 | ||
The benzyl alkynyl telluride 34 undergoes 1,1-carboboration with B(C6F5)3 to form the Te/B frustrated Lewis pair (FLP) 35 that e.g. undergoes 1,2-addition to phenylacetylene (see Scheme 10).31
Phosphanyl-substituted alkynes 36 underwent facile 1,1-carboboration reactions with the strongly Lewis acidic R-B(C6F5)2 reagents (see Scheme 11). There is evidence that the reactions proceed via zwitterionic phosphirenium borate intermediates 37, some of which were isolated under suitable reaction conditions and characterized by their very typical high field 31P NMR signals and by X-ray diffraction.32,33 The resulting P⋯B interacting Lewis pairs 3832–34 were rather unreactive, but several of them underwent a remarkable cooperative 1,1-addition reaction to isonitriles to give compounds 39,34 a reaction of these bifunctional main group element compounds that is reminiscent of the coordination behaviour typical of transition metal complexes.35
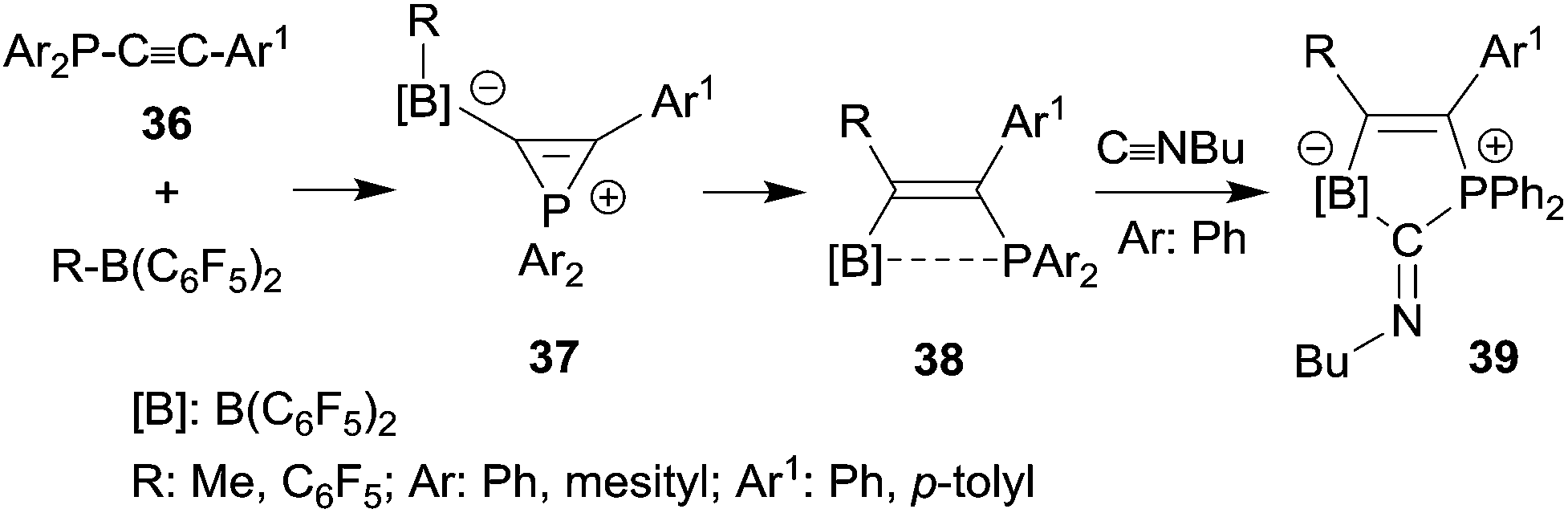 | ||
| Scheme 11 | ||
Phosphinoalkynes even undergo clean 1,1-carboboration reactions with alkenylboranes, provided their substitution pattern is not too sterically constrained. Thus, the trans-tert butylethenylborane 40, prepared by hydroboration of tert butyl acetylene with Piers' borane, underwent a clean 1,1-carboboration reaction with the diarylphosphinoalkyne 36a (Ar: mesityl, Ar1: Ph) to give the P/B functionalized conjugated diene product 41 (see Scheme 12).36 This reaction scheme can be extended: typically, the alkenylborane 40 reacted analogously with a variety of diarylphosphinoenynes (e.g.42) to give the respective hexatriene derivative 43 by 1,1 carboboration. Compound 43 eventually underwent electrocyclic ring closure upon heating to generate 44 which subsequently formed the product 45 by means of an intramolecular nucleophilic aromatic substitution reaction (see Scheme 12).37
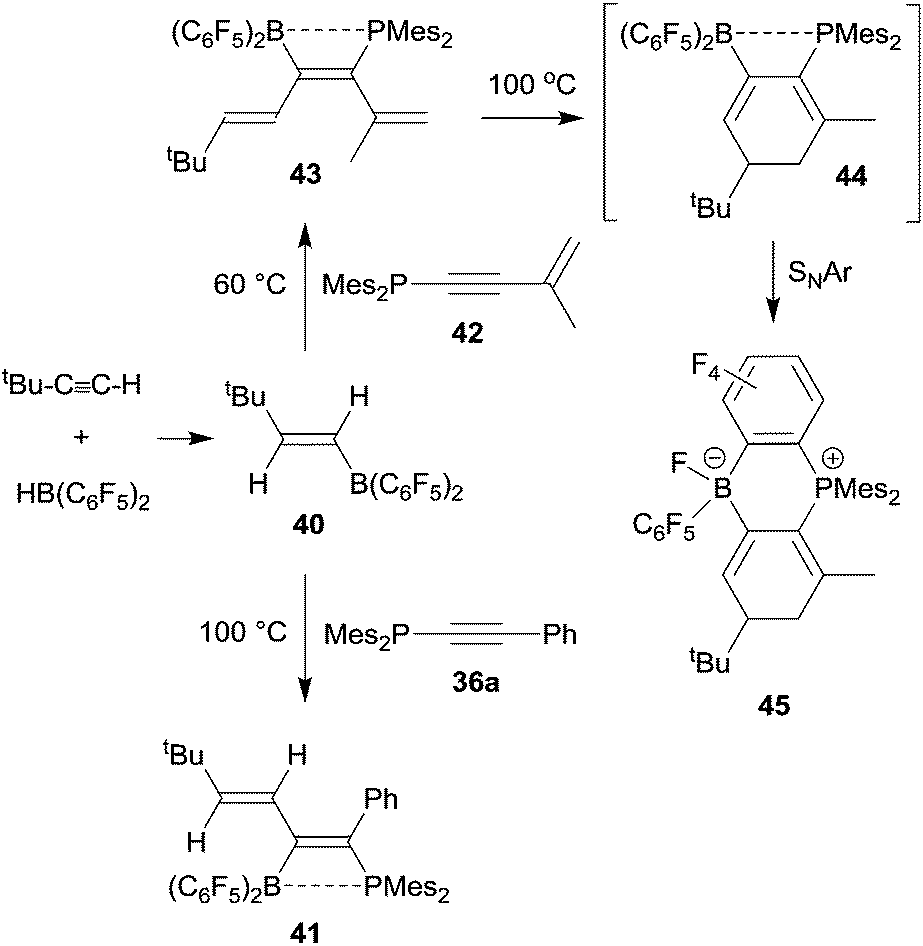 | ||
| Scheme 12 | ||
Since phosphanyl units turned out to be quite good migrating groups we developed these reactions into 1,1-carboboration phosphole syntheses. In a series of typical experiments we reacted several arylbis(alkynyl)phosphanes with B(C6F5)3. At slightly elevated temperatures the corresponding boryl substituted phospholes 49 were obtained in good yields (see Scheme 13).38 The reaction course was followed by temperature dependent 31P NMR spectroscopy. In this way we were able to observe some intermediates of the typical sequential 1,1-carboboration pathway along the way ranging from initial phosphane/borane adduct formation through the stages of the phosphirenium/borate zwitterion formation followed by the 1,1-carboboration product isomers at the first alkynyl unit, all the way to the final phosphole products. An example of the alkenylborane intermediates 48, several phosphirenium/borates 47 and a variety of the final phosphole products were isolated and eventually characterized by X-ray diffraction.
