
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructurally plastic peptide capsules for synthetic antimicrobial viruses†
Valeria
Castelletto
a,
Emiliana
de Santis
a,
Hasan
Alkassem
ab,
Baptiste
Lamarre
a,
James E.
Noble
a,
Santanu
Ray
a,
Angelo
Bella
a,
Jonathan R.
Burns
a,
Bart W.
Hoogenboom
b and
Maxim G.
Ryadnov
*a
aNational Physical Laboratory, Teddington, Middlesex TW11 0LW, UK. E-mail: max.ryadnov@npl.co.uk
bLondon Centre for Nanotechnology, Departments of Biochemical Engineering and Physics and Astronomy, University College London, London WC1E 6BT, UK
First published on 21st December 2015
Abstract
A conceptual design for artificial antimicrobial viruses is described. The design emulates viral assembly and function to create self-assembling peptide capsules that promote efficient gene delivery and silencing in mammalian cells. Unlike viruses, however, the capsules are antimicrobial, which allows them to exhibit a dual biological function: gene transport and antimicrobial activity. Unlike other antimicrobials, the capsules act as pre-concentrated antimicrobial agents that elicit rapid and localised membrane-disrupting responses by converting into individual pores at their precise landing positions on membranes. The concept holds promise for engineering virus-like scaffolds with biologically tuneable properties.
Introduction
The rise of antimicrobial resistance presents new challenges for supramolecular chemistry and design. A key driver here is the demand for smaller, non-toxic and biologically differential materials.1 This is particularly important for molecular therapy, which depends on adaptable nanoscale vehicles to safely handle and deliver therapeutic nucleic acids into human cells.2 However, gene delivery can be compromised by bacterial infections, which prompts the need for vehicles with antimicrobial properties.3 Viruses may be the most efficient gene-transfecting agents, but typically are not antimicrobial. Similarly, non-viral mimetics can emulate encapsulating and cell-penetrating properties of viruses, but lack the ability to selectively differentiate between bacterial and mammalian cells.4 A saving solution to this can come from virus-like assemblies of antimicrobial components that would induce localised toxic responses on binding to microorganisms. The innate immune system offers a rich repertoir of such components.5 Unlike conventional antibiotics, these are host-defense polypeptide sequences that recognise microbial surfaces, bind to them and at critical concentrations assemble into membrane-disrupting pores or channels.6 Nanoscale assembly may pre-concentrate their toxic conformations, which on microbial membranes would instantaneously convert into membrane-destabilising pores. The nanostructures are thus akin to antimicrobial projectiles that bypass the need for membrane-induced folding and destroy microbial membranes on direct contact. With this in mind, we set out to combine the efficiency of viral assembly and gene-delivery with the antimicrobial responsiveness of host-defense peptides into one self-assembling motif.Our design rationale uses three main principles. Firstly, the motif should assemble into nanoscale virus-like capsules with and without nucleic acids. This is to ensure the autonomous functioning of the capsules independent of cargo, which can vary. Secondly, the capsules have to be appreciably antimicrobial. This is necessary for fast and localised antimicrobial responses. Thirdly, the capsules must promote active gene transfer into mammalian cells without causing cytotoxic and haemolytic effects. An overarching principle for all three is the structural plasticity of the capsules without a priori requirements for a particular size.
Based on these conventions, we reviewed an antimicrobial and non-haemolytic protein lactoferrin. This is a major component of the innate immune system found in secretory fluids including saliva and milk and is notably responsible for providing antimicrobial protection to infants. The antimicrobial activity of this protein is attributed to a core sequence of six amino acids – RRWQWR (Fig. 1a).7 In its free, isolated form the peptide is moderately antimicrobial and does not assemble. The sequence is homologues to virus-derived tryptophan zippers, with a characteristic core motif WTW, which adopts β-turn and β-sheet conformations with cross-strand tryptophan rings packed tightly against one another.8 β-Strands, arranged antiparallel, foster interactions between amino-acid side chains of opposite termini.
