
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of new mutually orthogonal bioorthogonal cycloaddition pairs through computational screening†
Maruthi Kumar
Narayanam
 a,
Yong
Liang
b,
K. N.
Houk
*bc and
Jennifer M.
Murphy
*a
a,
Yong
Liang
b,
K. N.
Houk
*bc and
Jennifer M.
Murphy
*a
aCrump Institute for Molecular Imaging, David Geffen School of Medicine, University of California, Los Angeles, California 90095, USA. E-mail: jmmurphy@mednet.ucla.edu
bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA. E-mail: houk@chem.ucla.edu
cDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, California 90095, USA
First published on 11th November 2015
Abstract
Density functional theory (DFT) calculations and experiments in tandem led to discoveries of new reactivities and selectivities involving bioorthogonal sydnone cycloadditions. Dibenzocyclooctyne derivatives (DIBAC and BARAC) were identified to be especially reactive dipolarophiles, which undergo the (3 + 2) cycloadditions with N-phenyl sydnone with the rate constant of up to 1.46 M−1 s−1. Most significantly, the sydnone-dibenzocyclooctyne and norbornene-tetrazine cycloadditions were predicted to be mutually orthogonal. This was validated experimentally and used for highly selective fluorescence labeling of two proteins simultaneously.
Introduction
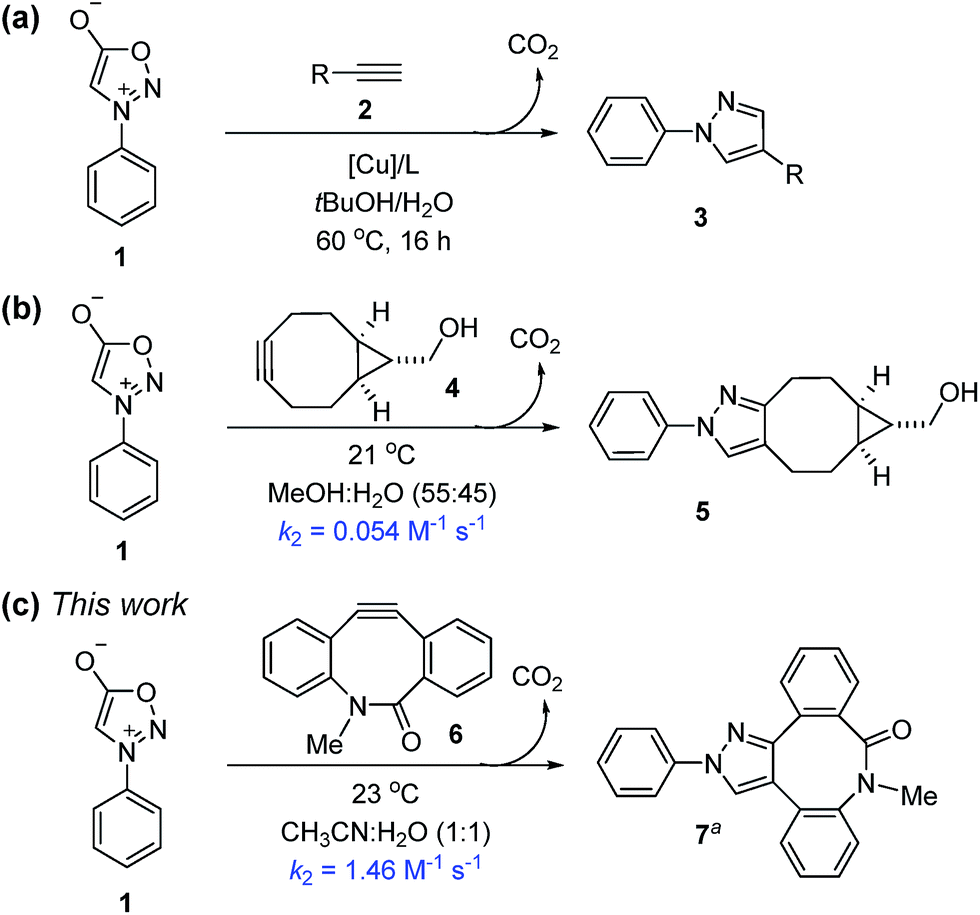
The interrogation of the molecular details of biological processes in living systems demands exquisite selectivity combined with high reactivity. Despite many challenges, the design and tuning of reaction partners has provided a myriad of bioorthogonal chemical reactions, which have had a major impact on the study of many biological processes in vivo.1 The discovery of useful bioorthogonal reactions is generally empirical. The modified Staudinger reaction, reported by Bertozzi in 2000,2 involves organic azides and triaryl phosphines, but is somewhat slow (typical second-order rate constant of 2 × 10−3 M−1 s−1), necessitating the use of high concentrations of phosphine reagents. A remarkable advance in this field was Bertozzi et al.'s azide-cyclooctyne (3 + 2) cycloaddition that has enabled cellular component labeling in living systems.3 Stimulated by this distortion-accelerated process, nearly all recent additions to the bioorthogonal reaction repertoire are cycloadditions,4 especially 1,3-dipolar5 and Diels–Alder6 reactions. A great deal of progress has been made towards developing new reactions for labeling biomolecules with extraordinarily high rate constants and monitoring multicomponent processes in complex environments.7First discovered in 1935, sydnones are remarkably stable mesoionic heterocycles