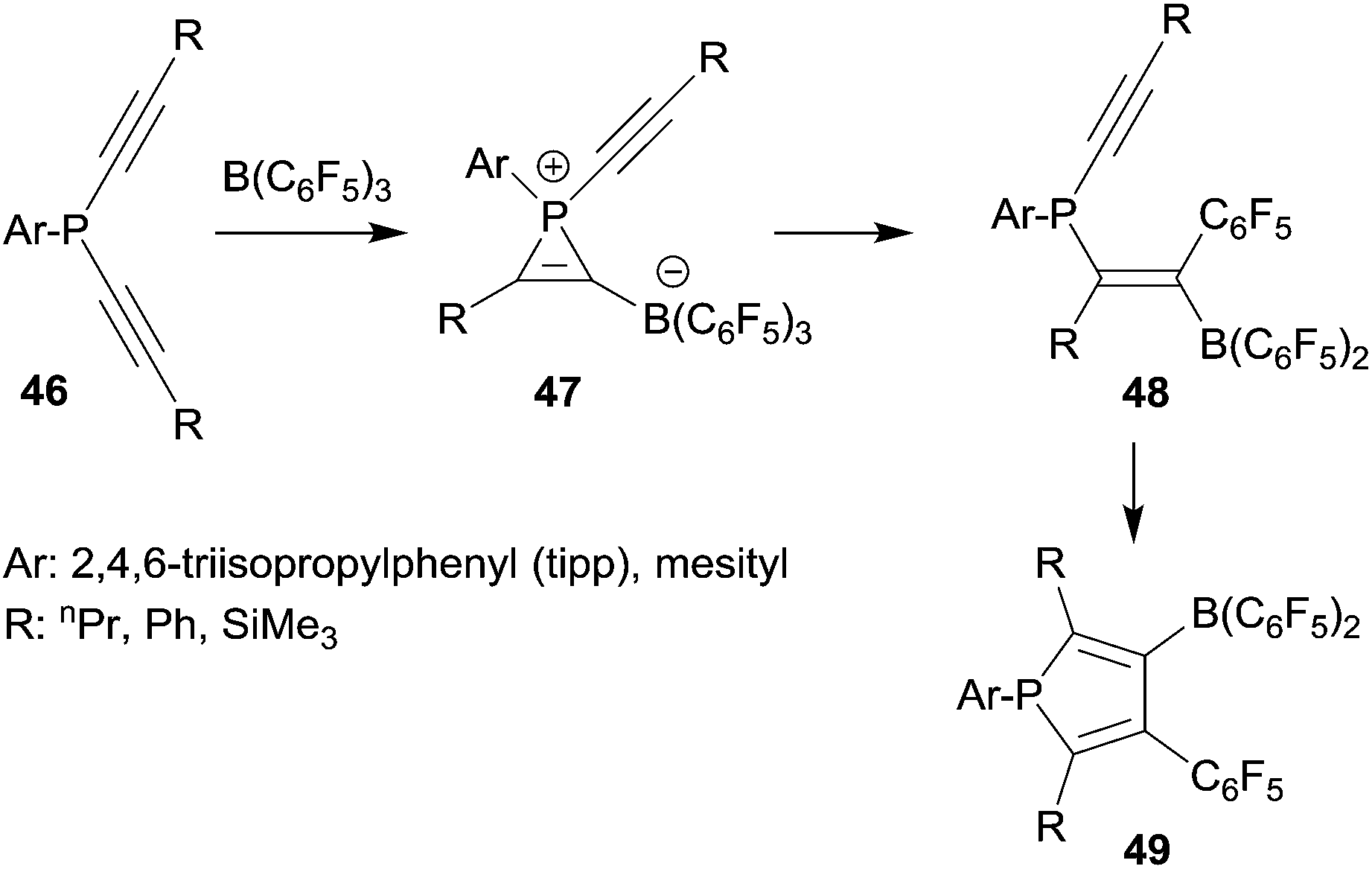 | ||
| Scheme 13 | ||
Phospholes with a variety of substituent patterns were prepared by the 1,1-carboboration route. Attachment of both the thienyl substituent isomers at the 2,5-positions was achieved via the respective thienyl substituted alkynyl-phosphane starting materials 46a (see Scheme 14). The B(C6F5)2 functionality of the respective phosphole 49a was later used for replacement by aryl groups by means of Pd-catalyzed cross-coupling. Scheme 14 shows typical examples. The photophysical properties of several of these phosphole derivatives (and of their corresponding phosphole-oxides) were investigated.39
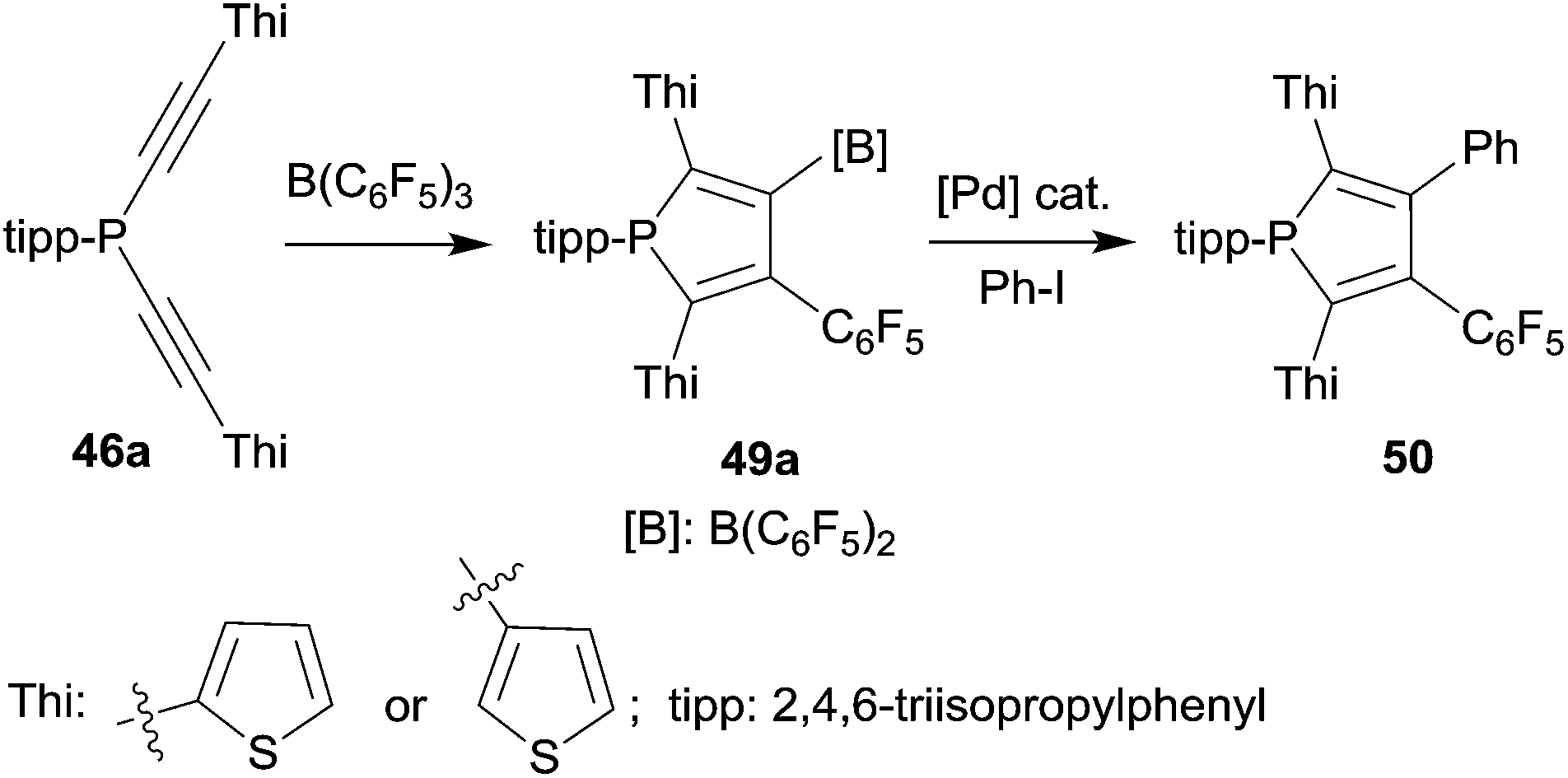 | ||
| Scheme 14 | ||
The alkenyl functionalized borylphosphole 49b, which was formed by the 1,1-carboboration sequence of 46b with the borane Ph–CH2CH2–B(C6F5)2 (and a few other RB(C6F5)2 reagents), showed a subsequent cyclization reaction to give the ring-closed isomer 51, characterized by NMR spectroscopy and by X-ray diffraction (see Scheme 15).40 This reaction represents a rare example of a rapidly proceeding thermally induced bora-Nazarov reaction.
