
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral Brønsted acid-catalyzed enantioselective Friedel–Crafts reaction of 2-methoxyfuran with aliphatic ketimines generated in situ†
Azusa
Kondoh
a,
Yusuke
Ota
b,
Takazumi
Komuro
b,
Fuyuki
Egawa
b,
Kyohei
Kanomata
b and
Masahiro
Terada
*ab
aResearch and Analytical Center for Giant Molecules, Graduate School of Science, Tohoku University, Aoba-ku, Sendai 980-8578, Japan. E-mail: mterada@m.tohoku.ac.jp
bDepartment of Chemistry, Graduate School of Science, Tohoku University, Aoba-ku, Sendai 980-8578, Japan
First published on 30th October 2015
Abstract
An enantioselective Friedel–Crafts reaction with aliphatic ketimines generated in situ from hemiaminal ethers catalyzed by a chiral Brønsted acid was investigated. The reaction of 2-methoxyfuran with (thio)hydantoin-derived hemiaminal methyl ether proceeded under the influence of a chiral phosphoric acid catalyst to afford the corresponding adduct possessing a quaternary stereogenic center in high yield with high enantioselectivity. Theoretical studies were also conducted to clarify the mechanism of the stereochemical outcome and the major factors contributing to the efficient enantioselection.
The Friedel–Crafts reaction is one of the most important carbon–carbon bond-forming reactions in organic chemistry. In recent years, the enantioselective Friedel–Crafts reaction using organocatalysis has emerged as a powerful method for the synthesis of enantio-enriched compounds.1 In particular, reactions employing unsymmetrical ketimines as electrophiles have received much attention because of the utility of the adducts, chiral amines possessing a quaternary stereogenic center. Although a number of reactions have been developed to date,2 limitations remain on the applicability for ketimines and nucleophiles. For instance, the reactions employing aliphatic ketimines have largely been unexploited. Hence the development of reactions applicable to a range of ketimines substituted by alkyl groups is still a challenging topic.3 In addition, indoles and pyrroles are the only nucleophiles that have achieved a high enantioselectivity so far. In this context, we envisaged to develop a new enantioselective Friedel–Crafts reaction of aliphatic ketimines that have a variety of alkyl substituents with the expanding scope of nucleophiles using chiral phosphoric acid (CPA) as a chiral Brønsted acid catalyst.4 From a mechanistic viewpoint of the chiral phosphoric acid-catalyzed reaction, the origin of the stereochemical outcome in the carbon–carbon (C–C) bond-forming reaction of the aliphatic ketimine has scarcely been investigated5 despite conducting detailed studies of the enantioselective reduction of ketimines.6 To establish the enantioselective Friedel–Crafts reaction of aliphatic ketimines followed by the acquisition of mechanistic insight into the stereo-determining step, we designed the reaction system shown in Scheme 1.
 | ||
| Scheme 1 Reaction design. | ||
For the development of the reaction employing aliphatic ketimines as a substrate, one often encounters a problem based not only on poor reactivity of aliphatic ketimines but also on their stability and synthetic difficulty. Thus we envisioned utilizing thiohydantoin derivatives 4 and hydantoin derivatives 5, which possess a hemiaminal ether moiety, as precursors for the aliphatic ketimines. With these substrates, the corresponding ketimines are generated in situ through the elimination of alcohols under the influence of a Brønsted acid catalyst,7 and an imine carbon is activated by an electron-withdrawing group and there is a sterically less congested environment around the imine carbon because of their planarity. The utilization of (thio)hydantoin derivatives as substrates is also attractive from the synthetic point of view. The products of the designed reaction are (thio)hydantoin derivatives that have a quaternary stereogenic center at the 5-position, these are known as an important class of biologically active molecules with broad medicinal and agrochemical applications,8–11 and can act as precursors of α-amino acid derivatives by hydrolysis. Furthermore, 2-methoxyfuran (6) was chosen as a reactant to expand the scope of nucleophiles, of which the product subunit can potentially function as a handle for further manipulation of the products.12 Herein we report the enantioselective Friedel–Crafts reaction of 2-methoxyfuran with aliphatic ketimines generated in situ catalyzed by chiral phosphoric acid to provide (thio)hydantoin derivatives containing a quaternary stereogenic center in a highly stereoselective manner. Theoretical studies were also conducted to clarify the mechanism of the stereochemical outcome and the major factors contributing to the efficient enantioselection.
The initial experiment was performed with racemic hemiaminal methyl ether 4a, having a methyl group as a substituent on the imine carbon, and 2-methoxyfuran (6) in the presence of a catalytic amount of chiral BINOL-derived phosphoric acid (R)-1a in toluene (Table 1, entry 1). Pleasingly, the reaction proceeded smoothly in the presence of molecular sieves (MS) 5A which were employed to scavenge methanol generated during the formation of the ketimine. The desired product 7a was obtained at a high yield albeit with moderate enantioselectivity. Evaluation of several phosphoric acids including chiral BINOL-derived (R)-1 having different substituents at the 3 and 3′-positions, chiral H8-BINOL-derived (R)-2, and chiral SPINOL-derived (S)-3, revealed that (S)-3c was the optimal phosphoric acid and resulted in a 94% yield with 92% ee (entry 7). Further improvement in both yield and ee value was achieved by using benzene as the solvent instead of toluene (entry 8).13
|
|
||||
|---|---|---|---|---|
| Entry | CPA | Time (h) | Yieldb (%) | eec (%) |
| a Reaction conditions: 4a (0.10 mmol), 6 (0.11 mmol), CPA (5.0 μmol), MS 5A (100 mg), toluene (0.50 mL). b Isolated yields. c Enantiomeric excess of 7a was determined by chiral stationary phase HPLC analysis. Absolute configuration of 7a was determined to be S by X-ray crystallographic analysis.14 See the ESI for detail. d Benzene was used as a solvent instead of toluene. | ||||
| 1 | (R)-1a | 4 | 92 | 57 |
| 2 | (R)-1b | 6 | 84 | 84 |
| 3 | (R)-1c | 4 | 68 | 75 |
| 4 | (R)-2b | 4 | 76 | 76 |
| 5 | (R)-2c | 6 | 68 | 70 |
| 6 | (S)-3b | 4 | 86 | 68 |
| 7 | (S)-3c | 4 | 94 | 92 |
| 8d | (S)-3c | 4 | >99 | 93 |
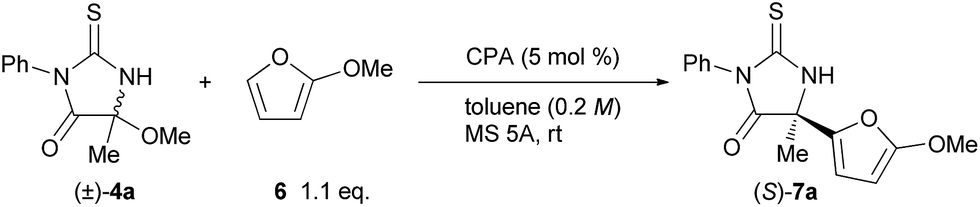
In order to clarify the origin of the stereochemical outcome, we then conducted theoretical studies of the transition states of the stereo-determining C–C bond-forming step. Four transition structures of the C–C bond-forming step were possible through the combination of the re- and si-faces of the pro-chiral reactants, the ketimine and 2-methoxyfuran (6) (Fig. 1). In the transition states TSss affording (S)-7a, the si-face of the ketimine reacts with the re- and si-faces of 2-methoxyfuran (6), generating TSs-re and TSs-si, respectively. Similarly, TSr-si and TSr-re were generated for TSrs, which results in the formation of (R)-7a. The geometries of the TSss and TSrs were fully optimized and characterized using frequency calculations at the B3LYP level of density functional theory with the 6-31G* basis set.15,16 After thorough screening of plausible transition structures to determine the relative location of the reactants and the chiral phosphoric acid catalyst, four transition structures of the corresponding configurations were localized. In each optimized structure, the ketimine and 2-methoxyfuran (6) interact with the catalyst through an O⋯H⋯N hydrogen bond and a C–H⋯O hydrogen bond, respectively.17 The TSs-si and TSr-re were energetically less favorable than the TSs-re and TSr-si, presumably due to the steric repulsion between the N-phenyl substituent of the ketimine and the methoxy group of 6 (dashed curves in Fig. 1). More importantly, the transition state TSs-re [which affords (S)-7a] was more stable than the TSr-si [which affords (R)-7a]. The (S)-selective pathway was energetically favorable for the reaction catalyzed by (S)-3c, which is consistent with the experimental results.