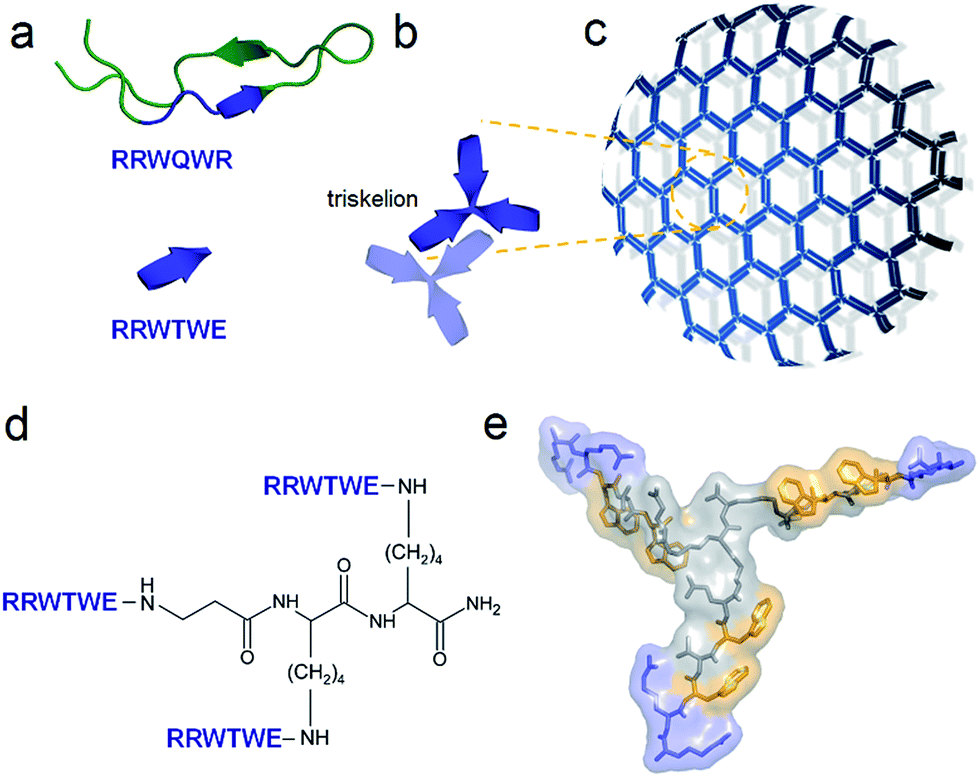 | ||
| Fig. 1 Capzip design. (a) Antimicrobial lactoferrin peptide (PDB entry 1LFC rendered by PyMol) with the RRWQWR motif highlighted in blue (top). The motif is converted to a self-complementary RRWTWE β-strand (bottom). (b) Three copies of RRWTWE are conjugated into a β-strand triskelion. For clarity only two triskelions forming a β-sheet are shown and highlighted in different colours. (c) Triskelions self-assemble via a β-sheet-formation following a trilateral honeycomb-like symmetry. (d) The chemical structure of the triskelion. (e) A β-strand triskelion model with arginine and tryptophan residues highlighted by blue and yellow, respectively. | ||
Results and discussion
To capitalize on this, the glutamine and C-terminal arginine of the lactoferrin motif were replaced with threonine and glutamate giving rise to a self-complementary sequence RRWTWE (Fig. 1a and b). With the virus architecture adopting an n-fold rotational symmetry, where n is usually 3 or 5 or both,9 a triskel conjugate of RRWTWE was generated to give a self-assembling motif with a trilateral symmetry reminiscent of native cage-like subunits (Fig. 1b–e and S1 in ESI†).The resulting construct, dubbed capzip, readily assembled into morphologically uniform and hollow capsules with dominating size ranges of 20–200 nm, as seen by transmission electron microscopy (TEM), atomic force microscopy (AFM) and cryo-scanning electron microscopy (SEM) (Fig. 2, S2a and b†). Larger assemblies were also observed suggesting thermodynamically equilibrated paracrystalline systems (Fig. 2a–c and S2c†).10 Paracrystals often exhibit regular nanoscale patterns such as striations or rings.11,12 Although fine structure could be observed on some capsules, this appeared irregular (Fig. 2a), which may imply a larger structural plasticity than that for perfectly crystalline materials.13 In the triskel conjugate, RRWTWE is prone to fold as a β-turn and pair into a β-sheet with another arm of another conjugate.7 Triskelions are then expected to propagate into two-dimensional sheets whose increasing curvature would facilitate the formation of three-dimensional capsules (Fig. 1c).