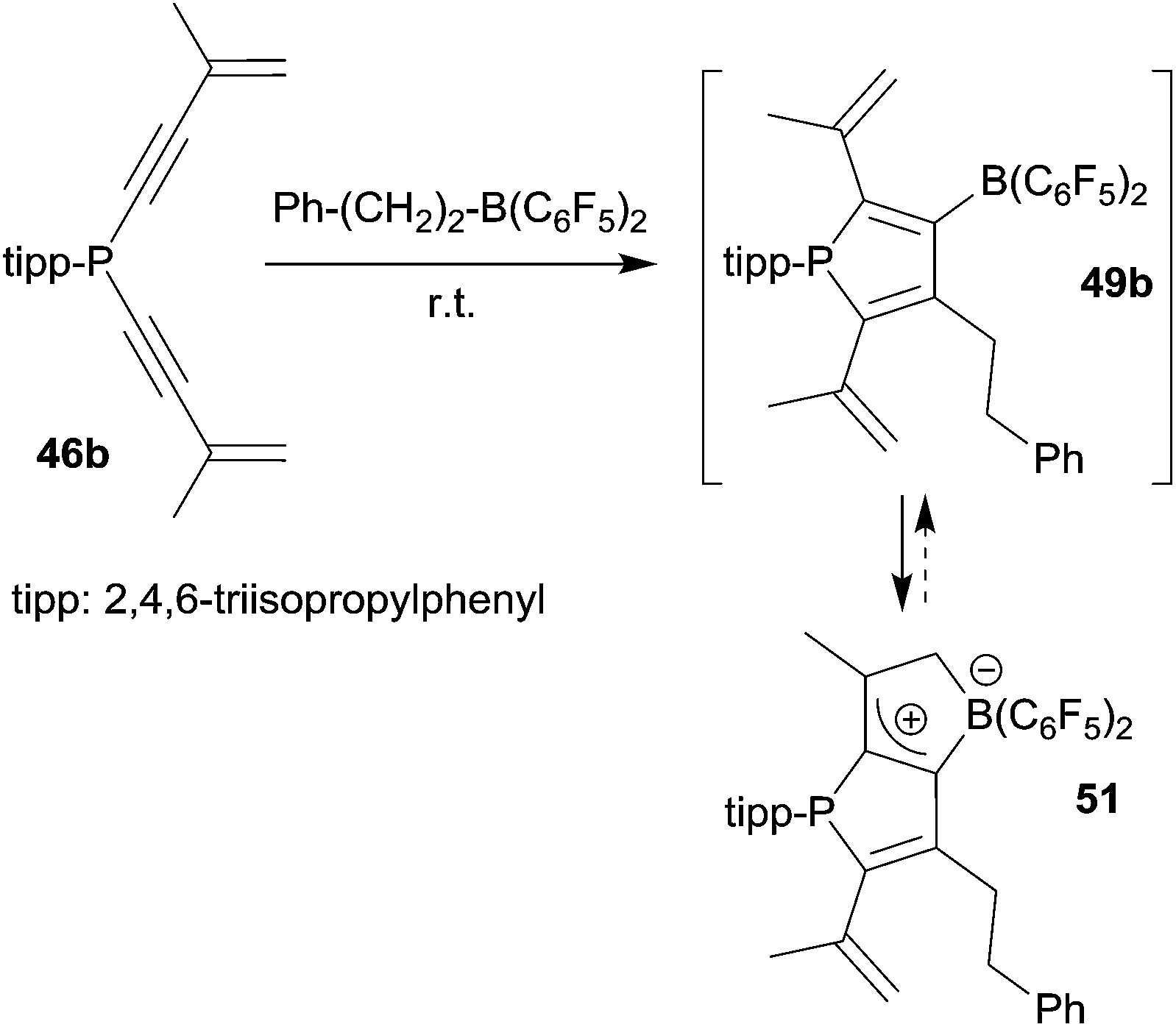 | ||
| Scheme 15 | ||
Boroles are formally antiaromatic compounds. They constitute 4π-electron systems that have a singlet ground state. The unsubstituted parent borole has only be synthetically obtained transition metal complex stabilized. Isolated borole examples are usually rather highly substituted. They are mostly prepared by reaction sequences involving metal–boron exchange reactions, often from the respective stannole which are available by means of transmetallation reactions from the corresponding dilithiobutadienes or the zirconacyclopentadienes.41 Wrackmeyer et al. had reported an early example of a borole synthesis by 1,1-carboboration using the superb ability of the SnMe3 substituent to serve as migrating group.42 Starting from 52a they were able to generate the borole system 53, which, however, rearranged at 60 °C to give 54, the product that was eventually isolated as an oil (see Scheme 16).
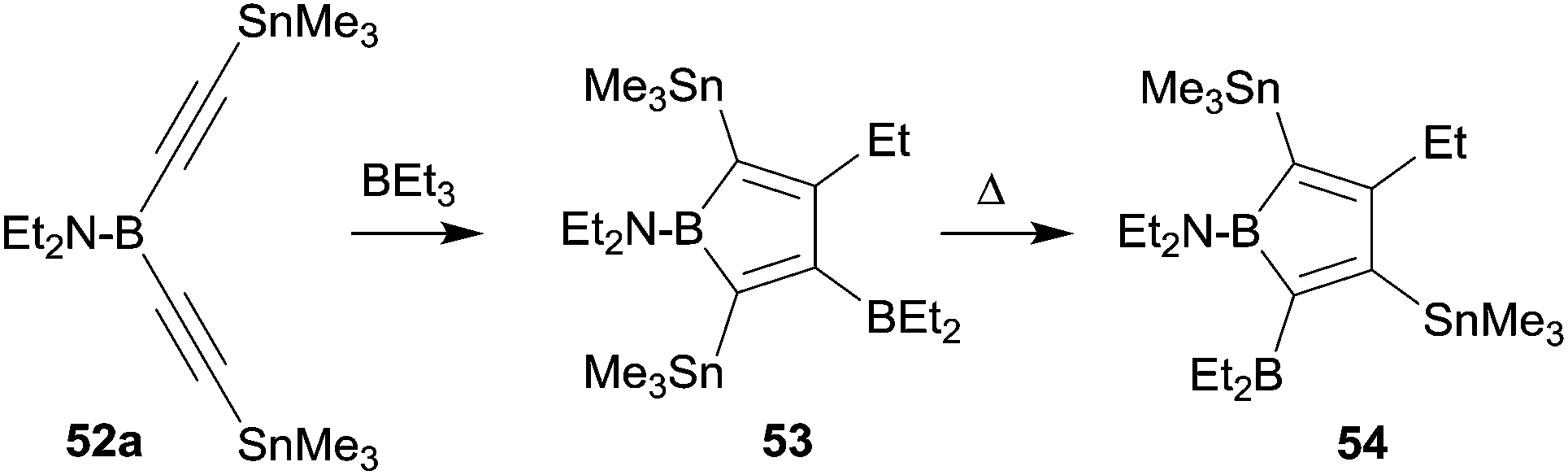 | ||
| Scheme 16 | ||
We investigated whether the borole formation by the typical 1,1-carboboration sequence starting from bis(alkynyl)boron compounds could be improved by using the strongly Lewis acidic R-B(C6F5)2 reagents. In a first attempt we treated the bis(isopropyl)amido boron acetylide 52b with B(C6F5)3, but this did not lead to the formation of the anticipated borole product. Instead it seemed that the sometimes observed typical carbocation pathway took over after the first 1,1-carboboration step and we eventually isolated the alternative product 57 (see Scheme 17).43
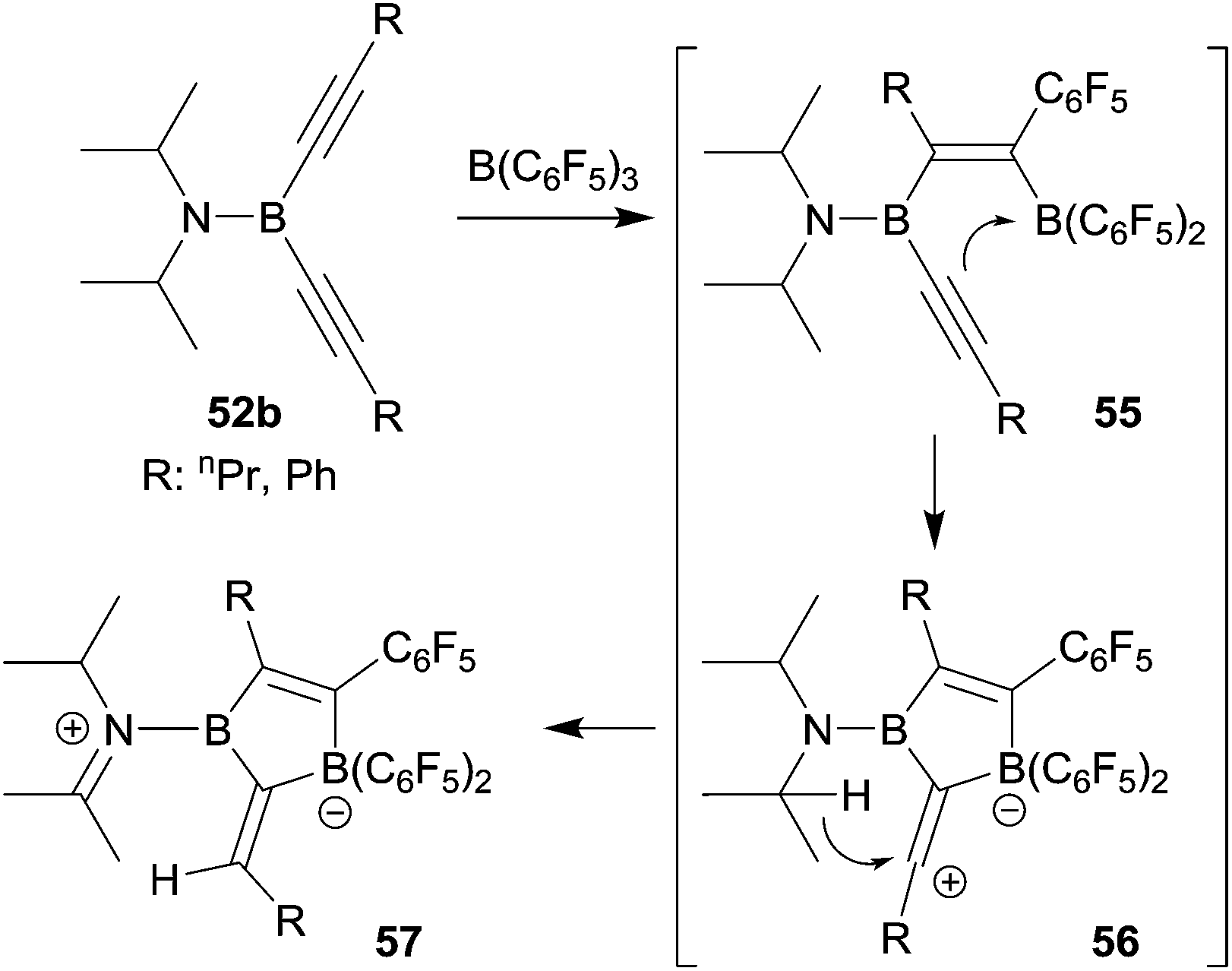 | ||
| Scheme 17 | ||
It was obvious that we should remove the hydride donor ability of the stabilizing substituent at the stage of the intermediate 56 and indeed the reaction of the corresponding diphenylamido substituted bis(alkynyl)borane 52c with B(C6F5)3 gave the expected borole derivative 58. Compound 58 was characterized spectroscopically. Its treatment with pyridine gave the pyridine addition product 59 of the pendant B(C6F5)2 functionality (see Scheme 18). In this step a substituent exchange reaction between the B(C6F5)2 group and the proximate SiMe3 substituent took place as it is sometimes observed in such situations (see above). Compound 59 was characterized by X-ray diffraction.44
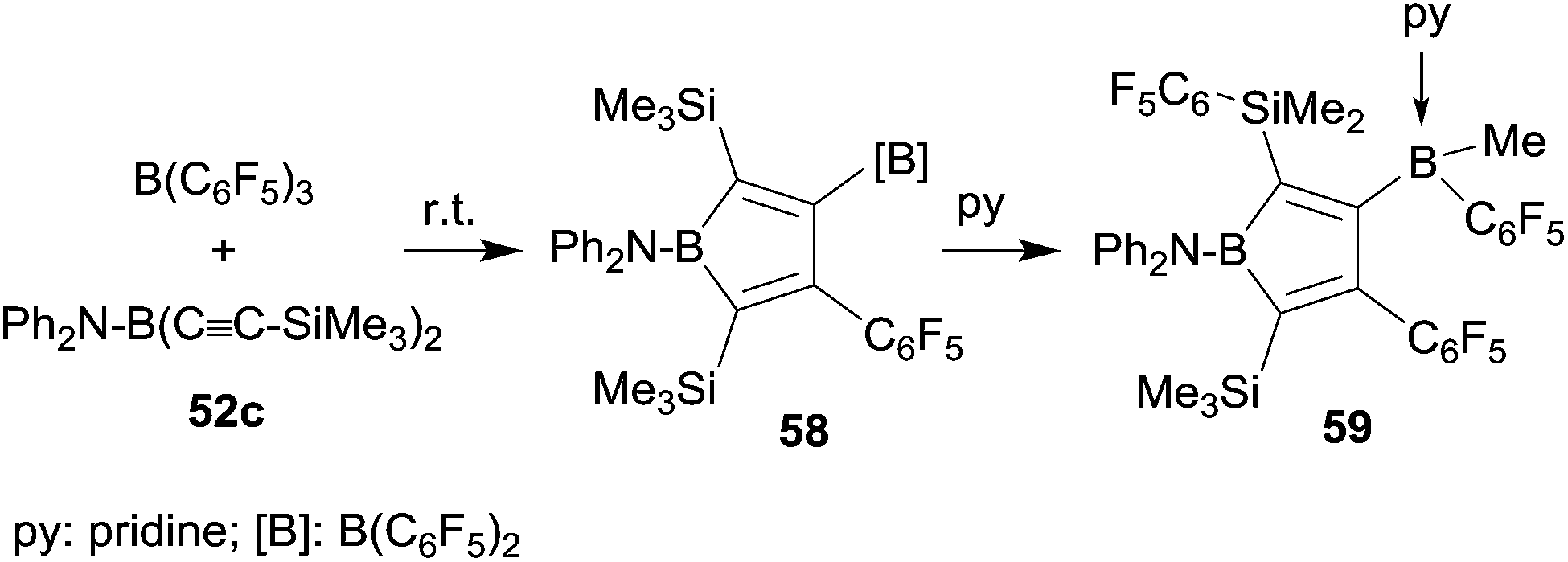 | ||
| Scheme 18 | ||
The phenylbis(alkynyl)borane 52d reacted cleanly with B(C6F5)3 to give the borole 60 by sequential 1,1-carboboration. It was isolated in 43% yield and characterized by X-ray diffraction (see Scheme 19).44 Compound 60 underwent [4 + 2] cyclo-addition with 3-hexyne to give 61. The electron-rich endocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in 61 shows a close contact to the apical boron atom, which gives a distorted square-pyramidal coordination geometry.44,45 A few [B]–H boranes had been shown to give borane carbonyls upon exposure to carbon monoxide.46 Piers et al. had shown that some pentaaryl boroles in a similar way form thermolabile [B]–CO adducts.47 We could show that the borole 60 reacts with carbon monoxide. We assume that initially a CO adduct at the borole boron atom was formed which then rapidly rearranged to yield the unusual ketenyl borane product 62 (see Scheme 19).45
C double bond in 61 shows a close contact to the apical boron atom, which gives a distorted square-pyramidal coordination geometry.44,45 A few [B]–H boranes had been shown to give borane carbonyls upon exposure to carbon monoxide.46 Piers et al. had shown that some pentaaryl boroles in a similar way form thermolabile [B]–CO adducts.47 We could show that the borole 60 reacts with carbon monoxide. We assume that initially a CO adduct at the borole boron atom was formed which then rapidly rearranged to yield the unusual ketenyl borane product 62 (see Scheme 19).45
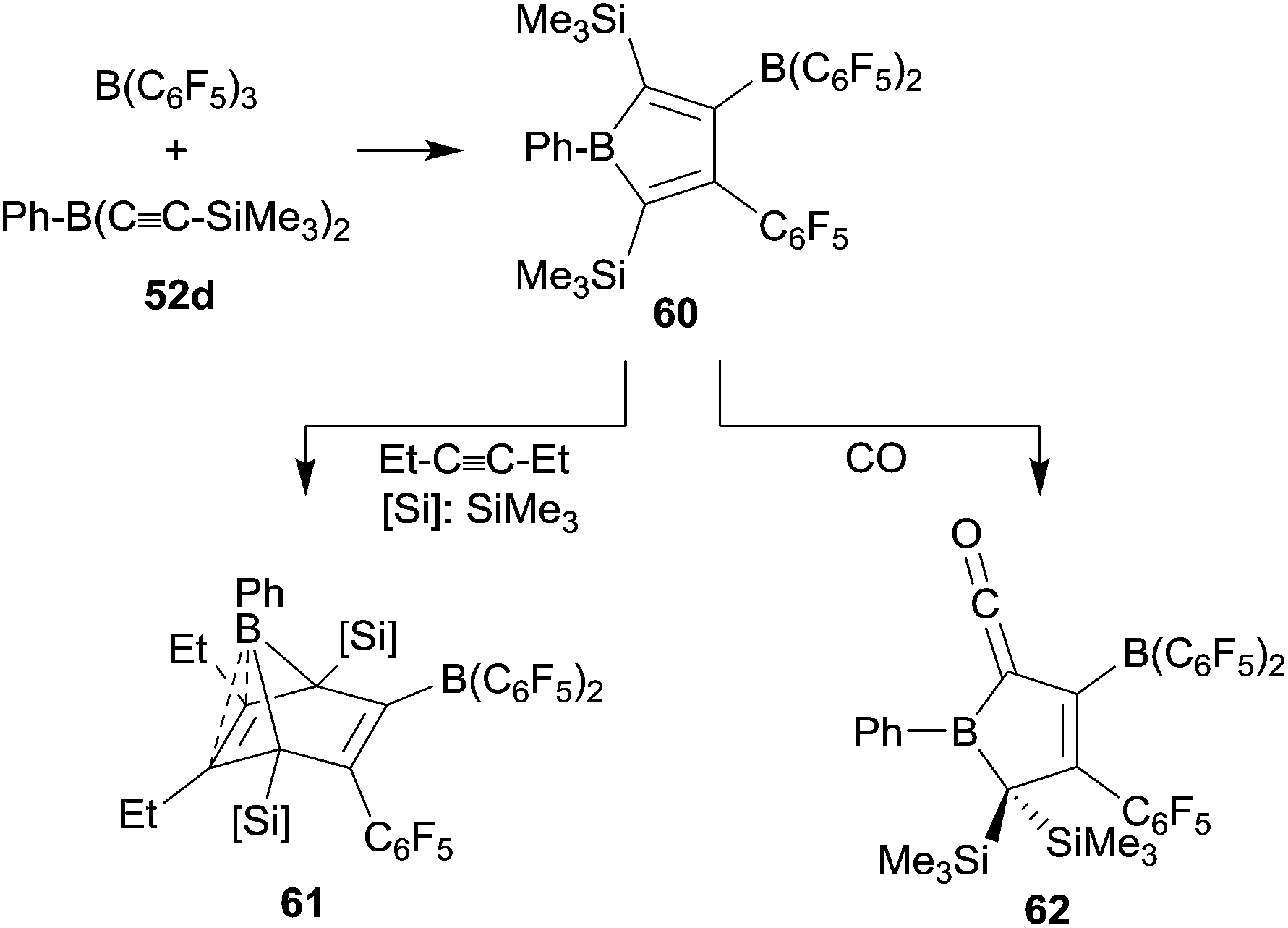 | ||
| Scheme 19 | ||
We have also prepared some silole derivatives by means of the R-B(C6F5)2 advanced 1,1-carboboration protocol although in this case the original Wrackmeyer procedure did not need much substantial improvement. However, silole formation by the reaction of the respective R2Si(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR1)2 reagents with B(C6F5)3 proceeded under milder conditions, typically forming the products in high yields at room temperature.20,48,49 The silole 64 formed from 63 and B(C6F5)3 is a typical example. Interestingly the silole 64 underwent isomerization to 65 upon photolysis. This reaction might be regarded as an example of the silicon analogue of a di-π-methane rearrangement (see Scheme 20).20,49
CR1)2 reagents with B(C6F5)3 proceeded under milder conditions, typically forming the products in high yields at room temperature.20,48,49 The silole 64 formed from 63 and B(C6F5)3 is a typical example. Interestingly the silole 64 underwent isomerization to 65 upon photolysis. This reaction might be regarded as an example of the silicon analogue of a di-π-methane rearrangement (see Scheme 20).20,49
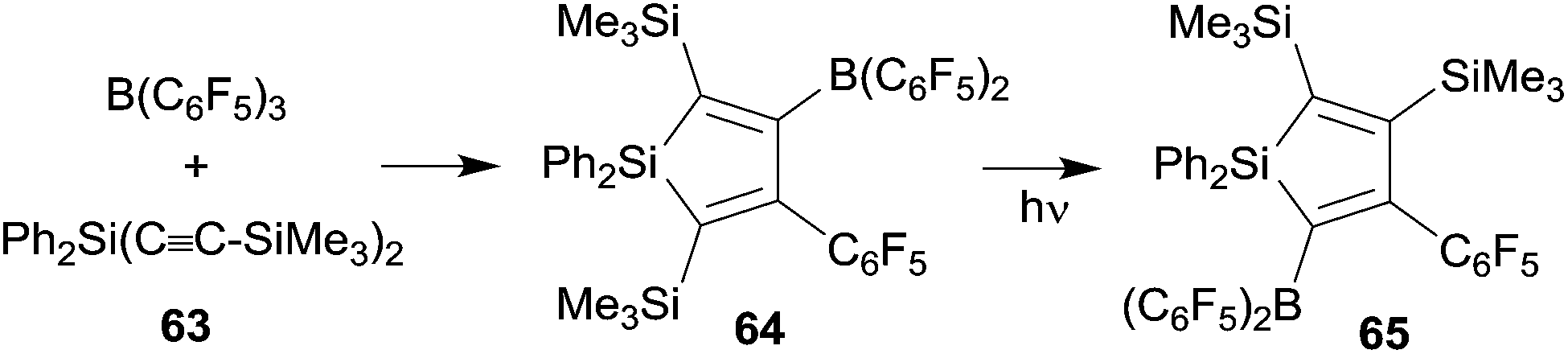 | ||
| Scheme 20 | ||
Functional group chemistry at frustrated Lewis pairs
Frustrated Lewis pair (FLP) chemistry has seen a remarkable development, especially in view of the ability of metal free small molecule binding and activation by these main group element pairs of active Lewis acids and bases.50 Intramolecular FLPs have made a significant contribution to this chemistry. Most intramolecular FLPs were very conveniently made by hydroboration of alkenyl phosphanes or enamines with HB(C6F5)2. However, once prepared the high reactivity and sensitivity of especially the boron Lewis acid component has substantially limited any further modification of these systems. 1,1-Carboboration chemistry has provided some solution of this synthetic problem by selectively using the borane functionality for chemical FLP modification. Here are a few examples (see Scheme 21).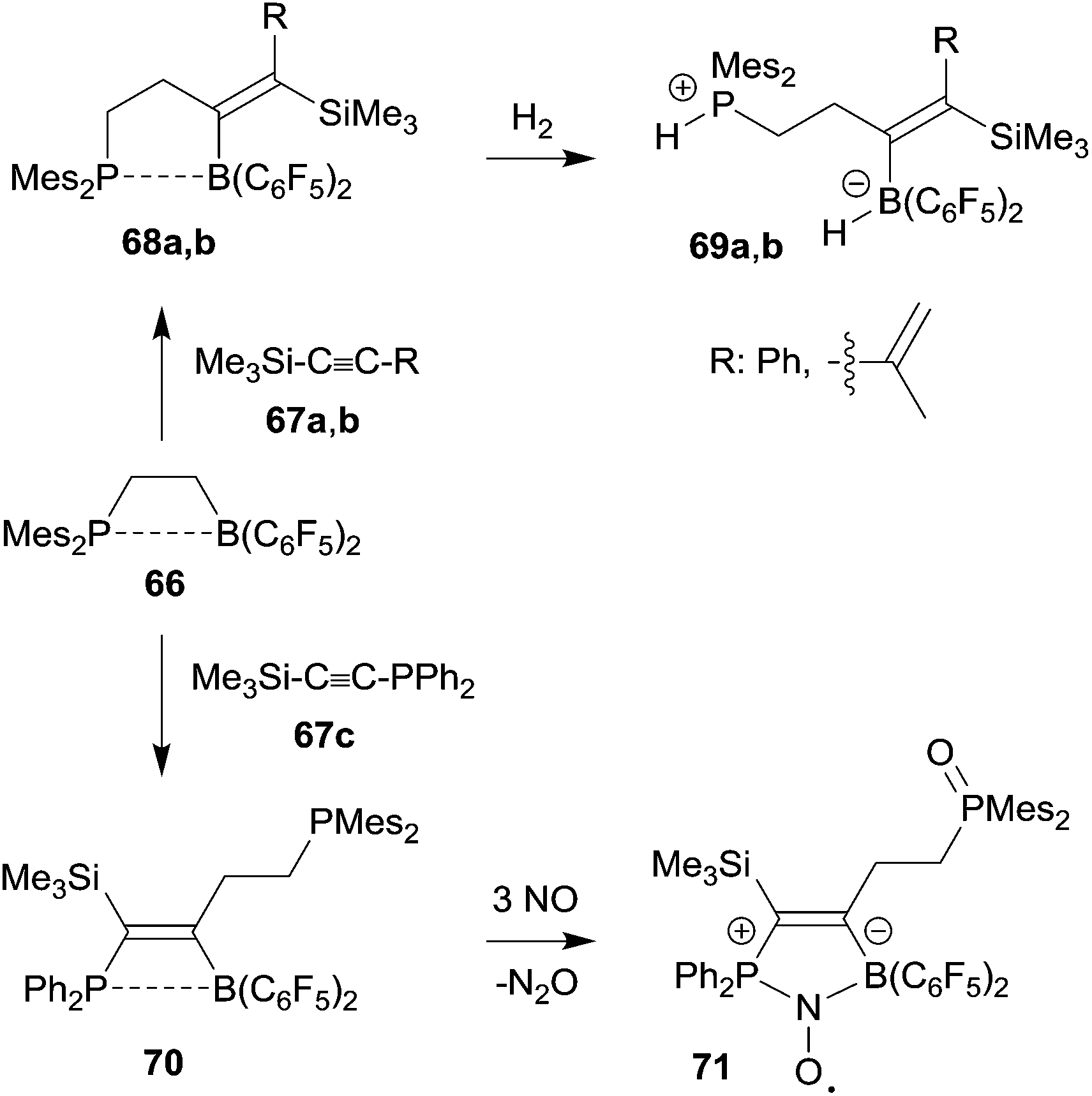 | ||
| Scheme 21 | ||
The ethylene-bridged intramolecular P/B FLP 66 undergoes a 1,1-carboboration reaction with trimethylsilylphenyl acetylene 67a to give the expected C3-bridged P/B FLP 68a. This features a weak phosphane/borane interaction. Nevertheless, the system 68a is able to heterolytically cleave dihydrogen with formation of 69a under mild conditions.51 The FLP 66 reacted in a similar way with the trimethylsilylenyne 67b to give the diene containing FLP 68b which is also a metal-free hydrogen activator.52 The 1,1-carboboration reaction of 66 with the phosphanyl substituted alkyne 67c gave a bifunctional P/P/B FLP 70. It reacted in very special way with nitric oxide (NO). First the pendant PMes2 group was oxidized and then NO cooperatively added to the remaining P/B FLP system to give the persistent P/B FLP NO radical 71.51 It underwent H-atom abstraction reactions typical for this class of compounds.53
1,1-Carbozirconation
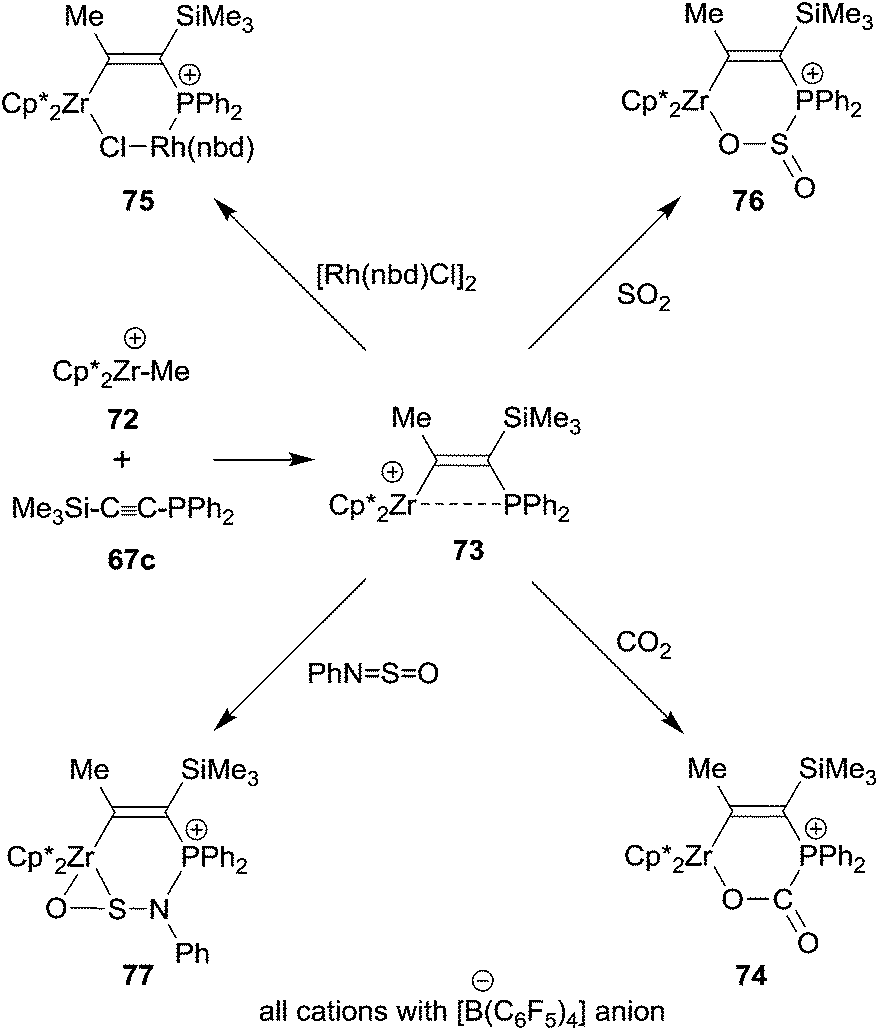
The general reaction scheme on which the 1,1-carboboration reaction is based has begun to stretch out to other areas. One is organometallic chemistry. Very reactive alkyl group 4 metallocene cations usually insert unsaturated organic substrates into their metal to carbon-σ-bond, i.e. they undergo a 1,2-carbometallation reaction. This reaction has provided the basis for the immensely important metallocene based homogeneous Ziegler–Natta olefin polymerization.54 We have recently found a deviation from this behaviour, namely the occurrence of a 1,1-carbozirconation reaction of a suitably substituted alkyne.55 We found that the bulky bis(pentamethylCp)ZrCH3+ cation 72 undergoes a clean 1,1-carbozirconation reaction with diphenylphosphinotrimethylsilylacetylene 67c to give the bulky β-phosphanyl substituted alkenyl zirconium cation product 73 (see Scheme 22). Complex 73 undergoes a variety of typical frustrated Lewis pair addition reactions to unsaturated substrates. The Zr+/P FLP adds to organic carbonyl compounds, including carbon dioxide (74). It even adds to some transition metal complexes (75), thereby taking the role of an ambiphilic ligand.56 It even adds to SO2 giving the heterocyclic product 76. With the related N-sulfinyl benzene amine reagent PhNSO compound 73 gave the unusually structured η2-O,S-addition product 77.55
 | ||
| Scheme 22 | ||
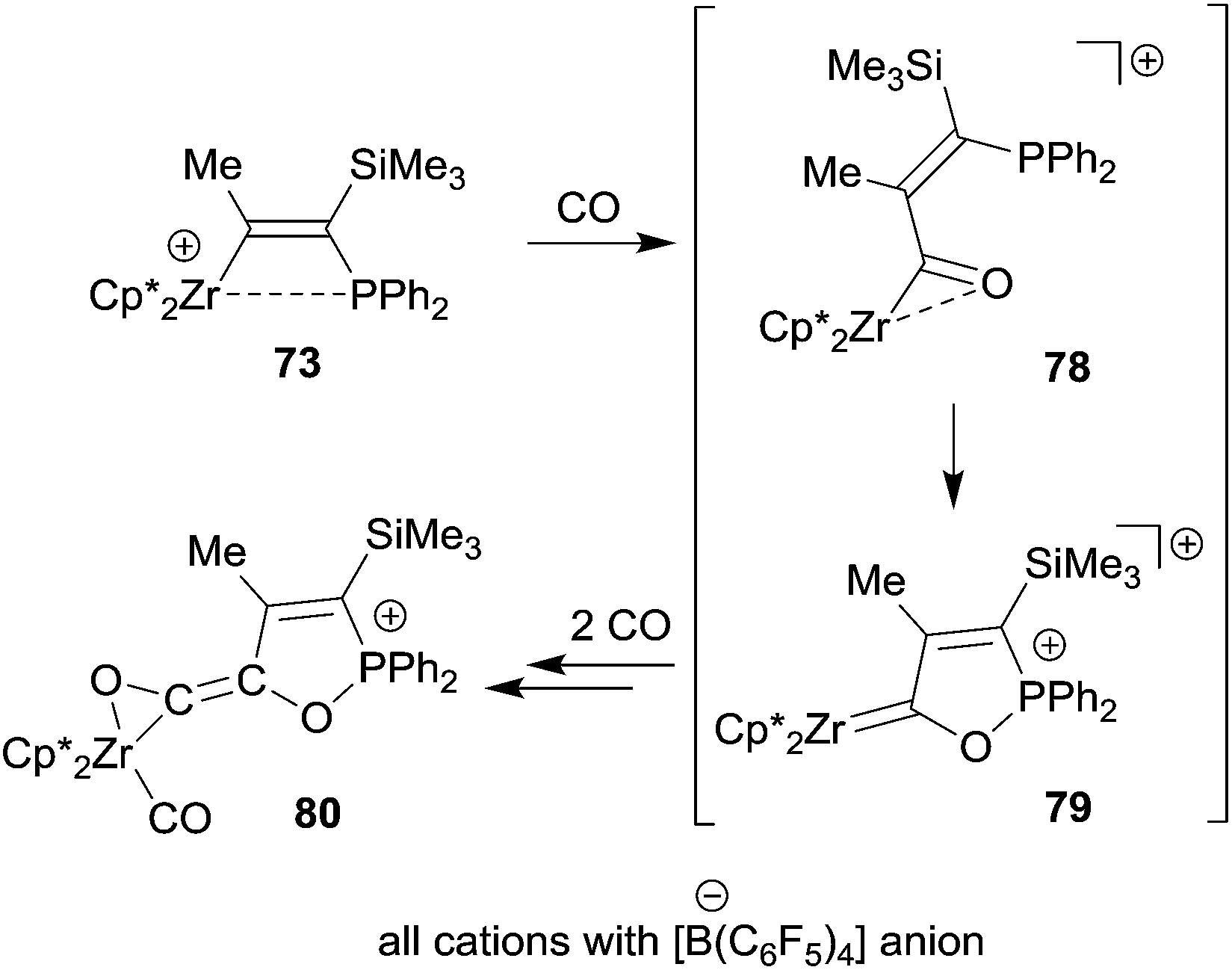
The Zr/P FLP 73 takes up three molar equivalents of carbon monoxide to give the (η2-ketene)zirconium carbonyl cation product 80. We assume that this reaction is initiated by CO insertion into the metal carbon bond followed by cooperative activation of the acyl moiety for further CO uptake to eventually give the product 80 (see Scheme 23). Compound 73 was also shown to be a good catalyst for the selective head to tail dimerization of phenylacetylene.55
 | ||
| Scheme 23 | ||
Conclusions
1,1-Carboboration chemistry had started with the work by Binger, Köster and their contemporaries on the reactions of the alkynylborate anions with electrophiles. However, the finding, development and many applications of the neutral 1,1-carboboration reactions and the establishment of this chemistry as a new method for synthesizing alkenylboranes is due to Wrackmeyer's seminal work. Wrackmeyer et al. have almost single handed developed the by now classical procedures of converting alkynes with suitable metal-containing migrating groups to often useful alkenylboranes by reacting them with e.g. triethylborane, triallylborane or similar simple borane reagents. This and the further development of this chemistry has surely justified the use of the term “Wrackmeyer reaction” for this general class of alkyne/borane transformations. They especially developed this into methods of carrying out sequential 