 | ||
| Fig. 1 Schematic representation models of TSss and TSrs. The relative energies of the optimized structures in the gas phase are shown in kcal mol−1, with relative Gibbs free energies (kcal mol−1) in parentheses. The relative energies (kcal mol−1) which were obtained by single-point energy calculations at the B3LYP/6-311+G** level using the SCRF method based on PCM (∈ = 2.2706 for benzene) are shown in brackets.18 Steric repulsions are indicated by dashed curves. | ||
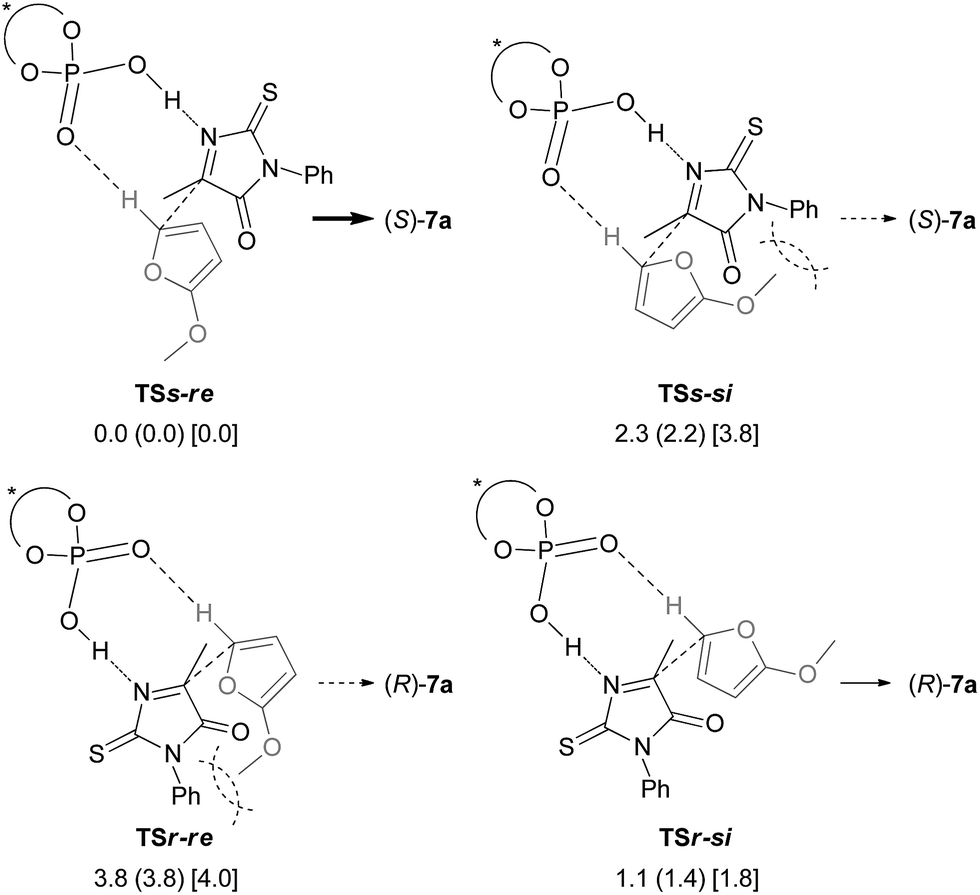
Further structural analyses of TSs-re and TSr-si allowed the identification of the major factors contributing to the efficient enantioselection. Three-dimensional transition structures of TSs-re and TSr-si are illustrated in Fig. 2. As pointed out in Fig. 1, the hydrogen atom at the 5-position of 2-methoxyfuran (6) interacts with chiral phosphoric acid (S)-3c through the C–H⋯O hydrogen bond (dashed blue lines in Fig. 2). In fact, the distances between the hydrogen and oxygen atoms (1.99 Å in TSs-re and 2.05 Å in TSr-si) are significantly shorter than the sum of the van der Waals radii of the hydrogen and oxygen atoms (ca. 2.7 Å). Furthermore, the ketimine is activated via protonation by chiral phosphoric acid (S)-3c to form the O⋯H⋯N hydrogen bond (dashed blue lines in Fig. 2). More interestingly, in both of the transition states, an additional C–H⋯O hydrogen bond forms between the α-hydrogen atom of the methyl group attached to the ketimine and the phosphoryl oxygen of (S)-3c (2.25 Å in TSs-re and 2.14 Å in TSr-si) (dashed red lines in Fig. 2). It can be considered that these two hydrogen bonds, O⋯H⋯N and C–H⋯O, fix the relative location between the ketimine and chiral phosphoric acid (S)-3c. It is obvious that the observed high enantioselectivity stems from the formation of the hydrogen bond network among the triad of components, resulting in a conformational fixation of the transition states. In the energetically favorable TSs-re, the ketimine and 6 are nearly parallel to the phenanthryl plane of the catalyst substituent to avoid steric congestion (Fig. 2a). In contrast, in the less-favorable TSr-si, both the ketimine and 6 are inserted perpendicularly between two phenanthryl planes (Fig. 2b), in which the methyl group of the ketimine locates close to the bottom phenanthryl substituent. This unfavorable interaction results in steric repulsion between the reactant and the catalyst (Fig. 2b), which would destabilize TSr-si.
 | ||
| Fig. 2 Three-dimensional structures of transition states (a) TSs-re and (b) TSr-si. Relative energies (in kcal mol−1) obtained by single-point energy calculations at the B3LYP/6-311+G** level and the M06-2X/6-311+G** level with the SCRF method based on PCM (∈ = 2.2706 for benzene) are shown in brackets and double parentheses, respectively.18 Bond lengths are shown in red (Å). | ||
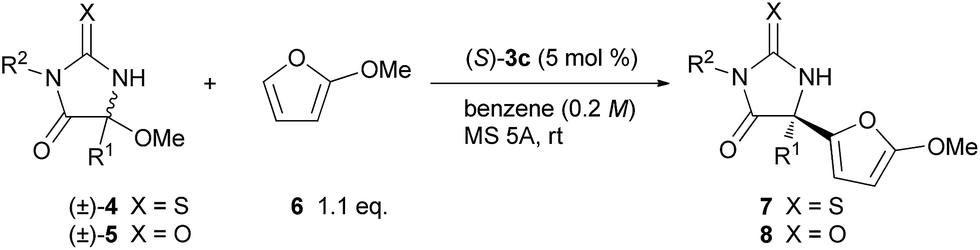
The scope of the thiohydantoin derivatives were further investigated under the optimized reaction conditions (Table 2, entries 1–12). Initially, different alkyl substituents at the 5-position were examined (entries 1–4). The reaction of isobutyl-substituted 4b proceeded smoothly to provide 7b in a high yield with a high ee (entry 1).19 In contrast, benzyl-substituted 4c required a longer reaction time for the full conversion of the substrate, and 7c was obtained in a moderate yield with a moderate enantioselectivity (entry 2). On the basis of the favorable transition state TSs-re as shown in Fig. 2a, the observed stereochemical outcome presumably arises from the steric repulsion between the phenanthryl substituent of catalyst 3c and the benzyl moiety introduced to substrate 4c. In this case, a substantial amount of enamide was formed via tautomerization of the imine generated in situ. The isopropyl-substituted 4d also could be applied to this reaction to yield 7d with high enantioselectivity;19 however, the reaction at room temperature did not achieve full conversion of the substrate, even after 24 h, and a considerable amount of 4d was recovered (entry 3). The ee of the recovered 4d showed that it was enantiomerically pure, indicating that kinetic resolution of 4d occurred during the elimination of methanol to generate ketimine under the influence of the chiral phosphoric acid catalyst.20 The higher temperature accelerated the reaction, however the ee was reduced (entry 4). Next, the effect of a substituent on the nitrogen at the 3-position was investigated. Substrates having an electron-donating group as well as an electron-withdrawing group at the para position of the phenyl group underwent a reaction to provide the corresponding products in high yields with high enantioselectivities (entries 5–8). meta-Bromophenyl-substituted 4i was also applicable to the reaction without any problem (entry 9). The reaction with ortho-bromo-substituted 4j provided a mixture of diastereomers due to the central chirality at the 5-position and the axial chirality around the C–N bond between the ortho-bromophenyl group and the nitrogen at the 3-position (entry 10). The ee of the major diastereomer was moderate while that of the minor diastereomer was very high. The benzyl group was also a suitable substituent on the nitrogen, and the corresponding product was obtained in a high yield with a high ee (entries 11 and 12). The scope of this reaction was expanded by using the hydantoin derivatives 5 in addition to the thiohydantoin derivatives 4 (entries 13–17). Although the reaction of 5 required a longer reaction time compared with that of 4, the corresponding products 8 were obtained in high yields with high enantioselectivities, except for 8c which has an isopropyl group (entry 15).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 4 or 5 | R1 | R2 | 7 or 8 | Time (h) | Yieldb (%) | eec (%) |
| a Reaction conditions: 4 or 5 (0.10 mmol), 6 (0.11 mmol), (S)-3c (5.0 μmol), MS 5A (100 mg), benzene (0.50 mL). b Isolated yields. c Enantiomeric excess of 7 and 8 were determined by chiral stationary phase HPLC analysis. d 35% of 4d was recovered with 99% ee. e Reaction was performed at 50 °C. f 1.0 mL of benzene was used. g 10 μmol of (S)-3c (10 mol%) was used. | |||||||
| 1 | 4b | iBu | Ph | 7b | 4 | 98 | 86 |
| 2 | 4c | Bn | Ph | 7c | 48 | 67 | 65 |
| 3 | 4d | iPr | Ph | 7d | 48 | 50d | 85 |
| 4e | 4d | iPr | Ph | 7d | 24 | 89 | 78 |
| 5f | 4e | Me | 4-MeOC6H4 | 7e | 4 | 87 | 90 |
| 6 | 4f | iBu | 4-MeOC6H4 | 7f | 4 | 98 | 90 |
| 7 | 4g | Me | 4-BrC6H4 | 7g | 4 | 97 | 90 |
| 8 | 4h | iBu | 4-BrC6H4 | 7h | 4 | 99 | 88 |
| 9 | 4i | Me | 3-BrC6H4 | 7i | 4 | 99 | 88 |
| 10 | 4j | Me | 2-BrC6H4 | 7j | 6 | 86 (dr = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) :2) |
67/94 |
| 11 | 4k | Me | Bn | 7k | 4 | 98 | 90 |
| 12 | 4l | iBu | Bn | 7l | 4 | 99 | 90 |
| 13 | 5a | Me | Ph | 8a | 24 | 86 | 93 |
| 14 | 5b | iBu | Ph | 8b | 24 | 98 | 92 |
| 15g | 5c | iPr | Ph | 8c | 48 | 51 | 89 |
| 16 | 5d | Me | Bn | 8d | 36 | 95 | 93 |
| 17 | 5e | iBu | Bn | 8e | 36 | 97 | 92 |
Finally, derivatization of the product based on the 2-methoxyfuryl moiety was performed (Scheme 2). The cleavage of the furan ring of 8a proceeded smoothly under Achmatowicz type reaction conditions,21 and subsequent chemoselective reduction of the keto moiety under Luche conditions resulted in the formation of butenolide 10 in a good yield over two steps. In the course of the derivatization, the loss of enantiomeric purity did not occur.22
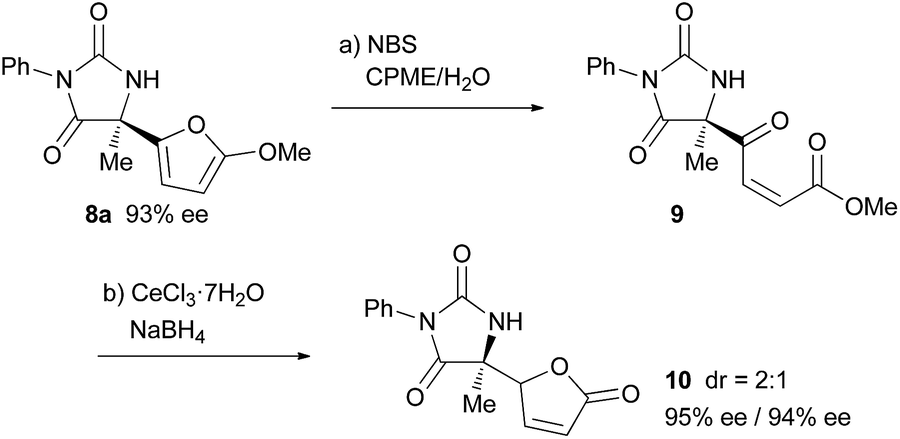 | ||
| Scheme 2 Derivatization of 8a. Reagents and conditions: (a) NBS (1.1 eq.), CPME/H2O, 0 °C, 30 min. (b) CeCl3·7H2O (1.5 eq.), NaBH4 (1.0 eq.), −78 °C to rt, 3 h, 78% (over 2 steps), dr = 2:1, 95% ee/94% ee. | ||
Conclusions
In conclusion, we have successfully developed an enantioselective Friedel–Crafts reaction of 2-methoxyfuran with aliphatic ketimines generated in situ from thiohydantoin- and hydantoin-derived hemiaminal methyl ether under the influence of a chiral phosphoric acid catalyst. This reaction is a rare example of the Friedel–Crafts reaction involving ketimines possessing alkyl substituents, such as isobutyl and isopropyl groups, and is also an attractive method for the synthesis of (thio)hydantoin derivatives which have a quaternary stereogenic center at the 5-position. In addition, theoretical studies were conducted to clarify the origin of the stereochemical outcome as well as the major factors contributing to the efficient enantioselection, which would contribute to developing new enantioselective reactions catalyzed by chiral phosphoric acid.Acknowledgements
This research was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” from MEXT (Japan) and a Grant-in-Aid for Scientific Research from the JSPS.Notes and references
- (a) V. Terrasson, R. M. de Figueiredo and J. M. Campagne, Eur. J. Org. Chem., 2010, 2635 CrossRef CAS; (b) S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC; (c) M. Zeng and S.-L. You, Synlett, 2010, 1289 CAS; (d) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed; (e) M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS PubMed.
- (a) M. Abid, L. Teixeira and B. Török, Org. Lett., 2008, 10, 933 CrossRef CAS PubMed; (b) M. Rueping, S. Raja and A. Núñez, Adv. Synth. Catal., 2011, 353, 563 CrossRef CAS; (c) R. Husmann, E. Sugiono, S. Mersmann, G. Raabe, M. Rueping and C. Bolm, Org. Lett., 2011, 13, 1044 CrossRef CAS PubMed; (d) Y. Qian, C. Jing, C. Zhai and W.-h. Hu, Adv. Synth. Catal., 2012, 354, 301 CrossRef CAS; (e) J. Feng, W. Yan, D. Wang, P. Li, Q. Sun and R. Wang, Chem. Commun., 2012, 48, 8003 RSC; (f) E. Aranzamendi, N. Sotomayor and E. Lete, J. Org. Chem., 2012, 77, 2986 CrossRef CAS PubMed; (g) K.-F. Zhang, J. Nie, R. Guo, Y. Zheng and J.-A. Ma, Adv. Synth. Catal., 2013, 355, 3497 CrossRef CAS; (h) T. Kano, R. Takechi, R. Kobayashi and K. Maruoka, Org. Biomol. Chem., 2014, 12, 724 RSC.
- (a) Y.-X. Jia, J. Zhong, S.-F. Zhu, C.-M. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2007, 46, 5565 CrossRef CAS PubMed; (b) M. Rueping and B. J. Nachtsheim, Synlett, 2010, 119 CrossRef CAS; (c) Q. Yin and S.-L. You, Chem. Sci., 2011, 2, 1344 RSC; (d) X. Yu, Y. Wang, G. Wu, H. Song, Z. Zhou and C. Tang, Eur. J. Org. Chem., 2011, 3060 CrossRef CAS; (e) M. Righi, F. Bartoccini, S. Lucarini and G. Piersanti, Tetrahedron, 2011, 67, 7923 CrossRef CAS.
- For reviews on chiral Brønsted acid catalysis, see: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed; (b) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS; (c) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS PubMed; (d) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed; (e) H. Yamamoto and N. Payette, in Hydrogen Bonding in Organic Synthesis, ed. P. M. Pihko, Wiley-VCH, Weinheim, 2009, p. 73 Search PubMed; (f) D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2010, 291, 395 CrossRef CAS PubMed; (g) M. Terada, Synthesis, 2010, 1929 CrossRef CAS; (h) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed; For seminal studies, see: (i) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566 CrossRef CAS PubMed; (j) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356 CrossRef CAS PubMed.
- L. Simón and J. M. Goodman, J. Org. Chem., 2010, 75, 589 CrossRef PubMed.
- (a) L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2008, 130, 8741 CrossRef PubMed; (b) T. Marcelli, P. Hammar and F. Himo, Chem.–Eur. J., 2008, 14, 8562 CrossRef CAS PubMed; (c) L. Simón and J. M. Goodman, J. Org. Chem., 2010, 75, 589 CrossRef PubMed; (d) Y. Shibata and M. Yamanaka, J. Org. Chem., 2013, 78, 3731 CrossRef CAS PubMed; (e) K. Saito, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 11740 CrossRef CAS PubMed; (f) K. Saito, K. Horiguchi, Y. Shibata, M. Yamanaka and T. Akiyama, Chem.–Eur. J., 2014, 20, 7616 CrossRef CAS PubMed.
- (a) M. Terada, K. Machioka and K. Sorimachi, Angew. Chem., Int. Ed., 2009, 48, 2553 CrossRef CAS PubMed; (b) M. Terada and Y. Toda, J. Am. Chem. Soc., 2009, 131, 6354 CrossRef CAS PubMed; (c) M. Terada, T. Komuro, Y. Toda and T. Korenaga, J. Am. Chem. Soc., 2014, 136, 7044 CrossRef CAS PubMed.
- Selected examples on thiohydantoin derivatives, see: (a) T. Mendgen, C. Steuer and C. D. Klein, J. Med. Chem., 2012, 55, 743 CrossRef CAS PubMed; (b) Y. Liu, J. Wu, P.-Y. Ho, L.-C. Chen, C.-T. Chen, Y.-C. Liang, C.-K. Cheng and W.-S. Lee, Cancer Lett., 2008, 271, 294 CrossRef CAS PubMed; (c) M. E. Jung, S. Ouk, D. Yoo, C. L. Sawyers, C. Chen, C. Tran and J. Wongvipat, J. Med. Chem., 2010, 53, 2779 CrossRef CAS PubMed; (d) A. Takahashi, H. Matsuoka, Y. Ozawa and Y. Uda, J. Agric. Food Chem., 1998, 46, 5037 CrossRef CAS; (e) A. A. El-Barbary, A. I. Khodair, E. B. Pedersen and C. Nielsen, J. Med. Chem., 1994, 37, 73 CrossRef CAS PubMed; (f) J. Marton, J. Enisz, S. Hosztafi and T. Tímár, J. Agric. Food Chem., 1993, 41, 148 CrossRef CAS; (g) J. E. Tompkins, J. Med. Chem., 1986, 29, 855 CrossRef CAS PubMed; (h) J. V. Marx, D. A. Richert and W. W. Westerfeld, J. Med. Chem., 1970, 13, 1179 CrossRef CAS PubMed.
- For reviews on hydantoin derivatives, see: (a) E. Ware, Chem. Rev., 1950, 46, 403 CrossRef CAS PubMed; (b) C. A. López and G. G. Trigo, Adv. Heterocycl. Chem., 1985, 38, 177 CrossRef; (c) M. Meusel and M. Gutschow, Org. Prep. Proced. Int., 2004, 36, 391 CrossRef CAS.
- For selected recent synthetic studies on thiohydantoin derivatives, see: (a) O. A. Attanasi, L. de Crescentini, P. Filippone, G. Giorgi, S. Nicolini, F. R. Perrulli and S. Santeusanio, Tetrahedron, 2014, 70, 7336 CrossRef CAS; (b) V. Mehra, P. Singh, N. Manhas and V. Kumar, Synlett, 2014, 25, 1124 CrossRef; (c) S. Gosling, C. E. Amri and A. Tatibouët, Synthesis, 2014, 46, 1079 CrossRef; (d) V. Ceban, K. Hands, M. Meazza, M. E. Light and R. Rios, Tetrahedron Lett., 2013, 54, 7183 CrossRef CAS; (e) F. Medda and C. Hulme, Tetrahedron Lett., 2012, 53, 5593 CrossRef CAS; (f) V. Kumar, H. Rana, R. Sankolli and M. P. Kaushik, Tetrahedron Lett., 2012, 53, 2377 CrossRef CAS; (g) G. Baccolini, C. Boga, C. Delpivo and G. Micheletti, Tetrahedron Lett., 2011, 52, 1713 CrossRef CAS; (h) F. Xiang, S. Zhang, C. Lu, Z. Chen and G. Yang, Synth. Commun., 2008, 38, 953 CrossRef CAS; (i) M. Carboni, J.-M. Gomis, O. Loreau and F. Taran, Synthesis, 2008, 417 CAS; (j) G. S. M. Sundaram, C. Venkatesh, H. Ila and H. Junjappa, Synlett, 2007, 251 CAS; (k) S. Porwal, R. Kumar, P. R. Maulik and P. M. S. Chauhan, Tetrahedron Lett., 2006, 47, 5863 CrossRef CAS; (l) S. Reyes and K. Burgess, J. Org. Chem., 2006, 71, 2507 CrossRef CAS PubMed.
- For selected recent synthetic studies on hydantoin derivatives, see: (a) H. Liu, Z. Yang and Z. Pan, Org. Lett., 2014, 16, 5902 CrossRef CAS PubMed; (b) M. C. Hillier, H.-H. Gong, D. S. Clyne and M. J. Babcock, Tetrahedron, 2014, 70, 9413 CrossRef CAS; (c) H. Rmedi and A. Baklouti, Tetrahedron Lett., 2014, 55, 3585 CrossRef CAS; (d) P. Ventosa-Andrés, J. A. González-Vera, M. T. García-López and R. Herranz, Tetrahedron, 2014, 70, 3407 CrossRef; (e) G. Chaubet, G. Cazals, A. Lebrun, J. Martinez and I. Parrot, Synlett, 2014, 25, 574 CrossRef CAS; (f) C. Menor-Salván and M. R. Marín-Yaseli, Chem.–Eur. J., 2013, 19, 6488 CrossRef PubMed; (g) M. C. Bellucci, M. Frigerio, T. Marcelli and A. Volonterio, Synlett, 2013, 24, 727 CrossRef CAS; (h) V. Mehra and V. Kumar, Tetrahedron Lett., 2013, 54, 6041 CrossRef CAS.
- D. Uraguchi, K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2004, 126, 11804 CrossRef CAS PubMed.
- A preliminary investigation of the reaction conditions, including the screening of solvents, was conducted. See the ESI† for details.
- CCDC no. 1405286 [(S)-7a]† contains the supplementary crystallographic data for this paper.
- All calculations were performed with the Gaussian 09 package. M. J. Frisch, et al., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed. See the ESI† for the full citation.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- The orientation of the phenanthryl group was thoroughly optimized in the transition states TSs-re and TSr-si. See the ESI† for details.
- (a) B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151 CrossRef CAS; (b) Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- The stereochemical outcome of the reactions of the other ketimines having isobutyl and isopropyl substituents can be also rationalized by similar transition states shown in Fig. 2. See the ESI† for details of three-dimensional structures and energies.
- In order to experimentally validate the proposed reaction mechanism involving the formation of imine from hemiaminal ethers, we carried out a control experiment. Thus, the possibility of the other reaction mechanism such as the SN2-type mechanism including dynamic kinetic resolution was examined. Treatment of enantiomerically-pure 4d with a catalytic amount of achiral diphenyl phosphate, (PhO)2P(
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OH,
provided the corresponding adduct 7d in a 55% yield in a completely racemic form. This result clearly reveals that the involvement of the SN2-type mechanism is ruled out and indirectly supports our proposed mechanism. For the kinetic resolution through the SN2 reaction catalyzed by chiral phosphoric acid, also see: I. Čorić, J. H. Kim, T. Vlaar, M. Patil, W. Thiel and B. List, Angew. Chem., Int. Ed., 2013, 52, 3490 CrossRef PubMed.
O)OH,
provided the corresponding adduct 7d in a 55% yield in a completely racemic form. This result clearly reveals that the involvement of the SN2-type mechanism is ruled out and indirectly supports our proposed mechanism. For the kinetic resolution through the SN2 reaction catalyzed by chiral phosphoric acid, also see: I. Čorić, J. H. Kim, T. Vlaar, M. Patil, W. Thiel and B. List, Angew. Chem., Int. Ed., 2013, 52, 3490 CrossRef PubMed. - For a review on the aza-Achmatowicz reaction, see: F. van der Pijl, F. L. van Delft and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2015, 4811 CrossRef CAS.
- The high acidity of the proton at the γ position of the butenolide moiety of 10 allowed the epimerization at that position to proceed easily, even during column purification, and a 2:1 diastereomixture was obtained from each attempt for the transformation of 9 to 10 under different reaction conditions. Thus the 2:1 ratio is assumed to be thermodynamically determined.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, exploratory investigation of the reaction conditions, characterization data and DFT studies. CCDC 1405286. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03175c |
| This journal is © The Royal Society of Chemistry 2016 |