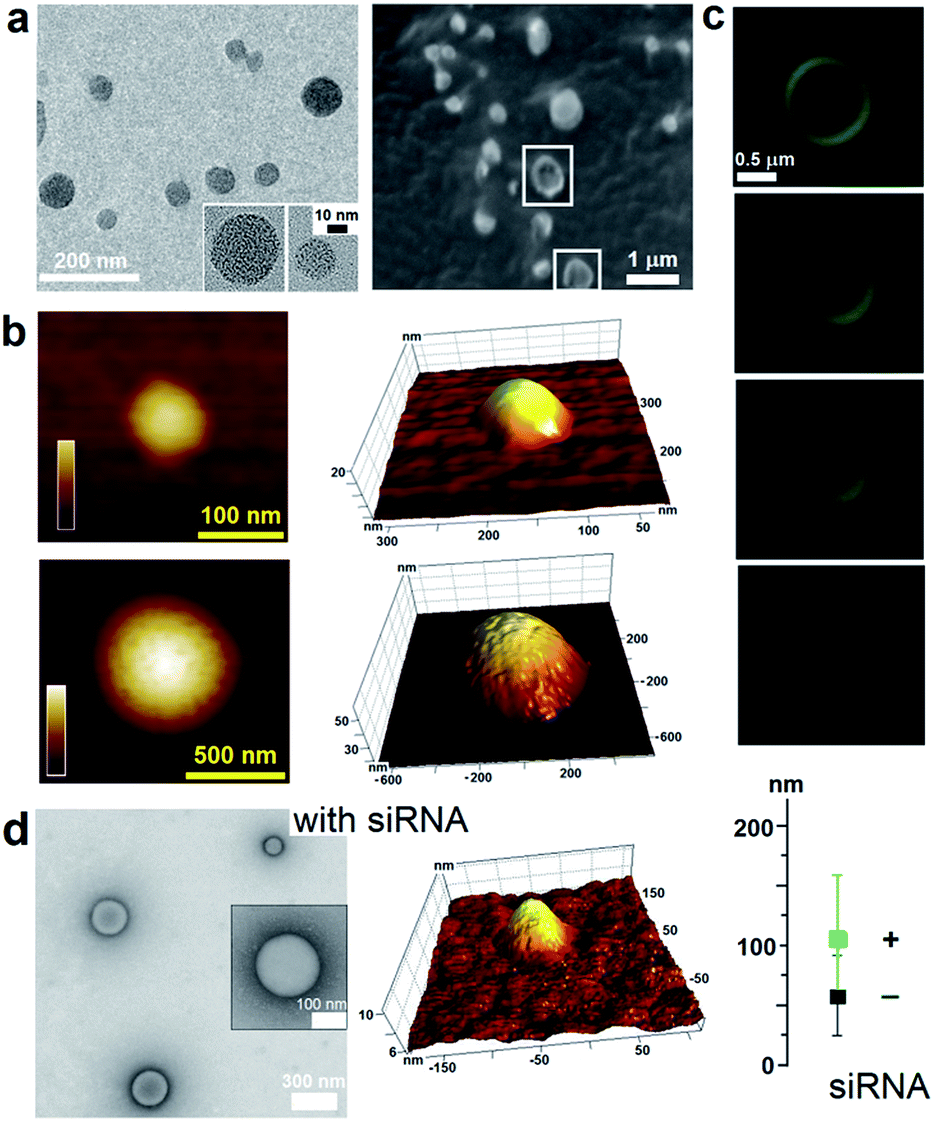 | ||
| Fig. 2 Capzip assembly with and without siRNA. (a) TEM (left) and cryo-SEM (right) images of assembled capsules. Collapsed capsules are highlighted by white squares. (b) In-air AFM topography images of capsules of different sizes and their 3D representations. Colour bars are 20 nm (upper) and 60 nm (lower). (c) Cross-section fluorescence micrographs of a large capsule with gradual depth changes (0.1–0.2 μm). (d) TEM (left) and AFM (middle) images of capsules assembled with siRNA. Capsule sizes with (+) and without (−) siRNA measure by high-res TEM. Assembly conditions: 100 μM peptide, 1/5 N/P (siRNA–peptide) molar ratio, pH 7.4, MOPS, 20 °C, overnight. | ||
Consistent with this, circular dichroism (CD) spectra for capzip were characteristic of β-turns, with minima at λ ∼ 200 nm and 214 nm, containing elements of a β-sheet structure and indole transitions of the tryptophan residues (Fig. 3a).7,8,14 Fourier transform infrared (FT-IR) spectra confirmed the structure with bands at 1660 and 1545 cm−1, and at 1535 and 1562 cm−1 for the de-convoluted amide II region corresponding to β-sheet and β-turn conformations, respectively (Fig. 3b).15 These findings infer that intra-molecular hydrogen bonds in the core of each conjugate (β-turn) complement inter-molecular hydrogen bonding between the arms of different conjugates (β-sheet).
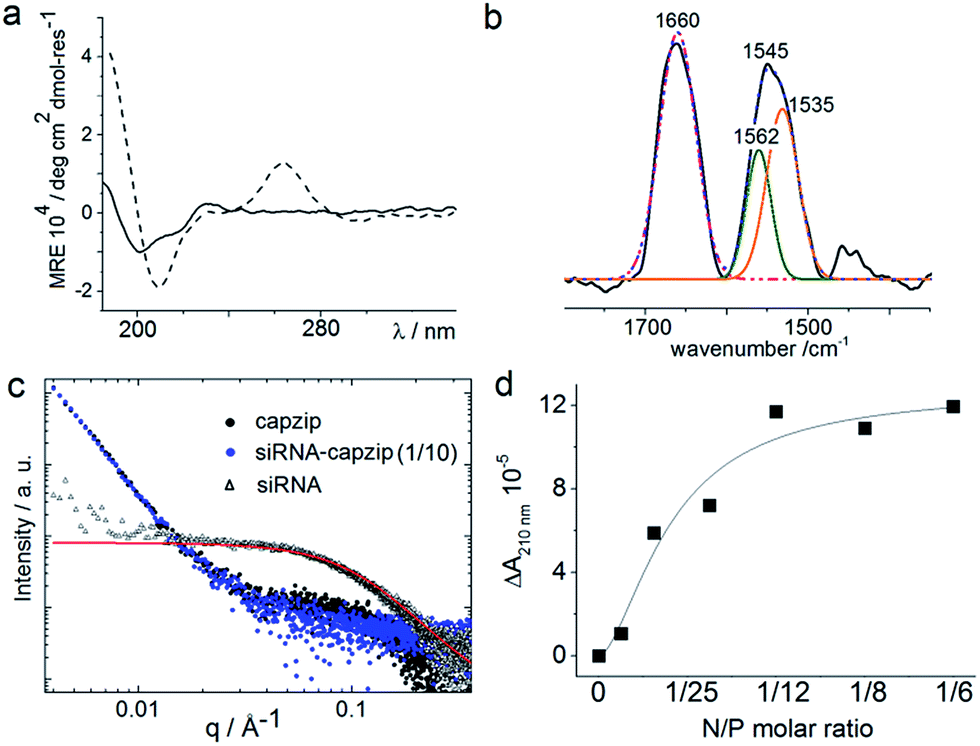 | ||
| Fig. 3 Capzip folding with and without siRNA. (a) CD spectra for capzip (solid line) and capzip with siRNA (30 μM, dashed line). (b) FT-IR spectra for capzip: raw spectrum (black line), cumulative fit (dotted blue line), deconvoluted amide I spectrum (pink line) and amide II spectra (green and orange lines). Folding conditions: 100 μM peptide, pH 7.4, 10 mM MOPS, 20 °C. (c) SAXS curves for capzip (200 μM, black), siRNA (20 μM, red) and capzip–siRNA (1/10 N/P molar ratio, blue). (d) Differential absorbance (ΔA) at 210 nm versus N/P molar ratio derived from CD titration spectra for capzip (100 μM). | ||
Such synergy between folding and assembly manifested in a compact globular morphology of the capsules observed by AFM in solution (Fig. S3†). Complementary evidence was provided by the cross-sectional analysis of the assembled capsules using fluorescence microscopy. Fluorescein, used to label capzip, incorporated exclusively in the walls of the capsules revealing an inner cavity (Fig. 2c and S2c†). Small angle X-ray scattering (SAXS) measurements, which can provide a direct probe of supramolecular assemblies in solution, re-emphasised the synergy: SAXS curves obtained with a characteristic Porod's dependence (I ∼ q−4) in the terminal regions were consistent with compact capsules interfacing with the solvent (Fig. 3c).16
Combined the results support the capsule-like assembly of capzip as per the design rationale. To assess the functional relevance of the design the capsules were probed for their ability to (i) enable gene transport and (ii) exert rapid and localized antimicrobial responses. Regarding (i), the capsules were probed to deliver small interfering RNAs (siRNAs), which are promising tools for RNA interference (RNAi) devised to inhibit targeted gene expression. However, siRNAs are easily degradable, cannot cross cellular membranes and require constant protection and guided delivery, which hampers further progress.17 Capzip offers an exploitable solution.
The design was found to readily co-assemble with model siRNAs (21 bp duplexes) into structures that were similar to bare capsules (Fig. 2d and S2d†). CD spectra of these assemblies contained characteristic features of the A-DNA form, which were more profound for higher siRNA ratios, including a positive Cotton effect at 260–270 nm and maxima at ∼210 nm and ∼190 nm (Fig. 3a). SAXS data was consistent with the CD results and for siRNA alone showed the expected double-helix dimensions of 2.3 × 5 nm (Fig. 3c). Signals recorded for capzip with and without siRNA were similar. However, the signals were distinctive from those for siRNA alone suggesting that siRNA incorporates without affecting the original capsule topology and is efficiently packed by the peptide assembly (Fig. 3c).18 siRNA affinity to capzip (Kd = 3.25 ± 0.88 μM) was in expected ranges for peptide–RNA complexes (Fig. 3d, S4a and b†),19,20 while N/P molar ratios at ∼1/10 matched those obtained as saturation points by agarose gel electrophoresis (Fig. S4c†).
At these ratios capzip promoted an active siRNA delivery within the first hours, with particulate cytoplasmic spreads of fluorescent siRNA suggesting an endocytic uptake. The spreads were stable over the first five hours of incubation whereupon fluorescence was not observed (Fig. 4a). These characteristics were comparable to those of other transfection reagents21 and conform to the RNAi rationale aimed at transient gene silencing.17 To gain a quantitative insight into this, we monitored knockdown at the mRNA level using a HeLa cell line with two housekeeping genes, ACTB (β-actin, targeted) and GAPDH (reference).22 The silencing of β-actin mRNA was detected at 22 ± 2 hours post-transfection. Knockdown “fitness” was expressed relative to cells treated with siRNA alone (background) and normalised against viable cell counts (Fig. 4b). Lipofectamine® used as a positive control showed higher knockdown levels compared to capzip but this was at the expense of substantially reduced cell numbers indicating apparent cytotoxicity.23 Indeed, Alamar Blue® cell proliferation and viability assays, which are quantitative redox indicators of metabolically active cells, revealed impaired cell viability for the control reagents (Fig. S5†). By contrast, cells transfected with capzip at different transfection concentrations remained viable over 24 and 48 hours (Fig. S5†). No obvious increases in knockdown were observed for capzip at higher peptide concentrations suggesting that N/P ratios close to equimolar are sufficient for siRNA uptake and that the peptide does not affect cell viability. In contrast to lipofectamine, which undergoes non-specific fusion with cell membranes, capzip is rich in arginines and tryptophans, known for profound cell-penetrating24 and membrane fusion25 properties, which render the capsules intrinsically transfecting. Because capzip proved to be non-toxic, but has antimicrobial propensity, it was deemed appropriate here to address the earlier point (ii).
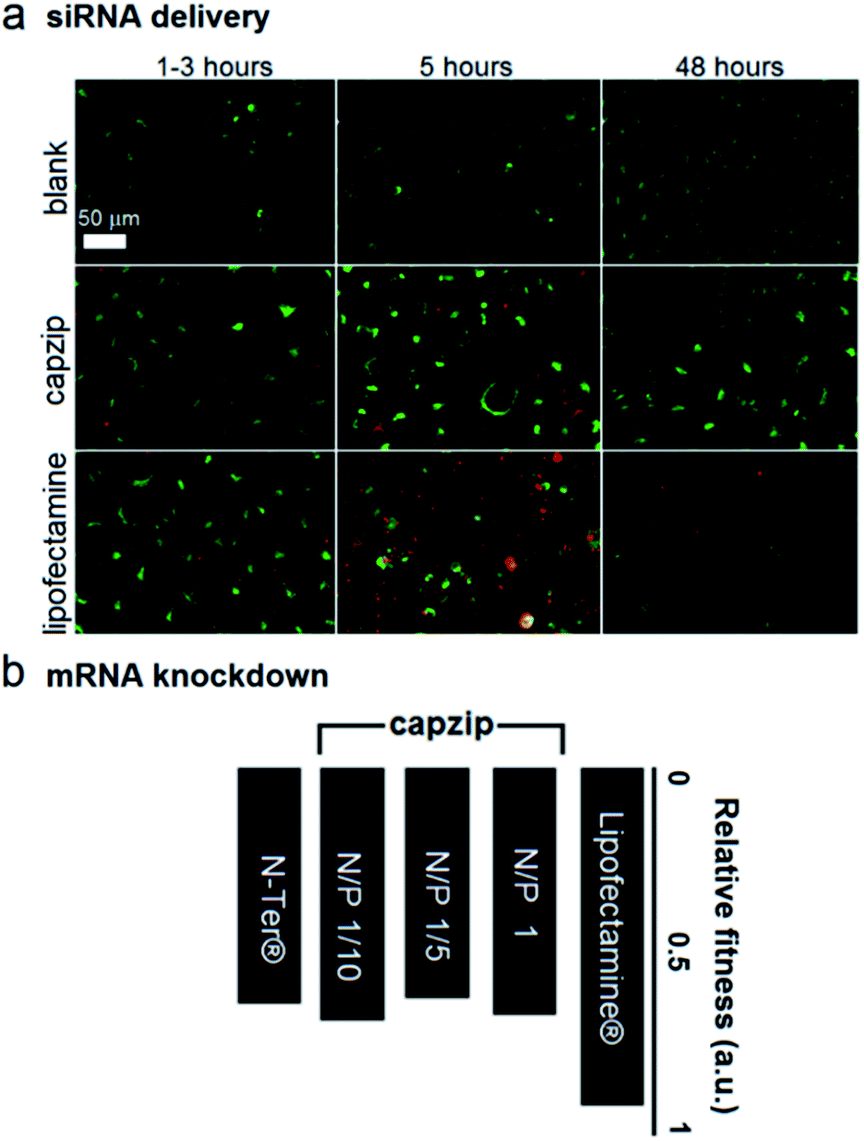 | ||
| Fig. 4 Capzip-promoted gene delivery and silencing. (a) Fluorescence micrographs of HeLa cells expressing green fluorescent protein used as internal background fluorescence for Alex647-labelled siRNA, at 1/5 N/P (siRNA–peptide) molar ratios. (b) Knockdown fitness of capzip and commercial Lipofectamine® RNAiMAX and N-TER® (positive controls) normalised against siRNA alone (negative control) and the total counts of viable cells at different N/P (siRNA–peptide) molar ratios. | ||
Capzip was found antimicrobial in bacterial culture, with minimum inhibitory concentrations (MICs) typical of other antimicrobial agents (Table S1†). The MICs were comparable to the transfection concentrations of capzip, non-toxic to human cells. To relate MICs to antimicrobial responses at the cellular level we sought complementary evidence from stain-dead assays using planktonic bacterial culture as a measure of total cell counts with and without capzip. Marginal counts for capzip-treated samples were in drastic contrast to substantial bacterial colonisation for incubations without peptide (Fig. 5a). The findings support sustained antimicrobial activity and, with no hemolytic effects detected (Table S1†), reflect the differential plasticity of capzip in cellular environments, likely involving fast and localised disruption of microbial membranes.
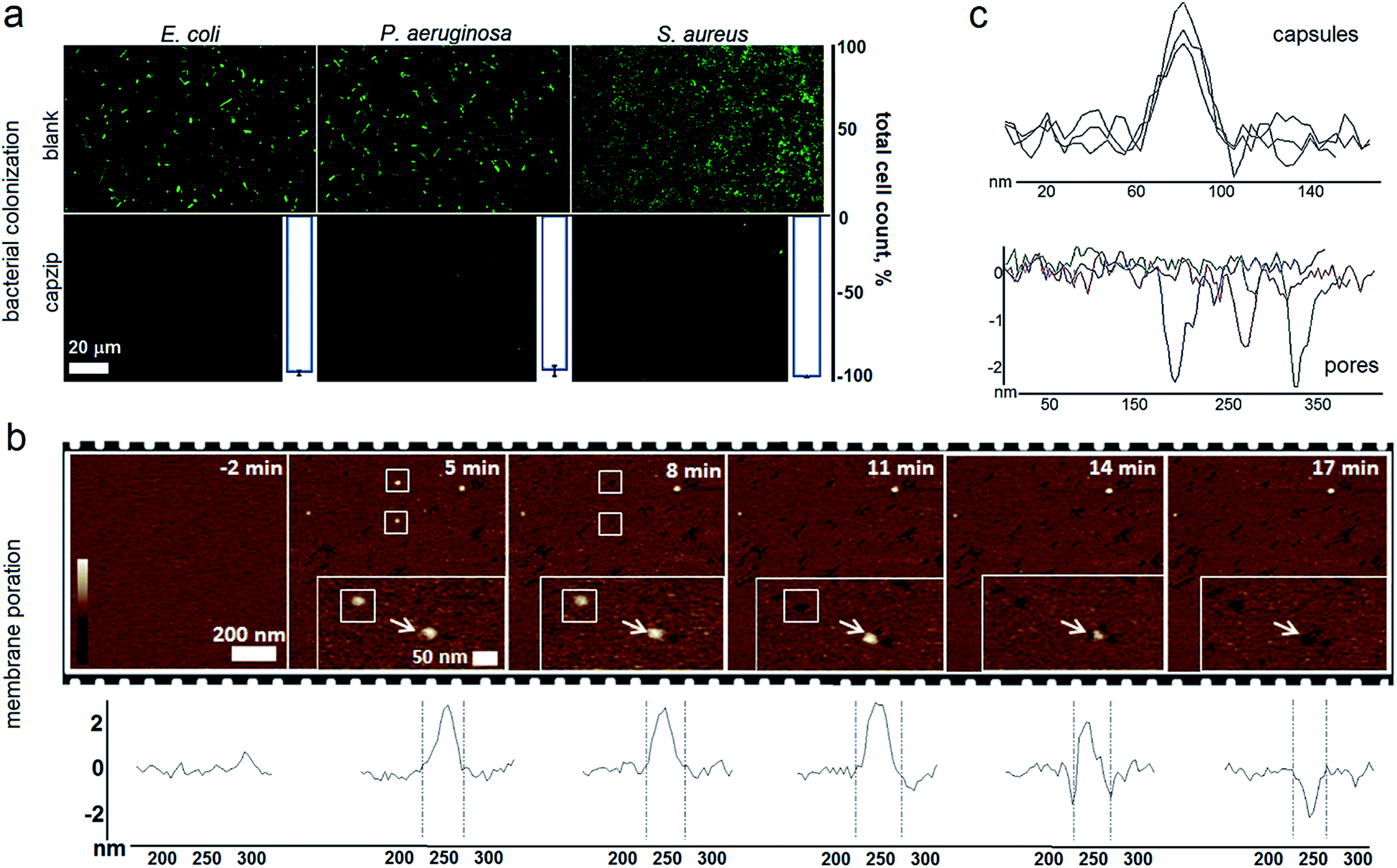 | ||
| Fig. 5 Capzip-promoted antimicrobial activity and mechanism. (a) Confocal micrographs of bacterial cells after 16 hour incubations with and without capzip, stained with membrane-permeant SYTO®9. White histogram bars denote total cell counts (%) for bacterial colonization with capzip after subtracting background adhesion taken as 100%. (b) AFM topography of SLBs during capzip incubation in solution. White boxes and arrows highlight individual capsule conversions into pores. Color scale is 6 nm. Cross sections show the evolution of a capsule (white arrow in inset images) into a pore in real time. (c) Representative cross-sections of capsules and pores. | ||
To probe this in sufficient detail, we measured membrane disruption in supported lipid bilayers (SLBs) used as models for fluid-phase bacterial membranes that can be imaged in solution and in real time.6 As gauged by AFM, capzip caused distinctive pore-like lesions in SLBs within just first minutes of incubation (Fig. 5b). The pore depths of 1.8 ± 0.4 nm were remarkably consistent, while their diameters correlated with those of observed capsules (31 ± 3 nm) (Fig. 5c). Notably, individual capsules tended to convert into individual pores at their precise landing positions, with conversion times being 3–17 min. Such fast and localised responses prompt a distinctive mechanism according to which arginines and tryptophans pre-arrange in the capsules into amphipathic cationic β-strands that penetrate lipid bilayers at one-strand depths, ∼2 nm (0.3 nm translation per β-sheet residue), as capsules land and collapse on membrane surfaces thereby converting into pore and lesions (Fig. 5b and Video S1†).
Conclusions
In summary, we have introduced a conceptual design for virus-like capsules with differential biological activity. Similar to viruses, the capsules self-assemble from individual subunits and promote gene delivery and silencing. Unlike viruses, for which monodispersity in size is a necessary constraint ensuring the encapsulation of viral genes, the capsules are not limited to hosting specific cargo and are structurally plastic. This allows them to attack bacterial cells on direct contact. With most bacteria having 0.2 × ≤2 μm dimensions, 0.02–1 μm capsules match the task perfectly. Rigid viral capsids cannot readily accommodate or adjust to encapsulate different cargo or more of the same cargo. Capzip capsules are free of such constraints and can be utilised to deliver a range of cargo while providing antimicrobial protection. Thus, the concept holds promise as a structural platform for engineering biologically differential nanomaterials and adds to the growing toolkit of nano-defined materials26–28 demonstrating the versatility of applied chemistry and design.Acknowledgements
We acknowledge funding from the UK's Department of Business, Innovation and Skills. We thank E. R. da Silva, K. Inoue and J. Doutch for support with SAXS experiments (B21 BioSAXS beamline; project No. SM10077-1) and R. Hussain and T. Javorfi with SRCD experiments (B23 beamline; project No. SM10083-1) (both Diamond Light Source, UK) and J. Mantel (University of Bristol) for support with the electron microscopy experiments.Notes and references
- X. Zhu, A. F. Radovic-Moreno, J. Wu, R. Langer and J. Shi, Nano Today, 2014, 9, 478 CrossRef CAS PubMed.
- B. Lamarre and M. G. Ryadnov, Macromol. Biosci., 2011, 11, 503 CrossRef CAS PubMed.
- I. Yacoby and I. Benhar, Nanomedicine, 2008, 3, 329 CrossRef CAS PubMed.
- E. Mastrobattista, M. A. van der Aa, W. F. Hennink and D. J. Crommelin, Nat. Rev. Drug Discovery, 2006, 5, 115 CrossRef PubMed.
- A. L. Hilchie, K. Wuerth and R. E. W. Hancock, Nat. Chem. Biol., 2013, 9, 761 CrossRef CAS PubMed.
- P. D. Rakowska, et al. , Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8918 CrossRef CAS PubMed.
- D. J. Schibil, P. M. Hwang and H. J. Vogel, FEBS Lett., 1999, 446, 213 CrossRef.
- A. G. Cochran, N. J. Skelton and M. A. Starovasnik, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5578 CrossRef CAS PubMed.
- T. Douglas and M. Young, Science, 2006, 312, 873 CrossRef CAS PubMed.
- N. Faruqui, A. Bella, J. Ravi, S. Ray, B. Lamarre and M. G. Ryadnov, J. Am. Chem. Soc., 2014, 136, 7889 CrossRef CAS PubMed.
- D. Papapostolou, A. M. Smith, E. D. Atkins, S. J. Oliver, M. G. Ryadnov, L. C. Serpell and D. N. Woolfson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10853 CrossRef CAS PubMed.
- M. G. Ryadnov, Angew. Chem., Int. Ed., 2007, 46, 969 CrossRef CAS PubMed.
- S. G. Tarasov, V. Gaponenko, O. M. Howard, Y. Chen, J. J. Oppenheim, M. A. Dyba, S. Subramaniam, Y. Lee, C. Michejda and N. I. Tarasova, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9798 CrossRef CAS PubMed.
- A. Perczel, M. Hollosi, B. M. Foxman and G. D. Fasman, J. Am. Chem. Soc., 1991, 113, 9772 CrossRef CAS.
- E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Biophys. J., 2006, 90, 2946 CrossRef CAS PubMed.
- C. Schmitt, C. Moitzi, C. Bovay, M. Rouvet, L. Bovetto, L. Donato, M. E. Leser, P. Schurtenberger and A. Stradner, Soft Matter, 2010, 6, 4876–4884 RSC.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- R. Ni and Y. S. Chau, J. Am. Chem. Soc., 2014, 136, 17902 CrossRef CAS PubMed.
- J. Pai, T. Yoon, N. D. Kim, I. S. Lee, J. Yu and I. Shin, J. Am. Chem. Soc., 2012, 134, 19287 CrossRef CAS PubMed.
- S. Goutelle, M. Maurin, F. Rougier, X. Barbaut, L. Bourguignon, M. Ducher and P. Maire, Fundam. Clin. Pharmacol., 2008, 22, 633 CrossRef CAS PubMed.
- S. A. Bustin, V. Benes, J. A. Garson, J. Hellemans, J. Huggett, M. Kubista, R. Mueller, T. Nolan, M. W. Pfaffl, G. L. Shipley, J. Vandesompele and C. T. Wittwer, Clin. Chem., 2009, 55, 611 CAS.
- L. Crombez, G. Aldrian-Herrada, K. Konate, Q. N. Nguyen, G. K. McMaster, R. Brasseur, F. Heitz and G. Divita, Mol. Ther., 2009, 17, 95 CrossRef CAS PubMed.
- M. Elsabahy, S. Zhang, F. Zhang, Z. J. Deng, Y. H. Lim, H. Wang, P. Parsamian, P. T. Hammond and K. L. Wooley, Sci. Rep., 2013, 3, 3313, DOI:10.1038/srep03313.
- I. Nakase, T. Takeuchi, G. Tanaka and S. Futaki, Adv. Drug Delivery Rev., 2008, 60, 598 CrossRef CAS PubMed.
- S. C. Harrison, Nat. Struct. Mol. Biol., 2008, 15, 690 CAS.
- A. Hernandez-Garcia, et al. , Nat. Nanotechnol., 2014, 9, 698 CrossRef CAS PubMed.
- M. T. Dedeo, K. E. Duderstadt, J. M. Berger and M. B. Francis, Nano Lett., 2010, 10, 181 CrossRef CAS PubMed.
- N. P. King, et al. , Nature, 2014, 510, 103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, microscopy, spectroscopy, electrophoretic and X-ray scattering data. See DOI: 10.1039/c5sc03260a |
| This journal is © The Royal Society of Chemistry 2016 |