that contain an azomethine imine moiety.8 In the early 1960s, Huisgen reported that, upon treatment with alkynes at 170 °C, sydnones undergo (3 + 2) cycloadditions to form pyrazoles, via a bicyclic intermediate which eliminates carbon dioxide.9 Although sydnones have been used for the synthesis of biologically interesting molecules,10 the potential of sydnones as bioorthogonal reaction partners was overlooked until recently. Using a high-throughput immunoassay screening, Taran and co-workers discovered that sydnones are biocompatible and undergo 1,3-dipolar cycloadditions with terminal alkynes in the presence of a copper phenanthroline complex to produce 1,4-pyrazoles in up to 99% yield (Scheme 1a).11 Chin developed a Cu-free strain-promoted (3 + 2) cycloaddition between N-phenyl sydnone and bicyclo-[6.1.0]-nonyne (BCN) at ambient temperature to afford the corresponding pyrazole in 30 minutes, with an isolated yield of 99% (Scheme 1b).12 The rate constant in MeOH–H2O (55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45) was determined to be 0.054 M−1 s−1 at 21 °C.13
:45) was determined to be 0.054 M−1 s−1 at 21 °C.13
 | ||
| Scheme 1 Reactivity of the (3 + 2) cycloadditions involving N-phenyl sydnone (aonly one regioisomer is depicted). | ||
These results inspired our investigation into the reactivity of sydnones as 1,3-dipoles for bioorthogonal cycloadditions. Since many strained alkenes and alkynes have been well established as bioorthogonal 2π cycloaddends, it is now efficient to evaluate reactivities towards N-phenyl sydnone through computational screening. Here we report the computation-guided discovery of two new bioorthogonal (3 + 2) cycloaddition reactions of N-phenyl sydnone with biarylazacyclooctynone (BARAC) and dibenzoazacyclooctyne (DIBAC), which demonstrate significant rate enhancement over the previously reported sydnone-BCN cycloaddition.12 In addition, with the aid of DFT calculations, we design and identify two mutually orthogonal reaction pairs, which enable highly selective fluorescence labeling of two proteins simultaneously.
Results and discussion
To better understand the sydnone-BCN cycloaddition and to accelerate the discovery of new bioorthogonal chemistry involving N-phenyl sydnone, DFT calculations were performed at the M06-2X/6-311+G(d,p)//M06-2X/6-31G(d) level of theory.14,15 We have previously established the reliability of prediction of rates in aqueous solution with this method.16 As shown in Fig. 1, the (3 + 2) cycloaddition of sydnone 1 with BCN 4via transition state TS1 requires an activation free energy of 22.2 kcal mol−1 in water, and the formation of cycloadduct 8 is exergonic by 22.6 kcal mol−1. The conversion of intermediate 8 to final pyrazole product 5 by the release of CO2via transition state TS2 has almost no barrier. The overall reaction is strongly exergonic by 111.1 kcal mol−1. This indicates that, after the 1,3-dipolar cycloaddition, the subsequent CO2 release occurs spontaneously, and that the reaction rate is entirely controlled by the (3 + 2) cycloaddition step.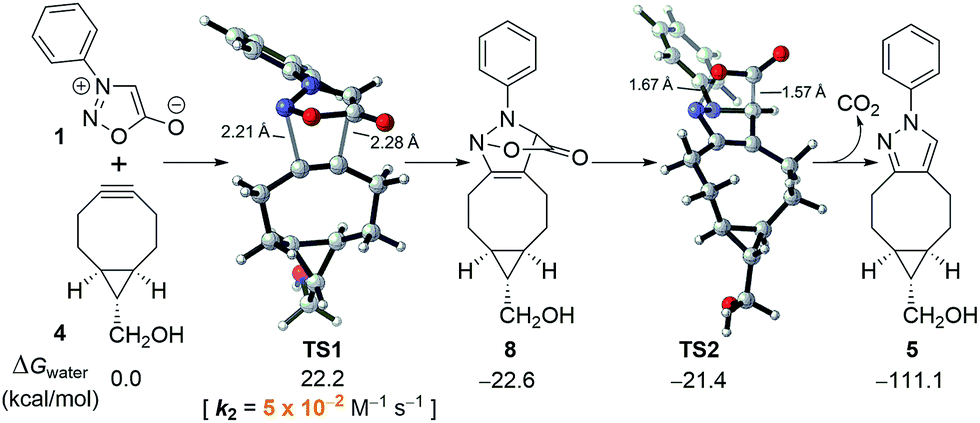 | ||
| Fig. 1 Energetics and transition states for the (3 + 2) cycloaddition of N-phenyl sydnone (1) with BCN 4 and subsequent CO2 release. | ||
As shown in Fig. 2a, we located transition-state structures TS3–10 for eight (3 + 2) cycloadditions of N-phenyl sydnone with strained alkenes and alkynes previously used as bioorthogonal reagents. On the basis of the DFT-computed activation free energies (TS1 and TS3–10, Fig. 1 and 2a) and the experimental rate constant of the sydnone-BCN cycloaddition (k2 = 5 × 10−2 M−1 s−1, Scheme 1b), we predicted a series of rate constants (k2) of unexplored bioorthogonal sydnone cycloadditions. It was found that the range of reactivities is enormous, with activation free energies varying from 30 to 19 kcal mol−1, which correspond to a 108 range in rate constants at room temperature. For cyclooctyne, 1,3-disubstituted cyclopropene (Cp(1,3)), difluorocyclooctyne (DIFO), and trans-cyclooctene (TCO) (TS5–8), the predicted rate constants range from 10−3 to 10−1 M−1 s−1 (shown in yellow, Fig. 2a). These data are very close to the reported rate constant for the sydnone-BCN cycloaddition. This suggests that these reactions may be employed for biomolecular labeling but may not be suitable for in vivo applications due to the moderate rate constants. To our delight, calculations predicted that when N-phenyl sydnone reacts with DIBAC and BARAC (TS9–10), the rate constants will be 1 and 10 M−1 s−1, respectively (shown in green, Fig. 2a). By contrast, sydnone 1 is predicted to be inert to norbornene and 3,3-disubstituted cyclopropene (Cp(3,3)) (TS3–4) under physiological conditions, according to the expected rate constants of 10−7 M−1 s−1 (shown in red, Fig. 2a). This dramatic reactivity difference between dibenzocyclooctyne derivatives (DIBAC and BARAC) and norbornene in the sydnone cycloaddition was then exploited for design of mutually orthogonal bioorthogonal reaction pairs.
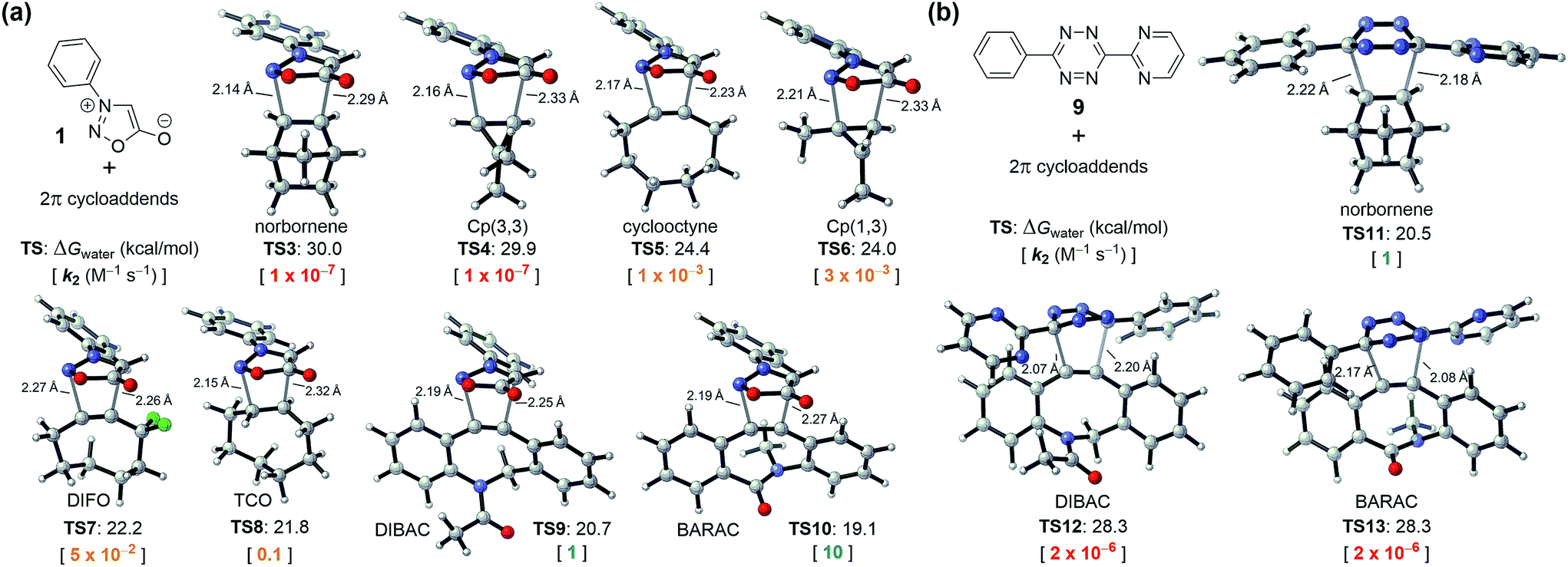 | ||
| Fig. 2 (a) DFT-computed activation free energies for the (3 + 2) cycloadditions of sydnone 1 with eight strained alkenes and alkynes at the CPCM(water)-M06-2X/6-311+G(d,p)//M06-2X/6-31G(d) level of theory and the predicted rate constants in water at 25 °C. (b) DFT-computed activation free energies and predicted rate constants for the (4 + 2) cycloadditions of tetrazine 9 with norbornene, DIBAC, and BARAC. | ||
Fig. 2b shows that diaryltetrazine 9 reacts smoothly with norbornene (TS11). However, the Diels–Alder reactions of DIBAC and BARAC (TS12–13) with diaryltetrazine 9 will not occur at room temperature, according to the predicted rate constants of 2 × 10−6 M−1 s−1 (shown in red, Fig. 2b). As a result, the sydnone-dibenzocyclooctyne and norbornene-tetrazine cycloadditions are predicted to be mutually orthogonal.
Our previous computational studies showed that the distortion energy of substrates is crucial to the reactivity of bioorthogonal cycloadditions.16,17 Usually, the more pre-distorted substrate is more reactive as it takes less energy to reach its transition-state geometry. Dibenzocyclooctyne derivatives (DIBAC and BARAC) have much smaller alkyne bond angles than normal alkynes (ca. 153° versus 180°),17a while norbornene is just pre-distorted by ca. 8° as compared to normal alkenes.17b Therefore, in sydnone cycloadditions, dibenzocyclooctyne derivatives (DIBAC and BARAC) are significantly more reactive than norbornene. However, DIBAC and BARAC exhibit steric hindrance due to the two aryl hydrogen atoms ortho to the alkyne,16 so their Diels–Alder reactions with bulky disubstituted tetrazines are dramatically slower than that of norbornene.
To test these predictions, we first measured the experimental rate constant for the 1,3-dipolar cycloaddition of N-phenyl sydnone (1) with BARAC 6 (for details, see the ESI†). As expected, a significantly enhanced second-order rate constant of 1.46 M−1 s−1 was obtained for the (3 + 2) cycloaddition in MeCN–H2O (1:1) at 23 °C (Scheme 1c). This is about 30 times larger than that for the sydnone-BCN cycloaddition.12
Scheme 2 summarizes experiments to test the predicted mutual orthogonality between sydnone-DIBAC and norbornene-tetrazine cycloadditions. The second-order rate constant for the (3 + 2) cycloaddition of DIBAC 10 with sydnone 1 in MeOH–H2O (55:45) was determined to be 0.902 M−1 s−1 (for details, see the ESI†). The Diels–Alder reaction of diaryltetrazine 13 with 5-norbornene-2-acetic acid (12) was found to have a k2 of 1.05 M−1 s−1 (for details, see the ESI†) in MeOH–H2O (55:45), which is comparable to rate constants reported in the literature for similar systems.18 To test potential cross-reactivity, sydnone 1 and norbornene 12 or diaryltetrazine 13 and DIBAC 10 were incubated in an NMR tube in deuterated methanol and monitored via NMR spectroscopy. As predicted, no reaction was observed between either reaction pair at room temperature after 24 hours.
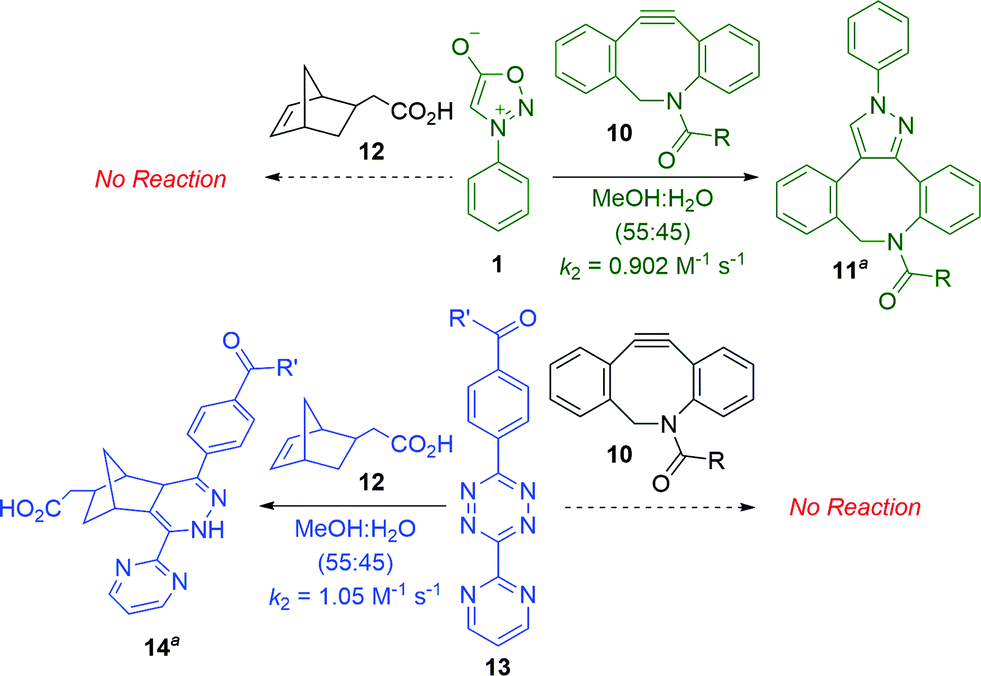 | ||
| Scheme 2 Mutual orthogonality between sydnone-DIBAC and norbornene-tetrazine cycloadditions (aR = CH2CH2NH2, R′ = NH(CH2)3NHBoc; only one regioisomer is depicted). | ||
Probing multiple biomolecules, within their native cellular habitat, requires the selective modification of each system with chemical moieties that are compatible with each other. A remarkable expansion of the bioorthogonal toolbox has equipped chemical biologists with a range of selective reactions.4,7 However, the highly complex nature of biological processes often demands concurrent modification and tracking of multiple targets within the same system. Clever manipulation of these various reactions has enabled the use of sequential click chemistry reactions to tag multiple targets in a single system without perturbations to the system.19 Hilderbrand and others have demonstrated the orthogonality between the Diels–Alder tetrazine ligation and the 1,3-dipolar azide cycloaddition to facilitate multicomponent labeling.20
Leveraging the excellent orthogonality between sydnone-DIBAC and norbornene-tetrazine cycloadditions, we examined the application of this chemistry towards dual fluorescence protein labeling. The DIBAC and norbornene moieties were appended to the surface of two ubiquitous proteins, bovine serum albumin (BSA) and ovalbumin (OVA) using standard coupling conditions (Fig. 3a). The modified proteins (BSA-DIBAC and OVA-Nor) were treated with either red fluorescent sydnone-BODIPY630 (Syd-630) or green fluorescent tetrazine-BODIPY504 (Tz-504) or both and analyzed via in gel fluorescence imaging. When the modified proteins were jointly treated with Syd-630, only BSA-DIBAC was fluorescently labeled (lane 7, Fig. 3b). Likewise, in the presence of Tz-504, OVA-Nor showed selective labeling without any detection of BSA-DIBAC fluorescence labeling (lane 8, Fig. 3b). Co-administration of both fluorophore conjugates in “one pot” resulted in the concurrent labeling of both proteins (lane 9, Fig. 3b). Unmodified proteins BSA and OVA showed no fluorescence labeling after exposure to Syd-630 and Tz-504 (lanes 4 and 5, Fig. 3b), indicating the absence of nonspecific reactivity. In a time-dependent experiment, the protein BSA-DIBAC underwent labeling and exhibited fluorescence within 1 min (lane 7, Fig. 3c), whereas the protein OVA-Nor exhibited fluorescence labeling after 15 min (lane 8, Fig. 3c). These experiments demonstrate the exquisite specificity of the sydnone and tetrazine imaging agents for DIBAC- and norbornene-modified proteins, respectively, as well as their mutual compatibility within a single environment. Moreover, for the new sydnone-DIBAC cycloaddition, the ability to undergo fluorescence labeling within 1 min using as little as 12 nM protein (with 500 μM dye) is very promising for in vivo imaging.
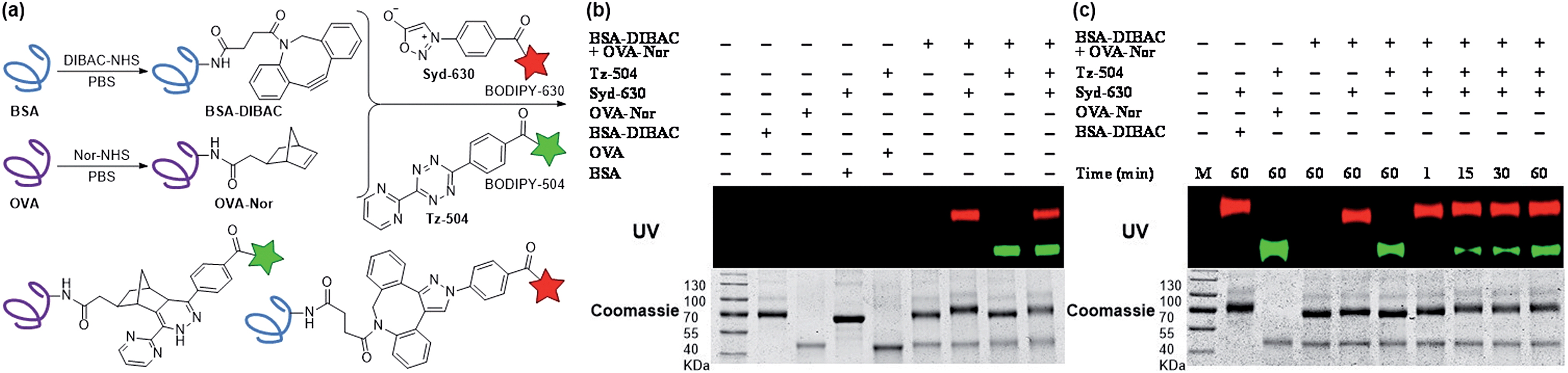 | ||
| Fig. 3 Modifications of protein surfaces via bioorthogonal cycloadditions. (a) Dibenzoazacyclooctyne (DIBAC) and 5-norbornene-2-acetic acid (Nor) were appended to BSA and OVA (20 mg mL−1 in PBS) respectively, via NHS ester-amine coupling conditions (20 mM labeling reagent). The labeled proteins BSA-DIBAC and OVA-Nor (2 mg mL−1) simultaneously react with sydnone-BODIPY (Syd-630) and tetrazine-BODIPY (Tz-504). (b) Gel analysis of BSA functionalized with DIBAC and OVA functionalized with Nor incubated for 1 h with either Syd-630, Tz-504, both reagents simultaneously, or no reagent (−). (c) Gel analysis of DIBAC-modified BSA and Nor-modified OVA with both Syd-630 and Tz-504 simultaneously for 1–60 min or no reagent (−). The protein loading on the gels was assessed with Coomassie stain. | ||
Conclusions
With the aid of computational screening, we have discovered two rapid cycloadditions between N-phenyl sydnone and dibenzocyclooctyne derivatives (DIBAC and BARAC) that proceed in aqueous media, at physiological temperature without the use of a catalyst. The predicted mutually orthogonal sydnone-DIBAC and norbornene-tetrazine cycloaddition pairs have been successfully applied to fluorescence labeling of two proteins simultaneously. DFT calculations shown in Fig. 2a also predicted that sydnone is inert to 3,3-disubstituted cyclopropene. Our previous study has proven that 3,3-disubstituted cyclopropenes react with nitrile imines via 1,3-dipolar cycloaddition but not with tetrazines via Diels–Alder reaction.19d This suggests that precise control over cycloaddition selectivity (dibenzocyclooctynes with sydnones, norbornenes with tetrazines, and 3,3-disubstituted cyclopropenes with nitrile imines) can enable labeling of three targets in a biological environment. We anticipate that these results will complement the expanding toolkit of bioorthogonal reactions and advance the efforts to simultaneously examine multiple processes within a biological system.Acknowledgements
This work was supported by the UCLA Center for In vivo Imaging in Cancer Biology (NIH P50 CA086306), the National Institutes of Health (R01GM109078), and the National Science Foundation (CHE-1361104). Calculations were performed on the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575). We thank Prof. James Wohlschlegel and Dr Ajay Vashisht for helpful discussions and assistance with protein labeling experiments. We thank Prof. Jennifer Prescher at UCI for helpful comments.Notes and references
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed; (b) M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475–484 CrossRef CAS; (c) C. S. McKay and M. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS; (d) A. Borrmann and J. C. van Hest, Chem. Sci., 2014, 5, 2123–2134 RSC.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- (a) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed; (b) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- (a) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC; (b) D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- (a) J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 16793–16797 CrossRef CAS PubMed; (b) J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS PubMed; (c) X. Ning, J. Guo, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS; (d) M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC; (e) J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS PubMed; (f) J. Dommerholt, S. Schmidt, R. Temming, L. J. Hendriks, F. P. Rutjes, J. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed; (g) T. Plass, S. Milles, C. Koehler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2011, 50, 3878–3881 CrossRef CAS; (h) M. F. Debets, S. S. Van Berkel, J. Dommerholt, A. J. Dirks, F. P. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed; (i) R. K. Lim and Q. Lin, Acc. Chem. Res., 2011, 44, 828–839 CrossRef CAS PubMed; (j) Z. Yu, Y. Pan, Z. Wang, J. Wang and Q. Lin, Angew. Chem., Int. Ed., 2012, 51, 10600–10604 CrossRef CAS PubMed; (k) Z. Yu, T. Y. Ohulchanskyy, P. An, P. N. Prasad and Q. Lin, J. Am. Chem. Soc., 2013, 135, 16766–16769 CrossRef CAS PubMed; (l) Z. Yu and Q. Lin, J. Am. Chem. Soc., 2014, 136, 4153–4156 CrossRef PubMed.
- (a) M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed; (b) N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed; (c) N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed; (d) D. S. Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 792–795 CrossRef CAS PubMed; (e) J. L. Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads, R. B. Cooley, C. Refakis, J. M. Fox and R. A. Mehl, J. Am. Chem. Soc., 2012, 134, 2898–2901 CrossRef CAS PubMed; (f) K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed; (g) K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298–304 CrossRef CAS PubMed; (h) W. Chen, D. Wang, C. Dai, D. Hamelberg and B. Wang, Chem. Commun., 2012, 48, 1736–1738 RSC; (i) J. Yang, J. Šečkutė, C. M. Cole and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 7476–7479 CrossRef CAS PubMed; (j) J. Šečkutė, J. Yang and N. K. Devaraj, Nucleic Acids Res., 2013, 41, e148 CrossRef PubMed; (k) Q. Li, T. Dong, X. Liu and X. Lei, J. Am. Chem. Soc., 2013, 135, 4996–4999 CrossRef CAS PubMed; (l) J. Yang, Y. Liang, J. Šečkutė, K. N. Houk and N. K. Devaraj, Chem.–Eur. J., 2014, 20, 3365–3375 CrossRef CAS PubMed; (m) H. Wu, J. Yang, J. Šečkutė and N. K. Devaraj, Angew. Chem., Int. Ed., 2014, 53, 5805–5809 CrossRef CAS PubMed; (n) H. Wu, B. T. Cisneros, C. M. Cole and N. K. Devaraj, J. Am. Chem. Soc., 2014, 136, 17942–17945 CrossRef CAS PubMed; (o) A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770–3776 RSC; (p) S. B. Engelsma, L. I. Willems, C. E. van Paaschen, S. I. van Kasteren, G. A. van der Marel, H. S. Overkleeft and D. V. Filippov, Org. Lett., 2014, 16, 2744–2747 CrossRef CAS PubMed; (q) D. N. Kamber, Y. Liang, R. J. Blizzard, F. Liu, R. A. Mehl, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2015, 137, 8388–8391 CrossRef CAS PubMed.
- (a) M. F. Debets, J. C. van Hest and F. P. Rutjes, Org. Biomol. Chem., 2013, 11, 6439–6455 RSC; (b) K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed; (c) H.-W. Shih, D. N. Kamber and J. A. Prescher, Curr. Opin. Chem. Biol., 2014, 21, 103–111 CrossRef CAS PubMed.
- J. C. Earl and A. W. Mackney, J. Chem. Soc., 1935, 899–900 RSC.
- R. Huisgen, R. Grashey, H. Gotthardt and R. Schmidt, Angew. Chem., Int. Ed. Engl., 1962, 1, 48–49 CrossRef.
- D. L. Browne and J. P. Harrity, Tetrahedron, 2010, 66, 553–568 CrossRef CAS.
- S. Kolodych, E. Rasolofonjatovo, M. Chaumontet, M. C. Nevers, C. Créminon and F. Taran, Angew. Chem., Int. Ed., 2013, 52, 12056–12060 CrossRef CAS PubMed.
- S. Wallace and J. W. Chin, Chem. Sci., 2014, 5, 1742–1744 RSC.
- Shortly after Chin's report (ref. 12), 4-chloro-sydnones were reported to react much faster with BCN than non-chlorinated analogues: L. Plougastel, O. Koniev, S. Specklin, E. Decuypere, C. Créminon, D.-A. Buisson, A. Wagner, S. Kolodych and F. Taran, Chem. Commun., 2014, 50, 9376–9378 RSC . For example, the rate constant for the (3 + 2) cycloaddition between N-phenyl 4-chloro-sydnone and BCN in PBS/DMSO was reported to be 0.872 M−1 s−1 at 25 °C.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- Y. Liang, J. L. Mackey, S. A. Lopez, F. Liu and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 17904–17907 CrossRef CAS PubMed.
- (a) C. G. Gordon, J. L. Mackey, J. C. Jewett, E. M. Sletten, K. N. Houk and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 9199–9208 CrossRef CAS PubMed; (b) S. A. Lopez and K. N. Houk, J. Org. Chem., 2013, 78, 1778–1783 CrossRef CAS PubMed; (c) F. Liu, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 11483–11493 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed.
- (a) B. C. Sanders, F. Friscourt, P. A. Ledin, N. E. Mbua, S. Arumugam, J. Guo, T. J. Boltje, V. V. Popik and G.-J. Boons, J. Am. Chem. Soc., 2010, 133, 949–957 CrossRef PubMed; (b) H. Stöckmann, A. A. Neves, H. A. Day, S. Stairs, K. M. Brindle and F. J. Leeper, Chem. Commun., 2011, 47, 7203–7205 RSC; (c) T. Plass, S. Milles, C. Koehler, J. Szymański, R. Mueller, M. Wießler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166–4170 CrossRef CAS PubMed; (d) D. N. Kamber, L. A. Nazarova, Y. Liang, S. A. Lopez, D. M. Patterson, H.-W. Shih, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2013, 135, 13680–13683 CrossRef CAS PubMed.
- (a) M. R. Karver, R. Weissleder and S. A. Hilderbrand, Angew. Chem., Int. Ed., 2012, 51, 920–922 CrossRef CAS PubMed; (b) L. I. Willems, N. Li, B. I. Florea, M. Ruben, G. A. van der Marel and H. S. Overkleeft, Angew. Chem., Int. Ed., 2012, 51, 4431–4434 CrossRef CAS PubMed; (c) D. M. Patterson, L. A. Nazarova, B. Xie, D. N. Kamber and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 18638–18643 CrossRef CAS PubMed; (d) C. M. Cole, J. Yang, J. Šečkutė and N. K. Devaraj, ChemBioChem, 2013, 14, 205–208 CrossRef CAS PubMed; (e) D. M. Patterson, K. A. Jones and J. A. Prescher, Mol. BioSyst., 2014, 10, 1693–1697 RSC; (f) I. Nikić, T. Plass, O. Schraidt, J. Szymański, J. A. G. Briggs, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2014, 53, 2245–2249 CrossRef PubMed; (g) A. Sachdeva, K. Wang, T. Elliott and J. W. Chin, J. Am. Chem. Soc., 2014, 136, 7785–7788 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: 10.1039/c5sc03259h |
| This journal is © The Royal Society of Chemistry 2016 |