1,1-carboboration reactions starting from geminal metal bis-acetylides or even oligo-alkynes to form various types of alkenylborane containing heterocycles, most noteworthy the five-membered siloles, stannoles, etc. The latter were most readily formed because of the superb migrating abilities of the organostannyl groups. Although the resulting tin compounds did not have a prime interest in themselves, they constituted valuable reagents for transmetallation and for transformation into other heterocyclic ring types.The use of the strongly Lewis acidic, strongly electrophilic R-B(C6F5)2 reagents has marked another more recent breakthrough in 1,1-carboboration chemistry. The use of these readily available reagents has allowed to carry out 1,1-carboboration reactions under much milder conditions and, consequently, has removed its restriction to the metal containing migrating groups (although silyl groups are still key fragments used for practical reasons). This has allowed to perform 1,1-carboboration reactions with simple alkynes carrying just conventional organic substituents. Many terminal alkynes have now been used since hydride migration along the alkynyl framework is often facile, but even internal alkynes have been successfully submitted to the advanced 1,1-carboboration reaction scheme, here proceeding with carbon–carbon bond cleavage. The use of the reactive R-B(C6F5)2 reagents has led to the development of sequential reactions for benzannulation. Since sulphur, phosphorus and even boron containing groups are migrating well, this has resulted in the development of interesting heterocycle synthesis of thiophenes, phospholes, and new boroles. The alkenyl-B(C6F5)2 functionalities in many of these compounds have been found to serve as useful reagents in cross-coupling reactions giving the respective B(C6F5)2 free organic products. In some cases the bulky alkenyl-B(C6F5)2 products found a high interest in themselves, e.g. as Lewis acid components in frustrated Lewis pair chemistry. Overall is seems that with these recent developments the 1,1-carboboration reaction is becoming more and more a viable synthetic alternative to the ubiquitous hydroboration reaction of alkynes, especially in cases where regioselectivity and bulkiness of substituents is critical. We also note that the general scheme of the 1,1-carboboration reaction is beginning to reach out to other areas of chemistry, as our recent example of the newly found 1,1-carbozirconation reaction illustrates. We will see which interesting new developments in this dynamic and rapidly developing field is lying ahead of us.
Acknowledgements
Gerhard Erker thanks his group of very talented coworkers for their remarkable contributions to the studies cited in this article. It has been a pleasure to work with them in this fascinating area. He gratefully acknowledges financial support of his work by the Deutsche Forschungsgesellschaft, the Fonds der Chemischen Industrie, the Alexander von Humboldt-Stiftung and the European Research Council.Notes and references
- (a) P. Binger and R. Köster, Tetrahedron Lett., 1965, 24, 1901 CrossRef; (b) R. Köster, Pure Appl. Chem., 1977, 49, 765 CrossRef; (c) A. Pelter and K. Smith, Compr. Org. Chem., 1979, 3, 892 Search PubMed.
- (a) P. Binger and R. Köster, J. Organomet. Chem., 1974, 73, 205 CrossRef CAS; (b) L. A. Hagelee and R. Köster, Synth. React. Inorg. Met.-Org. Chem., 1977, 7, 53 CrossRef CAS PubMed.
- For contemporary examples of this reaction see: (a) A. Suzuki, Acc. Chem. Res., 1982, 15, 178 CrossRef CAS; (b) N. Ishida, T. Shinmoto, S. Sawano, T. Miura and M. Murakami, Bull. Chem. Soc. Jpn., 2010, 83, 1380 CrossRef CAS; (c) X. Zhao, L. Liang and D. W. Stephan, Chem. Commun., 2012, 48, 10189 RSC.
- B. Wrackmeyer, Coord. Chem. Rev., 1995, 145, 125 CAS.
- (a) G. Menz and B. Wrackmeyer, Z. Naturforsch., B: J. Chem. Sci., 1977, 32, 1400 Search PubMed; (b) C. Bihlmayer and B. Wrackmeyer, Z. Naturforsch., B: J. Chem. Sci., 1981, 36, 1265 Search PubMed; (c) B. Wrackmeyer, C. Bihlmayer and M. Schilling, Chem. Ber., 1983, 116, 3182 CrossRef CAS PubMed.
- (a) A. Sebald and B. Wrackmeyer, J. Chem. Soc., Chem. Commun., 1983, 309 RSC; (b) A. Sebald and B. Wrackmeyer, J. Organomet. Chem., 1997, 544, 105 CrossRef; (c) B. Wrackmeyer, A. Pedall and J. Weidinger, J. Organomet. Chem., 2002, 649, 225 CrossRef CAS.
- (a) R. Köster, G. Seidel and B. Wrackmeyer, Chem. Ber., 1989, 122, 1825 CrossRef PubMed for a recent BH variant see (b) A. Boussonnière, X. Pan, S. J. Geib and D. P. Curran, Organometallics, 2013, 32, 7445 CrossRef.
- (a) B. Wrackmeyer, K. Horchler and R. Boese, Angew. Chem., Int. Ed. Engl., 1989, 28, 1500 CrossRef PubMed; (b) B. Wrackmeyer, G. Kehr and R. Boese, Angew. Chem., Int. Ed. Engl., 1991, 30, 1370 CrossRef PubMed; (c) B. Wrackmeyer, S. Kundler and R. Boese, Chem. Ber., 1993, 126, 1361 CrossRef CAS PubMed; (d) B. Wrackmeyer, P. Thoma, S. Marx, T. Bauer and R. Kempe, Eur. J. Inorg. Chem., 2014, 2103 CrossRef CAS PubMed.
- (a) B. Wrackmeyer, J. Chem. Soc., Chem. Commun., 1986, 397 RSC; (b) B. Wrackmeyer, G. Kehr and J. Süβ, Chem. Ber., 1993, 126, 2221 CrossRef CAS PubMed; (c) E. Khan, S. Bayer and B. Wrackmeyer, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 995 CAS; (d) E. Khan and B. Wrackmeyer, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 1098 CAS.
- (a) B. Wrackmeyer, Heteroat. Chem., 2006, 17, 188 CrossRef CAS PubMed; (b) B. Wrackmeyer and O. L. Tok, Comprehensive Heterocyclic Chemistry III, Elsevier Ltd., 2008, p. 1181 Search PubMed.
- (a) L. Killian and B. Wrackmeyer, J. Organomet. Chem., 1977, 132, 213 CrossRef CAS; (b) L. Killian and B. Wrackmeyer, J. Organomet. Chem., 1978, 148, 137 CrossRef CAS; (c) B. Wrackmeyer, P. Thoma, S. Marx, G. Glatz and R. Kempe, Z. Anorg. Allg. Chem., 2013, 639, 1205 CrossRef CAS PubMed.
- (a) B. Wrackmeyer, G. Kehr, J. Süβ and E. Molla, J. Organomet. Chem., 1998, 562, 207 CrossRef CAS; (b) B. Wrackmeyer, G. Kehr, J. Süβ and W. Molla, J. Organomet. Chem., 1999, 577, 82 CrossRef CAS; (c) B. Wrackmeyer, O. L. Tok, E. Klimkina and W. Milius, Eur. J. Inorg. Chem., 2010, 2276 CrossRef CAS PubMed; (d) B. Wrackmeyer, E. V. Klimkina, W. Milius, C. Butterhof and K. Inzenhofer, Z. Naturforsch., B: J. Chem. Sci., 2014, 69, 1269 CAS.
- R.-J. Binnewirtz, H. Klingenberger, R. Welte and P. Paetzold, Chem. Ber., 1983, 116, 1271 CrossRef CAS PubMed.
- (a) A. G. Massey, A. J. Park and F. G. A. Stone, Proc. Chem. Soc., 1963, 212 CAS; (b) A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245 CrossRef CAS.
- D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492 CrossRef CAS.
- C. Chen, F. Eweiner, B. Wibbeling, R. Fröhlich, S. Senda, Y. Ohki, K. Tatsumi, S. Grimme, G. Kehr and G. Erker, Chem.–Asian J., 2010, 5, 2199 CrossRef CAS PubMed.
- C. Jiang, O. Blacque and H. Berke, Organometallics, 2010, 29, 125 CrossRef CAS.
- C. Chen, T. Voss, R. Fröhlich, G. Kehr and G. Erker, Org. Lett., 2011, 13, 62 CrossRef CAS PubMed.
- J. S. Reddy, B.-H. Xu, T. Mahdi, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 5638 CrossRef CAS.
- G. Kehr and G. Erker, Chem. Commun., 2012, 48, 1839 RSC.
- (a) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (b) Topics in Current Chemistry, C-C Bond Activation, ed. G. Dong, Springer, Berlin, Heidelberg, 2014, vol. 346 Search PubMed.
- C. Chen, G. Kehr, R. Fröhlich and G. Erker, J. Am. Chem. Soc., 2010, 132, 13594 CrossRef CAS PubMed.
- E. Jahnke and R. T. Tykwinski, Chem. Commun., 2010, 46, 3235 RSC.
- (a) C. Fan, W. E. Piers, M. Parvez and R. McDonald, Organometallics, 2010, 29, 5132 CrossRef CAS.
- (a) R. G. Bergman, Acc. Chem. Res., 1973, 6, 25 CrossRef CAS; (b) B. P. Warner, S. P. Millar, R. D. Broene and S. L. Buchwald, Science, 1995, 269, 814 CrossRef CAS PubMed; (c) Y. Luo, X. Pan, X. Yu and J. Wu, Chem. Soc. Rev., 2014, 43, 834 RSC.
- R. Liedtke, M. Harhausen, R. Fröhlich, G. Kehr and G. Erker, Org. Lett., 2012, 14, 1448 CrossRef CAS PubMed.
- (a) R. Liedtke, G. Kehr, R. Fröhlich, C. G. Daniliuc, B. Wibbeling, J. L. Peterson and G. Erker, Helv. Chim. Acta, 2012, 95, 2515 CrossRef CAS PubMed see also: (b) B. Wrackmeyer, M. H. Bhatti, S. Ali, O. L. Tok and Y. N. Bubnov, J. Organomet. Chem., 2002, 657, 146 CAS.
- R. Liedtke, F. Tenberge, C. G. Daniliuc, G. Kehr and G. Erker, J. Org. Chem., 2015, 80, 2240 CrossRef CAS PubMed.
- (a) C. Eller, G. Kehr, C. G. Daniliuc, R. Fröhlich and G. Erker, Organometallics, 2013, 32, 384 CrossRef CAS; (b) C. Eller, G. Kehr, C. G. Daniliuc, D. W. Stephan and G. Erker, Chem. Commun., 2015, 51, 7226 RSC.
- F. A. Tsao, A. J. Lough and D. W. Stephan, Chem. Commun., 2015, 51, 4287 RSC.
- (a) F. A. Tsao and D. W. Stephan, Dalton Trans., 2015, 44, 71 RSC see also: (b) C. Eller, K. Bussmann, G. Kehr, B. Wibbeling, C. G. Daniliuc and G. Erker, Chem. Commun., 2014, 50, 1980 RSC.
- (a) O. Ekkert, G. Kehr, R. Fröhlich and G. Erker, Chem. Commun., 2011, 47, 10482 RSC; (b) O. Ekkert, G. Kehr, C. G. Daniliuc, R. Fröhlich, B. Wibbeling, J. L. Peterson and G. Erker, Z. Anorg. Allg. Chem., 2013, 639, 2455 CrossRef CAS PubMed see also: (c) P. Feldhaus, G. Kehr, R. Fröhlich, C. G. Daniliuc and G. Erker, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 666 CAS.
- O. Ekkert, G. Kehr, R. Fröhlich and G. Erker, J. Am. Chem. Soc., 2011, 133, 4610 CrossRef CAS PubMed.
- O. Ekkert, G. G. Miera, T. Wiegand, H. Eckert, B. Schirmer, J. L. Peterson, C. G. Daniliuc, R. Fröhlich, S. Grimme, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 2657 RSC.
- (a) M. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C71 Search PubMed; (b) J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939 RSC.
- O. Ekkert, O. Tuschewitzki, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2013, 49, 6992 RSC.
- G.-Q. Chen, G. Kehr, C. G. Daniliuc and G. Erker, Org. Biomol. Chem., 2015, 13, 764 CAS.
- (a) J. Möbus, Q. Bonnin, K. Ueda, R. Fröhlich, K. Itami, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2012, 51, 1954 CrossRef PubMed; (b) J. Möbus, K. Malessa, H. Frisch, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, Heteroat. Chem., 2014, 25, 396 CrossRef PubMed.
- J. Möbus, A. Galstyan, A. Feldmann, C. G. Daniliuc, R. Fröhlich, C. A. Strassert, G. Kehr and G. Erker, Chem.–Eur. J., 2014, 20, 11883 CrossRef PubMed.
- (a) J. Möbus, G. Kehr, C. G. Daniliuc, C. Mück-Lichtenfeld and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 12366 CrossRef PubMed see also: (b) A. Iida, S. Saito, T. Sasamori and S. Yamaguchi, Angew. Chem., Int. Ed., 2013, 52, 3760 CrossRef CAS PubMed.
- H. Braunschweig, I. Krummenacher and J. Wahler, Adv. Organomet. Chem., 2013, 61, 1 CrossRef CAS PubMed.
- (a) A. Sebald and B. Wrackmeyer, J. Organomet. Chem., 1986, 307, 157 CrossRef CAS see also (b) Ref. 11b ; (c) B. Wrackmeyer, Organometallics, 1984, 3, 1 CrossRef CAS.
- F. Ge, G. Kehr, C. G. Daniliuc and G. Erker, Chem.–Asian J., 2013, 8, 2227 CrossRef CAS PubMed.
- (a) F. Ge, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2014, 136, 68 CrossRef CAS PubMed see also: (b) F. Ge, G. Kehr, C. G. Daniliuc, C. Mück-Lichtenfeld and G. Erker, Organometallics, 2015, 34, 4205 CrossRef CAS.
- F. Ge, G. Kehr, C. G. Daniliuc and G. Erker, Organometallics, 2015, 34, 229 CrossRef CAS.
- (a) A. B. Burg and H. I. Schlesinger, J. Am. Chem. Soc., 1937, 59, 780 CrossRef CAS; (b) H. C. Brown, Acc. Chem. Res., 1969, 2, 65 CrossRef CAS; (c) M. Sajid, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2014, 53, 1118 CrossRef CAS PubMed.
- A. Fukazawa, J. L. Dutton, C. Fan, L. G. Mercier, A. Y. Houghton, Q. Wu, W. E. Piers and M. Parvez, Chem. Sci., 2012, 3, 1814 RSC.
- (a) G. Dierker, J. Ugolotti, G. Kehr, R. Fröhlich and G. Erker, Adv. Synth. Catal., 2009, 351, 1080 CrossRef CAS PubMed; (b) J. Ugolotti, G. Dierker, R. Fröhlich, G. Kehr and G. Erker, J. Organomet. Chem., 2011, 696, 1184 CrossRef CAS PubMed.
- J. Ugolotti, G. Kehr, R. Fröhlich and G. Erker, Chem. Commun., 2010, 46, 3016 RSC.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed.
- R. Liedtke, F. Scheidt, J. Ren, B. Schirmer, A. J. P. Cardenas, C. G. Daniliuc, H. Eckert, T. H. Warren, S. Grimme, G. Kehr and G. Erker, J. Am. Chem. Soc., 2014, 136, 9014 CrossRef CAS PubMed.
- A. Feldmann, G. Kehr, C. G. Daniliuc, C. Mück-Lichtenfeld and G. Erker, Chem.–Eur. J., 2015, 21, 12456 CrossRef CAS PubMed.
- (a) M. Sajid, A. Stute, A. J. P. Cardenas, B. J. Culotta, J. A. M. Hepperle, T. H. Warren, B. Schirmer, S. Grimme, A. Studer, C. G. Daniliuc, R. Fröhlich, J. L. Petersen, G. Kehr and G. Erker, J. Am. Chem. Soc., 2012, 134, 10156 CrossRef CAS PubMed see also: L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034 CrossRef CAS PubMed.
- (a) H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed., 1995, 34, 1143 CrossRef CAS PubMed; (b) E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391 CrossRef CAS PubMed.
- (a) X. Xu, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 13629 CrossRef CAS PubMed; (b) X. Xu, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2014, 136, 12431 CrossRef CAS PubMed.
- G. Bouhadir, A. Amgoune and D. Bourissou, Adv. Organomet. Chem., 2010, 5, 1 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |