
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unprecedentedly targeted customization of molecular energy levels with auxiliary-groups in organic solar cell sensitizers†
Yongshu
Xie‡
,
Wenjun
Wu‡
,
Haibo
Zhu
,
Jingchuan
Liu
,
Weiwei
Zhang
,
He
Tian
and
Wei-Hong
Zhu
*
Key Laboratory for Advanced Materials and Institute of Fine Chemicals, Shanghai Key Laboratory of Functional Materials Chemistry, Collaborative Innovation Center for Coal Based Energy (i-CCE), East China University of Science & Technology, Shanghai 200237, China. E-mail: whzhu@ecust.edu.cn
First published on 9th October 2015
Abstract
In dye-sensitized solar cells (DSSCs), the HOMO–LUMO energy gap of organic sensitizers should be large enough to enable efficient electron injection and dye regeneration. However, the LUMOs of most practical organic dyes are always too high, making energy “waste”. In order to deepen the LUMOs, we focus on the targeted modulation of the molecular energy levels by embedding an electron donor or acceptor into the skeleton of a typical D–π–A model. The electron-rich group of 3,4-ethylenedioxythiophene (EDOT) lifts up the HOMO level with little influence on the LUMO, while the electron-deficient group of benzothiadiazole (BTD) or benzooxadiazole (BOD) mainly lowers the customized LUMO level. As a consequence, the auxiliary group change from EDOT (dye WS-53) to BOD (dye WS-55) brings forth a huge photoelectric conversion efficiency (PCE) increase by 38 fold from 0.24 to 9.46% based on an I−/I3− redox couple, and even reaching a high PCE of 10.14% with WS-55 under 0.3 sunlight irradiation.
Introduction
Dye sensitized solar cells (DSSCs) have received considerable attention due to their relatively high power conversion efficiency, low cost and high stability.1–3 Enormous research passion has also been devoted to metal-free organic dyes because of their excellent photophysical properties.4 Up to now, the donor–π bridge–acceptor (D–π–A) motif has been widely exploited for tailoring organic sensitizers.5 Among them, introducing auxiliary groups to the skeleton of the D–π–A system can exhibit a significant influence on the energy levels, light response, and dye stability as well as the photovoltaic performance of organic sensitizers.6,7Generally, the LUMO and HOMO energy levels of organic sensitizers play important roles in electron injection and dye regeneration for DSSCs. Specifically, the driving force for electron injection from the excited dyes to the TiO2 conduction band (−0.5 V vs. NHE) should be greater than 0.2 V, and that for efficient dye regeneration from an iodine electrolyte (0.4 V vs. NHE) should be greater than 0.3 V.8 That is, the ideal LUMO and HOMO for an organic sensitizer should lie around −0.7 V and 0.7 V, respectively, with an appropriate band gap of about 1.4 eV. However, the LUMOs of most organic sensitizers actually used in DSSCs are too high, resulting in energy “waste”. Given the basic injection dynamics, lowering the LUMO level and lifting the HOMO level in organic dyes can be expected to narrow the band gap (E0–0) and extend the light response, thus efficiently optimizing the photovoltaic performance of DSSCs. In this regard, the targeted customization of molecular energy levels is still a challenge.
With this in mind, we herein present a new series of dithieno[3,2-b:2′,3′-d]pyrrole (DTP)-based organic sensitizers (Scheme 1) using different auxiliary groups in the π-bridge. Based on the reference dye WS-52, an electron-rich unit of 3,4-ethylenedioxythiophene (EDOT) and the electron-deficient groups of benzothiadiazole (BTD) and benzooxadiazole (BOD) were specifically introduced into WS-53, WS-54 and WS-55, respectively. Interestingly, the embedding electron donor or acceptor can customize the molecular energy levels well. In distinct contrast with EDOT, the electron-deficient groups of BTD or BOD mainly lower the LUMO level, being capable of preventing the energy waste in electron injection. Typically, the auxiliary group change from EDOT (dye WS-53) to BOD (dye WS-55) induces a large PCE increase of 38 fold from 0.24% to 9.46% based on an I−/I3− redox couple, even reaching a high PCE of 10.14% with WS-55 under 0.3 sunlight irradiation.
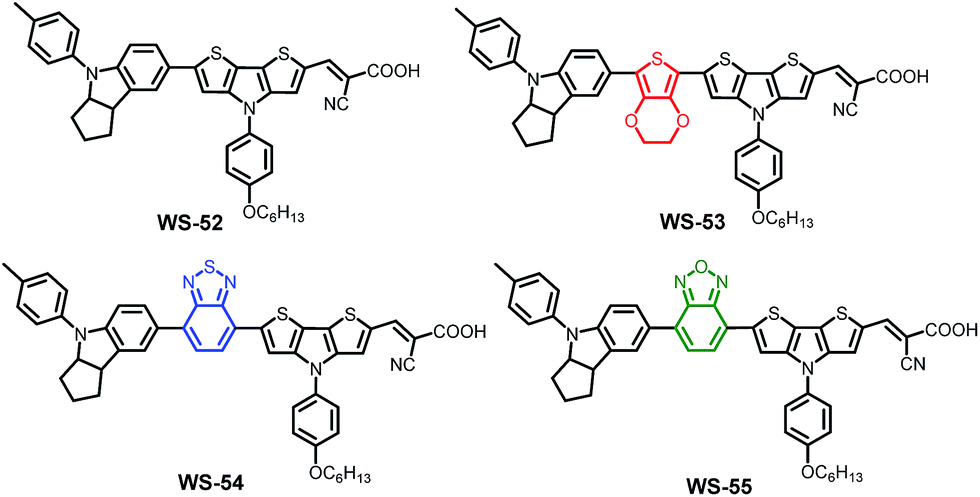 | ||
| Scheme 1 Molecular structures of dyes WS-52, WS-53, WS-54 and WS-55 containing different auxiliary groups for the targeted customization of molecular energy levels. | ||
Results and discussion
The syntheses of the dyes WS-52, WS-53, WS-54 and WS-55 are straightforward and described in the ESI.† Their absorption spectra in CH2Cl2 are depicted in Fig. 1a, and the corresponding data are summarized in Table 1. The reference dye WS-52 shows two distinct absorption bands around 360 and 549 nm, corresponding to the π–π* and intramolecular charge transfer (ICT) transition bands, respectively. WS-53 exhibits a significant bathochromic shift in the ICT band from 549 to 570 nm due to the extended π-conjugation with the EDOT unit. Through inserting the strong electron-withdrawing units of BTD and BOD, WS-54 and WS-55 exhibit bathochromic shifts of 14 and 9 nm, respectively. Compared with WS-52, the insertion of the auxiliary group (electron-rich or deficient group) into the π-spacer leads to an obvious red-shift of the ICT band in CH2Cl2. Upon adsorption onto TiO2 films (Fig. 1b), all four dyes show hypsochromic shifts due to the deprotonation of the cyanoacrylic acid group (Table 1). WS-52 and WS-53 show large hypsochromic shifts of 88 nm, from 549 to 461 nm, and 78 nm, from 570 to 492 nm, respectively. In contrast, WS-54 and WS-55, containing strong electron-withdrawing auxiliary-groups, bestow much smaller hypsochromic shifts of 33 nm, from 563 to 530 nm, and 13 nm, from 558 to 545 nm, respectively. Obviously, upon the incorporation of strong electron withdrawing groups like BTD or BOD, the D–A–π–A featuring WS-54 and WS-55 bring forth a broader light response, which contributed to the smaller hypsochromic shift onto TiO2 and the presence of an additional sub-absorption band in the region of 400–450 nm.6b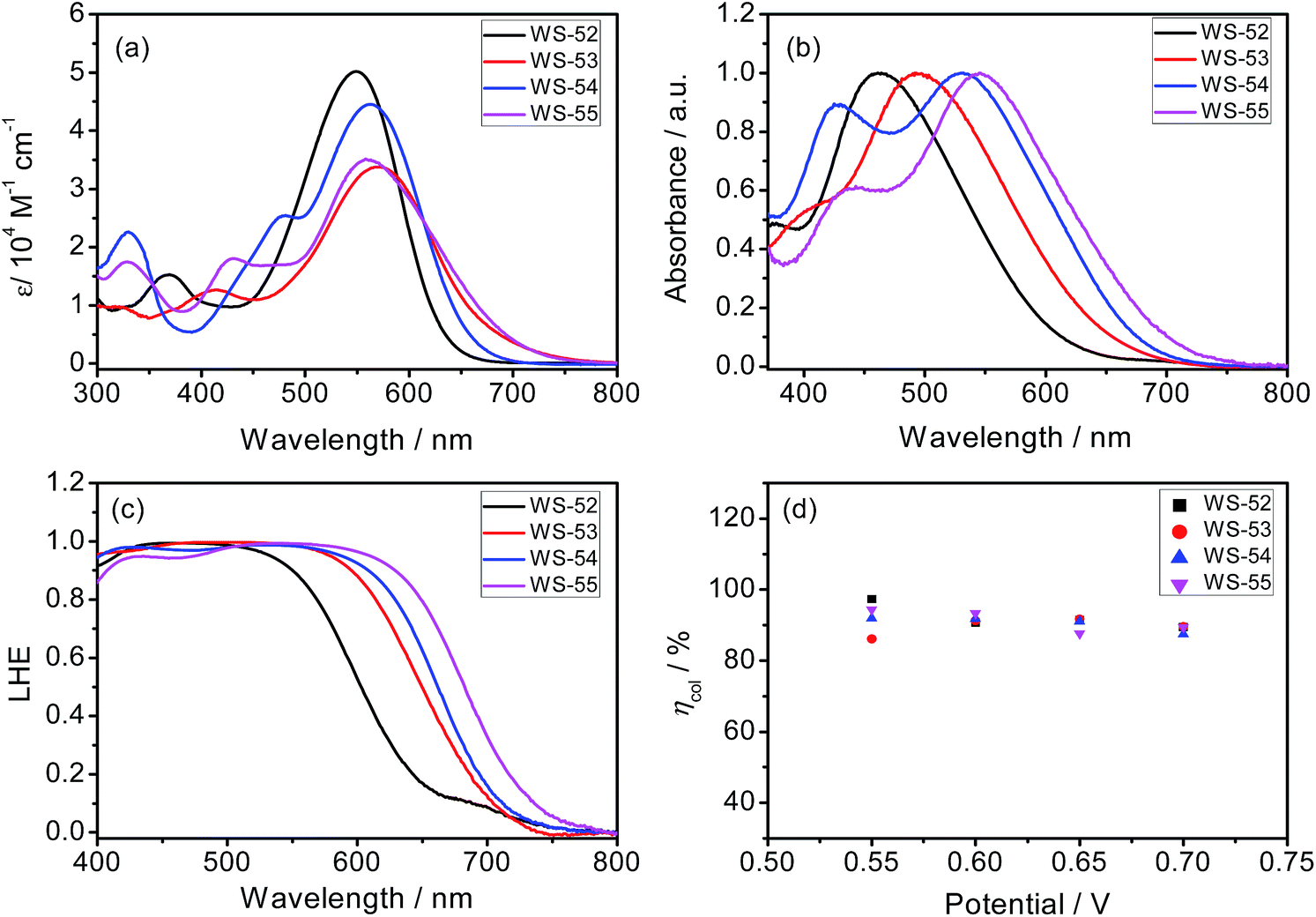 | ||
| Fig. 1 Absorption spectra of WS-52, WS-53, WS-54 and WS-55 in CH2Cl2 solution (a) and coated onto 3 μm TiO2 film (b), LHE spectra calculated from the absorption spectra of dye-loaded TiO2 film (c), and charge collection efficiency (ηcoll) in DSSCs as a function of bias potentials during the EIS measurement (d). | ||
| Dyes | λ max (nm) | ε (M−1 cm−1) | λ max (nm) | HOMOc (V) | E 0–0 (V) | LUMOe (V) | J SC (mA cm−2) | V OC (mV) | FF | η (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Absorption parameters were obtained in CH2Cl2. b Absorption parameters were obtained on 3 μm nanocrystalline TiO2 film. c The HOMO was obtained in CH2Cl2 with ferrocene (0.63 V vs. NHE) as an external reference. d E 0–0 values were estimated from the wavelength at 10% maximum absorption intensity for the dye-loaded 3 μm nanocrystalline TiO2 film. e The LUMO was calculated according to LUMO = HOMO − E0–0. f The efficiency was obtained from the average value of five devices. g The photovoltaic parameters were obtained under 0.3 sunlight irradiation. | ||||||||||
| WS-52 | 549 | 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 160 160 |
461 | 0.75 | 2.02 | −1.27 | 7.88 ± 0.06 | 640 ± 5 | 0.68 ± 0.01 | 3.44 ± 0.11 |
| WS-53 | 570 | 33788 |
492 | 0.57 | 1.89 | −1.32 | 1.22 ± 0.12 | 440 ± 8 | 0.48 ± 0.03 | 0.24 ± 0.06 |
| WS-54 | 563 | 44514 |
530 | 0.81 | 1.83 | −1.02 | 15.84 ± 0.06 | 660 ± 3 | 0.68 ± 0.01 | 7.14 ± 0.09 |
| WS-55 | 558 | 35105 |
545 | 0.90 | 1.77 | −0.87 | 19.66 ± 0.47 | 678 ± 5 | 0.70 ± 0.02 | 9.46 ± 0.19 |
| WS-55 | 6.74 ± 0.08 | 643 ± 3 | 0.73 ± 0.01 | 10.05 ± 0.09 | ||||||
Next, we focus on the customized modulation of the molecular energy levels by embedding the auxiliary electron donor or acceptor into the skeleton of a typical D–π–A model. Based on the cyclic voltammetry measurements (Fig. 2a and Table 1), the first redox potentials corresponding to the HOMO values are 0.75, 0.57, 0.81 and 0.90 V (vs. NHE) for dyes WS-52, WS-53, WS-54 and WS-55, respectively. Due to the electron donating property of EDOT, the HOMO of WS-53 is lifted by 0.18 V with respect to WS-52, and there exists only a 0.17 V driving force for dye regeneration from the iodine electrolyte (Fig. 2b).8b As estimated from the HOMO and E0–0 (Table 1), the LUMO values of WS-52, WS-53, WS-54 and WS-55 are −1.27, −1.32, −1.02 and −0.87 V, respectively. Interestingly, the auxiliary electron-rich EDOT group predominantly lifts up the HOMO level with little influence on the LUMO, while the electron-deficient BTD or BOD group mainly lowers the LUMO level. It is noteworthy that the stronger electron-withdrawing capability of BOD in WS-55 dramatically lowers the LUMO orbital from −1.27 V (WS-52) to −0.87 V. With regard to these four dyes, the insertion of different pull or push auxiliary groups can provide an efficient channel to realize the targeted customization of the HOMO and LUMO energy levels. Generally, the driving force for TiO2 electron injection is always much larger than the minimum requirement because the LUMOs of most organic sensitizers are always higher than −1.0 V. Comparing these four dyes, the customized LUMO orbital change from −1.27 V (reference WS-52) to −1.02 V (WS-54) to −0.87 V (WS-55) gives an unprecedented preferable modulation, in which we can efficiently decrease the “waste” in the electron-injection driving force, and thus efficiently decrease the HOMO–LUMO energy gap, resulting in a desirable light response in the long-wavelength range. Indeed, WS-55 exhibited a long absorption onset wavelength as well as a promising PCE of 9.46% (Table 1), which is around 38 fold higher than the dye WS-53 (0.24%). In the following, we obtain insight into how the incorporated auxiliary groups of EDOT, BTD and BOD play such a different role in the photovoltaic performance, especially focusing on the short-circuit current density (JSC) and open-circuit voltage (VOC).
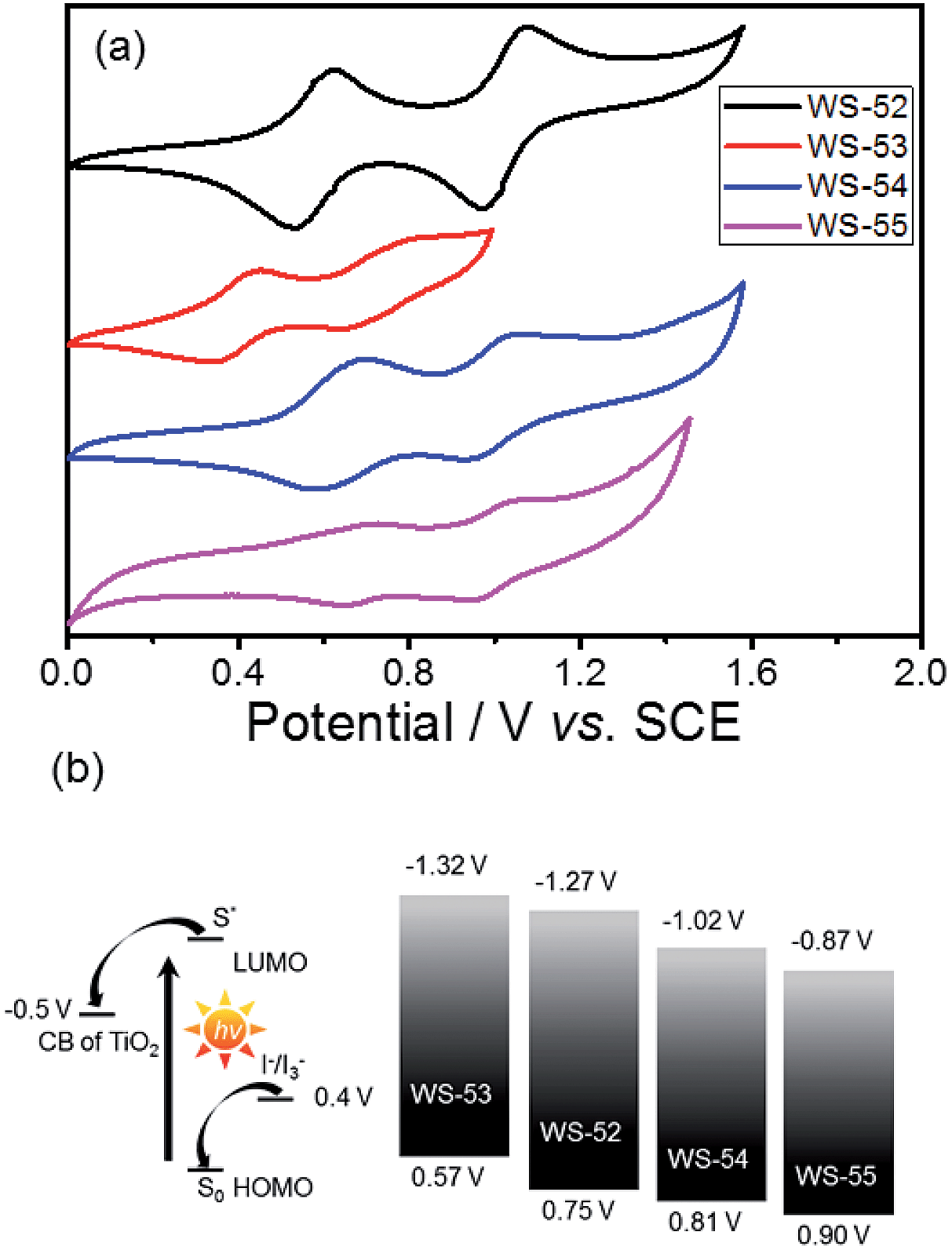 | ||
| Fig. 2 (a) Cyclic voltammograms of WS-52, WS-53, WS-54 and WS-55 in CH2Cl2, and (b) schematic diagram of the energy levels of the TiO2 conduction band, dyes, and I−/I3− redox couple. | ||
Generally, the photocurrent JSC can be estimated from the incident photon-to-electron conversion efficiency (IPCE). Fig. 3a shows the IPCE curves as a function of the excitation wavelengths for these four dyes, which is critically dependent upon the inserted auxiliary group. To our great surprise, although the inserted EDOT unit can distinctly shift absorption to a long wavelength, WS-53 exhibited very disappointing IPCE values (as low as 5%) across the whole visible range from 300–800 nm. In contrast, it is impressive that WS-54 and WS-55 bestow very broad and relatively high IPCE values. Upon increasing the electron-withdrawing capability of the auxiliary group, the IPCE onset wavelength was extended step by step (Fig. 3a), from 730 nm for the reference dye WS-52 and 800 nm for WS-54 to 840 nm for WS-55, which is very uncommon for pure organic sensitizers. Among these four dyes, WS-55 also showed the highest IPCE plateau with a maximum value of 85.9%.
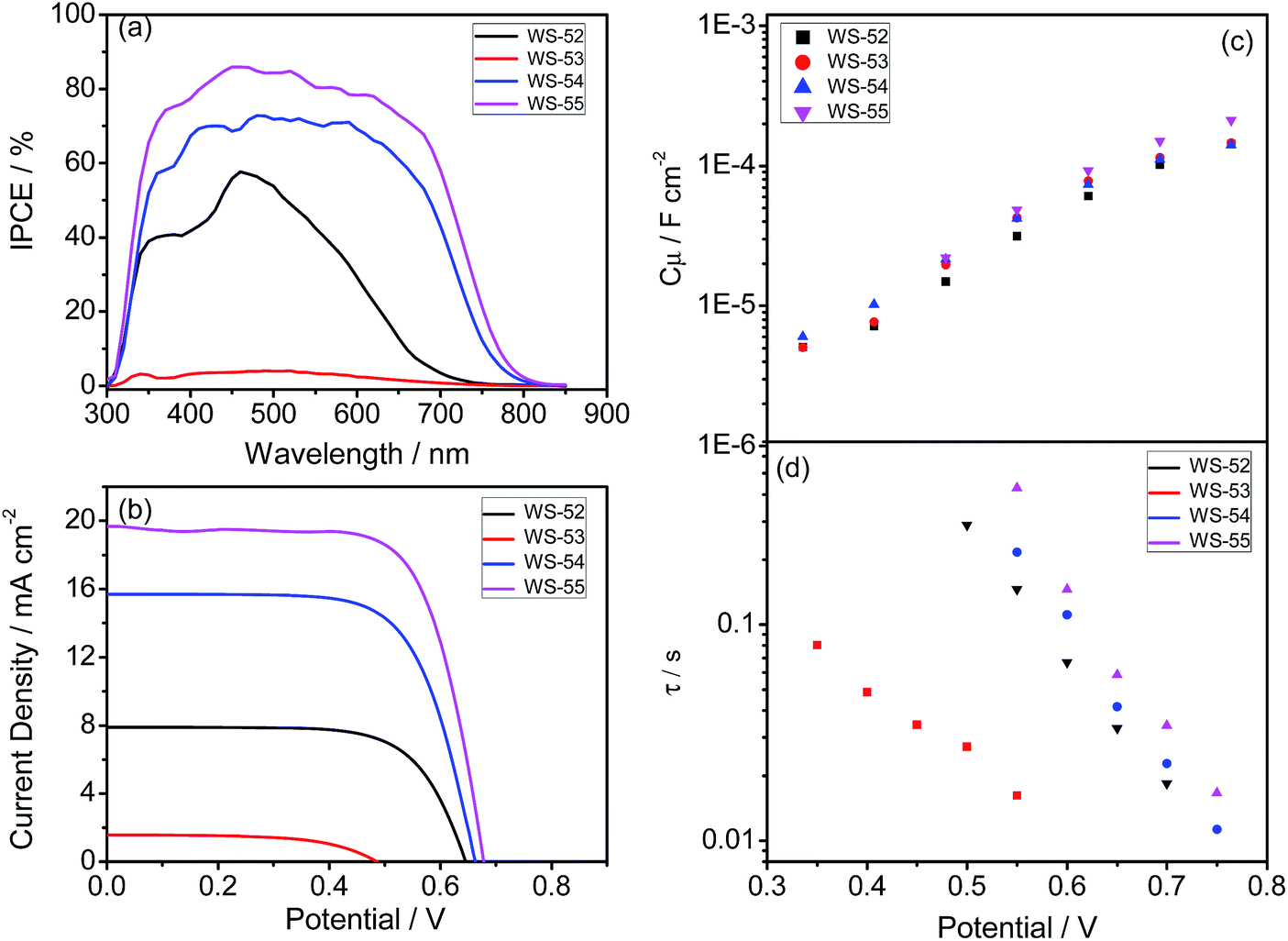 | ||
| Fig. 3 IPCE (a) and J–V curves (b), and bias potential against TiO2 capacitance (c) and electron lifetime (d) in DSSCs sensitized by WS-52, WS-53, WS-54 and WS-55. | ||
As is known, the IPCE value is determined on the basis of four factors as follows:5a
| IPCE = LHE × φinj × φreg × ηcoll | (1) |
Moreover, based on the abovementioned eqn (1), assuming that the φinj is unity, the obtained φregvs. wavelength curves are shown in Fig. S2.† In the 480–640 nm visible region, the electron-deficient auxiliary groups (BTD or BOD) have a powerful effect on the φreg, which almost reaches unity over 640 nm. With the enhancement of the electron-withdrawing capability, it is very advantageous to the regeneration of the oxidation state dyes, which makes the dye WS-55 exhibit very good regeneration efficiency in this region, within 0.9–1. However, the electron-rich group EDOT makes the φreg of WS-53 sharply drop with the increase in wavelength. This result is extremely consistent with the low driving force to dye regeneration (0.17 V) for WS-53, along with the very poor photocurrent (1.22 mA cm−2).
Apparently, among these four dyes, the insertion of the electron-donating EDOT unit undesirably lifts up the HOMO energy level, resulting in a detrimentally insufficient driving force for dye regeneration. In contrast, the incorporation of electron-withdrawing BTD and BOD units can dramatically lower or deepen the LUMO orbital levels, resulting in narrow HOMO–LUMO gaps with a preferable broad light response range. Given that BOD has stronger electron-withdrawing capability than BTD, we can decrease the LUMO orbital level step-by-step, and extend the light response range (Fig. 1b and 2b). In this way, upon the targeted modulation of the LUMO levels, the photocurrent JSC for WS-53, WS-52, WS-54 and WS-55 increased stepwise from 1.22 to 7.88 to 15.84 to 19.66 mA cm−2 (Table 1), respectively, which corresponds well to the integrals of the IPCE curves (1.17, 7.10, 14.43 and 19.56 mA cm−2, Fig. S3†). In addition to the efficient level regulation action and high efficiency, the DSSCs based on WS-55 also presented satisfactory photostability, remaining at 92% of the initial conversion efficiency after 500 h under visible-light irradiation (Fig. S4†).
Besides JSC, WS-53 also exhibits a low photovoltage (VOC) of 440 mV, which is almost 200 mV lower than WS-52, WS-54 and WS-55. As is known, the alternation of the photovoltage VOC originates from a displacement of the electron quasi-Fermi-level (Ef) in TiO2, which intrinsically stems from a change in the TiO2 conduction band edge (ECB) and/or a fluctuation in electron density (the charge recombination rate in DSSCs).11 Considering that the chemical capacitance (Cμ) stands for the density of states in the bandgap of TiO2, we plot the variation of capacitance at different bias potentials with the fitting of electrochemical impedance spectra (EIS, Fig. S5†) for illustrating the shift in the ECB of TiO2. Since these four dyes exhibited almost identical Cμ values (Fig. 3c), we can rule out a shift in the TiO2 conduction band as the main reason for the rather low photovoltage of WS-53. On the other hand, the fluctuation in TiO2 electron density can also induce a difference in VOC, which is closely related to the recombination resistance.12Fig. 3d illustrates the plots of the electron lifetime under a series of potential biases, and the calculated electron lifetimes lie in the order of WS-55 > WS-54 > WS-52 > WS-53, which is exactly consistent with the sequence of VOC. Among these four dyes, the targeted LUMO and HOMO energy levels of WS-55 are the most desirable. As a matter of fact, the change of auxiliary groups can distinctly increase the photovoltaic efficiency from EDOT (dye WS-53, 0.24 ± 0.06%) to BDT (dye WS-54, 7.14 ± 0.09%) to BOD (dye WS-55, 9.46 ± 0.19%, obtained from the average of five cells listed in Fig. S6†). Under 0.3 sunlight irradiation, WS-55 can even achieve a high PCE of 10.14% (Table 1 and Fig. S7†).
Conclusions
In summary, we have unprecedentedly targeted the customization of molecular energy levels by introducing auxiliary groups from an electron donor or acceptor into D–π–A featuring organic sensitizers. Dyes WS-52, WS-53, WS-54 and WS-55 exhibit well-modulated molecular energy levels in a stepwise manner, thus distinctly lowering the LUMOs, which are always too high and make energy “waste” in most practical organic dyes. As demonstrated, the photovoltaic efficiency is greatly improved when changing the auxiliary group from EDOT (dye WS-53, 0.24%) to BDT (dye WS-54, 7.14%) or BOD (dye WS-55, 9.46%). Moreover, WS-55 can even achieve a high PCE of 10.14% under 0.3 sunlight irradiation. It shows how the delicate balance of LUMO energy levels and HOMO–LUMO energy gaps guarantees the optimizing of photovoltaic properties.Acknowledgements
This work was supported by NSFC for Creative Research Groups (21421004) and Distinguished Young Scholars (21325625), NSFC/China, Oriental Scholarship, Fundamental Research Funds for the Central Universities (WJ1416005 and WJ1315025), and the Scientific Committee of Shanghai (14ZR1409700 and 15XD1501400) for financial support.Notes and references
- (a) B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef PubMed; (b) D. J. Lipomi and Z. N. Bao, Energy Environ. Sci., 2011, 4, 3314–3328 RSC; (c) J. W. Shiu, Y. C. Chang, C. Y. Chan, H. P. Wu, H. Y. Hsu, C. L. Wang, C. Y. Lin and E. W. G. Diau, J. Mater. Chem. A, 2015, 3, 1417–1420 RSC; (d) C. C. Chou, K. L. Wu, Y. Chi, W. P. Hu, S. J. Yu, G. H. Lee, C. L. Lin and P. T. Chou, Angew. Chem., Int. Ed., 2011, 50, 2054–2058 CrossRef CAS PubMed; (e) L. Gao, J. Zhang, C. He, Y. Zhang, Q. J. Sun and Y. F. Li, Sci. China: Chem., 2014, 57, 966–972 CrossRef CAS; (f) M. Hoesel, H. F. Dam and F. C. Krebs, Energ. Tech., 2015, 3, 293–304 CrossRef PubMed; (g) S. Ahmad, M. K. Nazeeruddin and J. Bisquert, ChemPhysChem, 2014, 15, 987–989 CrossRef CAS PubMed; (h) Y. Q. Wang, B. Chen, W. J. Wu, X. Li, W. H. Zhu, H. Tian and Y. S. Xie, Angew. Chem., Int. Ed., 2014, 53, 10779–10783 CrossRef CAS PubMed; (i) S. W. Wang, K. L. Wu, E. Ghadiri, M. G. Lobello, S. T. Ho, Y. Chi, J. E. Moser, F. D. Angelis, M. Grätzel and M. K. Nazeeruddin, Chem. Sci., 2013, 4, 2423–2433 RSC; (j) Z. Sun, M. Liang and J. Chen, Acc. Chem. Res., 2015, 48, 1541–1550 CrossRef CAS PubMed.
- (a) L. Y. Han, A. Islam, H. Chen, C. Malapaka, B. Chiranjeevi, S. F. Zhang, X. D. Yang and M. Yanagida, Energy Environ. Sci., 2012, 5, 6057–6060 RSC; (b) A. Agrestia, S. Pescetellia, E. Gattob, M. Venanzib and A. D. Carloa, J. Power Sources, 2015, 287, 87–95 CrossRef PubMed; (c) J. Zeng, T. L. Zhang, X. F. Zang, D. B. Kuang, H. Meier and D. R. Cao, Sci. China: Chem., 2013, 56, 505–513 CrossRef CAS; (d) Z. Y. Yao, M. Zhang, R. Z. Li, L. Yang, Y. N. Qiao and P. Wang, Angew. Chem., Int. Ed., 2015, 54, 5994–5998 CrossRef CAS PubMed; (e) T. Higashino, Y. Fujimori, K. Sugiura, Y. Tsuji, S. Ito and H. Imahori, Angew. Chem., Int. Ed., 2015, 54, 9052–9056 CrossRef CAS PubMed; (f) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- (a) S. Ito, H. Miura, S. Uchida, M. Takata, K. Sumioka, P. Liska, P. Comte, P. Pechy and M. Grätzel, Chem. Commun., 2008, 5194–5196 RSC; (b) B. Xu, H. Tian, L. Lin, D. Qian, H. Chen, J. Zhang, N. Vlachopoulos, G. Boschloo, Y. Luo, F. Zhang, A. Hagfeldt and L. Sun, Adv. Energy Mater., 2015, 5, 1401185 Search PubMed; (c) R. Lin, H. Lin, Y. Yen, C. Chang, H. Chou, P. Chen, C. Hsu, Y. Chen, J. T. Lin and K. Ho, Energy Environ. Sci., 2013, 6, 2477–2486 RSC; (d) J. Liu, Y. Numata, C. J. Qin, A. Islam, X. D. Yang and L. Y. Han, Chem. Commun., 2013, 49, 7587–7589 RSC; (e) S. H. Kim, H. W. Kim, C. Sakong, J. Namgoong, S. W. Park, M. J. Ko, C. H. Lee, W. I. Lee and J. P. Kim, Org. Lett., 2011, 13, 5784–5787 CrossRef CAS PubMed.
- (a) Z. J. Ning, Q. Zhang, W. J. Wu, H. C. Pei, B. Liu and H. Tian, J. Org. Chem., 2008, 73, 3791–3797 CrossRef CAS PubMed; (b) M. D. Zhang, H. Xie, X. H. Ju, L. Qin, Q. Yang, H. G. Zheng and X. F. Zhou, Phys. Chem. Chem. Phys., 2013, 15, 634–641 RSC; (c) S. Namuangruk, R. Fukuda, M. Ehara, J. Meeprasert, T. Khanasa, S. Morada, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, J. Phys. Chem. C, 2012, 116, 25653–25663 CrossRef CAS; (d) L. L. Tan, J. F. Huang, Y. Shen, L. M. Xiao, J. M. Liu, D. B. Kuang and C. Y. Su, J. Mater. Chem. A, 2014, 2, 8988–8994 RSC; (e) B. Liu, B. Wang, R. Wang, L. Gao, S. H. Huo, Q. B. Liu, X. Y. Li and W. H. Zhu, J. Mater. Chem. A, 2014, 2, 804–812 RSC.
- (a) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed; (b) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed; (c) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC; (d) Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903–2934 CrossRef CAS PubMed; (e) L. L. Li and E. W. G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC; (f) W. J. Ying, J. B. Yang, M. Wielopolski, T. Moehl, J. E. Moser, P. Comte, J. L. Hua, S. M. Zakeeruddin, H. Tian and M. Grätzel, Chem. Sci., 2014, 5, 206–214 RSC.
- (a) Y. Z. Wu and W. H. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC; (b) W. H. Zhu, Y. Z. Wu, S. T. Wang, W. Q. Li, X. Li, J. Chen, Z. S. Wang and H. Tian, Adv. Funct. Mater., 2011, 21, 756–763 CrossRef CAS PubMed; (c) Y. Z. Wu, X. Zhang, W. Q. Li, Z. S. Wang, H. Tian and W. H. Zhu, Adv. Energy Mater., 2012, 2, 149–156 CrossRef CAS PubMed; (d) Q. P. Chai, W. Q. Li, J. C. Liu, Z. Y. Geng, H. Tian and W. H. Zhu, Sci. Rep., 2015, 5, 11330 CrossRef CAS PubMed; (e) W. Q. Li, B. Liu, Y. Z. Wu, S. Q. Zhu, Q. Zhang and W. H. Zhu, Dyes Pigm., 2013, 99, 176–184 CrossRef CAS PubMed.
- (a) X. F. Lu, Q. Y. Feng, T. Lan, G. Zhou and Z. S. Wang, Chem. Mater., 2012, 24, 3179–3187 CrossRef CAS; (b) X. W. Kang, J. X. Zhang, D. O'Neil, A. J. Rojas, W. Chen, P. Szymanski, S. R. Marder and M. A. El-Sayed, Chem. Mater., 2014, 26, 4486–4493 CrossRef CAS; (c) J. Shi, J. N. Chen, Z. F. Chai, H. Wang, R. L. Tang, K. Fan, M. Wu, H. W. Han, J. G. Qin, T. Y. Peng, Q. Q. Li and Z. Li, J. Mater. Chem., 2012, 22, 18830–18838 RSC; (d) J. B. Yang, P. Ganesan, J. Teuscher, T. Moehl, Y. J. Kim, C. Yi, P. Comte, K. Pei, T. W. Holcombe, M. K. Nazeeruddin, J. L. Hua, S. M. Zakeeruddin, H. Tian and M. Grätzel, J. Am. Chem. Soc., 2014, 136, 5722–5730 CrossRef CAS PubMed; (e) K. D. Seo, I. T. Choi, Y. G. Park, S. Kang, J. Y. Lee and H. K. Kim, Dyes Pigm., 2012, 94, 469–474 CrossRef CAS PubMed.
- (a) K. Hara, T. Sato, R. Katoh, A. Furube, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara and H. Arakawa, J. Phys. Chem. B, 2003, 107, 597–606 CrossRef CAS; (b) T. Daeneke, A. J. Mozer, Y. Uemura, S. Makuta, M. Fekete, Y. Tachibana, N. Koumura, U. Bach and L. Spiccia, J. Am. Chem. Soc., 2012, 134, 16925–16928 CrossRef CAS PubMed.
- (a) X. Xu, H. Wang, F. Gong, G. Zhou and Z. S. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 3219–3223 CrossRef CAS PubMed; (b) Y. Tachibana, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem., 1996, 100, 20056–20062 CrossRef CAS; (c) Y. Tachibana, S. A. Haque, I. P. Mercer, J. R. Durrant and D. R. Klug, J. Phys. Chem. B, 2000, 104, 1198–1205 CrossRef CAS; (d) J. B. Asbury, E. Hao, Y. Wang and T. Lian, J. Phys. Chem. B, 2000, 104, 11957–11957 CrossRef CAS; (e) B. O'Regan, J. Moser, M. Anderson and M. Grätzel, J. Phys. Chem., 1990, 94, 8720–8726 CrossRef.
- S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, J. Teuscher, J. Moser, R. Humphry-Baker, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, Adv. Funct. Mater., 2012, 22, 1291–1302 CrossRef CAS PubMed.
- (a) K. Pei, Y. Z. Wu, A. Islam, S. Q. Zhu, L. Y. Han, Z. Y. Geng and W. H. Zhu, J. Phys. Chem. C, 2014, 118, 16552–16561 CrossRef CAS; (b) W. Q. Li, Y. Z. Wu, Q. Zhang, H. Tian and W. H. Zhu, ACS Appl. Mater. Interfaces, 2012, 4, 1822–1830 CrossRef CAS PubMed.
- F. Fabregat-Santiago, G. Garcia-Belmonte, I. Mora-Sero and J. Bisquert, Phys. Chem. Chem. Phys., 2011, 13, 9083–9118 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, and additional photovoltaic data. See DOI: 10.1039/c5sc02778k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |