
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-delivery of nitric oxide and antibiotic using polymeric nanoparticles†
Thuy-Khanh
Nguyen
a,
Ramona
Selvanayagam
a,
Kitty K. K.
Ho
b,
Renxun
Chen
b,
Samuel K.
Kutty
b,
Scott A.
Rice
 cd,
Naresh
Kumar
b,
Nicolas
Barraud
*ce,
Hien T. T.
Duong‡
*a and
Cyrille
Boyer
*a
cd,
Naresh
Kumar
b,
Nicolas
Barraud
*ce,
Hien T. T.
Duong‡
*a and
Cyrille
Boyer
*a
aCentre for Advanced Macromolecular Design (CAMD) and Australian Centre for NanoMedicine (ACN), School of Chemical Engineering, UNSW Australia, Sydney, NSW 2052, Australia. E-mail: cboyer@unsw.edu.au; hien.duong@sydney.edu.au
bSchool of Chemistry, UNSW Australia, Sydney, NSW 2052, Australia
cCentre for Marine-Innovation, School of Biological, Earth and Environmental Sciences, University of New South Wales, Sydney, Australia 2052. E-mail: n.barraud@melix.org
dThe Singapore Centre for Environmental Life Sciences Engineering and The School of Biological Sciences, Nanyang Technological University, Singapore
eDepartment of Microbiology, Genetics of Biofilms Unit, Institute Pasteur, Paris, France
First published on 10th November 2015
Abstract
The rise of hospital-acquired infections, also known as nosocomial infections, is a growing concern in intensive healthcare, causing the death of hundreds of thousands of patients and costing billions of dollars worldwide every year. In addition, a decrease in the effectiveness of antibiotics caused by the emergence of drug resistance in pathogens living in biofilm communities poses a significant threat to our health system. The development of new therapeutic agents is urgently needed to overcome this challenge. We have developed new dual action polymeric nanoparticles capable of storing nitric oxide, which can provoke dispersal of biofilms into an antibiotic susceptible planktonic form, together with the aminoglycoside gentamicin, capable of killing the bacteria. The novelty of this work lies in the attachment of NO-releasing moiety to an existing clinically used drug, gentamicin. The nanoparticles were found to release both agents simultaneously and demonstrated synergistic effects, reducing the viability of Pseudomonas aeruginosa biofilm and planktonic cultures by more than 90% and 95%, respectively, while treatments with antibiotic or nitric oxide alone resulted in less than 20% decrease in biofilm viability.
Introduction
Nosocomial infections are the fourth leading cause of disease in the U.S.A. and Europe with over 3.5 million cases annually,1 resulting in significant increases in healthcare costs. More importantly, the number of cases (and by consequence deaths) is rapidly increasing due to the emergence of bacteria resistant to antibiotics.2,3 One key adaptive process used by bacteria that leads to their survival and development of resistance after antibiotic treatments is the ability to form multicellular communities of cells encased in a matrix of secreted polymeric substances known as microbial biofilms.4 Their formation and persistence have a considerable impact for patient health, as many biofilm infections are difficult to resolve, and often result in chronic or recurrent infections.5–7 Indeed, bacteria in biofilms show significantly increased resistance to external stresses, including antimicrobials and host immune defenses, compared with free-living single bacterial cells.8,9 Biofilms can also favor gene transfer between bacteria, thus spreading antibiotic resistance or converting a previously non-virulent commensal organism into a virulent pathogen.10 Consequently, biofilm infections present a number of clinical challenges,11–17 including diseases involving uncultivable species, chronic inflammation, impaired wound healing, rapidly acquired antibiotic resistance, and the spread of infections. Accordingly, there is an urgent need for novel therapeutics and treatment strategies that are effective against biofilms and biofilm-related infections.Biofilm researchers have now established that most bacteria follow a lifecycle in which the biofilm mode of growth is the main phase. Bacterial cells can alternate between the biofilm and the planktonic lifestyles via transition stages of either attachment or dispersal that involve the expression of specific genes and are highly regulated.18 In 2006, the biologically ubiquitous nitric oxide (NO) gas was found to be a major signal for biofilm dispersal in the important human pathogen Pseudomonas aeruginosa,19,20 which was found to account for up to 30% of hospital-acquired infectious diseases (nosocomial).21 Follow-up studies showed that exposure to NO in the pM and low nM range can induce dispersal in several other single- and multi-species bacterial and yeast biofilms and that the effects correlate with increases in bacterial phosphodiesterase activity and associated decreases in intracellular levels of the secondary messenger cyclic di-GMP.22,23 After the onset of dispersal induced by NO, both released cells and the remaining biofilms display enhanced sensitivity towards a range of antibiotics, including aminoglycosides.19,24,25 Furthermore, it was recently shown for the first time that inducing a full-scale dispersal event, by means of a genetically modified organism, can clear infections in animal models without killing the host.26 Similar results were obtained in vitro using NO donor compounds, which under a specific condition release NO gas. Therefore, all these combined results have unveiled a new anti-biofilm strategy, which uses low concentrations of NO-donor compounds in combination with antibiotics to eradicate bacterial biofilm infections.27
However, NO donors present a poor stability in biological media, resulting in fast release of NO, which severely limits any potential applications. To overcome these problems, the design of NO donor specifically by enzymatic reactions and the encapsulation of NO donor into polymeric nanoparticles or inorganic materials have been proposed by our group and others.24,28–41 It is well known that the encapsulation of therapeutic compounds into nanoparticles enhances their stability and solubility as well as increases their local concentration.42–45 In previous studies, we and others have developed polymeric and organic/inorganic nanoparticles for the delivery of NO, facilitating its application in dispersion or eradication of biofilms.28,46–48 For instance, we made core cross-linked star polymers containing N-diazeniumdiolate (NONOate) compounds that were capable of releasing NO in a controlled manner for several days, and these polymeric materials were able to prevent and disperse biofilms.48 In addition, NO at high concentration (typically mM) can have a killing effect on several types of bacteria as demonstrated by Schoenfisch and co-workers.46,49–54 The authors have investigated a range of nano-scaled objects with various shapes, and studied their efficacy as bactericidal agents.53 Very recently, the combination of cationic polymers presenting antimicrobial activity with NO for the treatment of biofilms was reported in recent papers by Schoenfisch,55,56 significantly enhanced the killing ability of the antibacterial polymers.
In this study, we combined in one polymeric nanoparticle a NO donor and an aminoglycoside antibiotic, gentamicin. More importantly, in this approach, the NO donor was directly obtained by reaction of gentamicin with NO gas to yield gentamicin-NONOate complex. By engineering the nanoparticles (i.e. placing gentamicin-NONOate in the core), we aimed to obtain a simultaneous and sustainable release of gentamicin and NO, with both released agents acting synergistically on biofilms. The gentamicin-NONOate nanoparticles were found to effectively disperse biofilms of the model organism P. aeruginosa, and, at concentrations of 10–50 μM, strongly decreased the viability of both biofilm and planktonic cells by more than 90% and 95%, respectively. In contrast, gentamicin and NO donor separately presented a lower efficiency against biofilm and planktonic cells.
Experiments and methods
Materials
All chemicals were used as received from Ajax and Sigma-Aldrich, unless otherwise specified. Monomer oligo(ethylene glycol) methyl ether methacrylate with an average Mn of 300 g mol−1 (OEGMA) and 3-vinylbenzaldehyde (VBA) were de-inhibited by passing them through a column of basic alumina. 2,2′-Azobisisobutyronitrile (AIBN) was purified by recrystallization from methanol.Synthesis of POEGMA macro-RAFT agent
The RAFT agent, 4-cyanopentanoic acid dithiobenzoate (CPADB) was prepared according to a published procedure.57 OEGMA (2.58 g, 8.60 × 10−3 mol), CPADB RAFT agent (5.33 × 10−2 g, 1.91 × 10−4 mol) and AIBN (6.27 × 10−3 g, 3.82 × 10−5 mol) were dissolved in 20 mL of toluene in a round-bottom flask equipped with a magnetic stirrer bar. The flask was then sealed with a rubber septum and purged with nitrogen gas for 30 min. The reaction mixtures were then immersed in a preheated oil bath at 70 °C. After 17 h, the polymerization was terminated by quenching the samples in an ice bath for 5 min. The POEGMA polymer was purified three times by precipitation with excess petroleum spirits (boiling range of 40–60 °C) followed by centrifugation (7000 rpm for 15 min) and the polymer was dried under vacuum at room temperature. The samples were stored at 4 °C until required for further chain extension. By comparing the intensity of vinyl proton peaks (6.1 and 5.6 ppm) to that of ester –OCH2 proton peaks (4.1 ppm), the conversion of monomer during the course of polymerization was determined using 1H NMR. After 17 h, a conversion of 80% was obtained. The molecular weight of the POEGMA macro-RAFT agent was measured to be 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1 (PDI = 1.08) by DMAC SEC and Mn, NMR = 10800 g mol−1 by 1H NMR.
200 g mol−1 (PDI = 1.08) by DMAC SEC and Mn, NMR = 10800 g mol−1 by 1H NMR.
Synthesis of POEGMA-b-PVBA
POEGMA with 36 repeating units (Mn, NMR = 10800 g mol−1, Mn, SEC = 11200 g mol−1) was used as a macro-RAFT agent for chain extension with VBA. The number of repeating units of POEGMA was calculated from the monomer conversion obtained from 1H NMR. The POEGMA macro-RAFT agent (1 g, 9.25 × 10−5 mol) was dissolved in 5 mL of toluene containing VBA (1.84 × 10−1 g, 1.39 × 10−3 mol). The reaction mixture was purged with nitrogen gas for 30 min in an ice bath. The polymerization was carried out in an oil bath at 70 °C overnight. The polymerization was terminated by placing the samples on ice for 5 min. The POEGMA polymer was purified three times by precipitation in excess of diethyl ether followed by centrifugation (7000 rpm for 15 min), and the polymer was then dried under reduced pressure at room temperature. Block copolymer with 36 repeating units of OEGMA, 7 repeating units of VBA (as confirmed by 1H NMR in ESI, Fig. S1†) was chosen for further conjugation with gentamicin (Mn, theo = 11700 g mol−1, Mn, SEC = 13700 g mol−1).
Conjugation of POEGMA-b-PVBA to gentamicin
Gentamicin sulfate (Enzo Life Sciences, Sapphire Bioscience Pty. Ltd., Australia) (0.3 g, 2.02 × 10−4 mol) and 100 μL triethylamine (TEA) were dissolved in 2.5 mL of distilled water. The solution was left in an incubator at 37 °C whilst being shaken at 140 rpm for 1 h. Upon completion, drug solution was added into 2.5 mL of POEGMA-b-PVBA (0.3 g, 2.04 × 10−5 mol) in distilled water and the mixture of polymer and drug was incubated at 37 °C with shaking at 100 rpm for a further 48 h. The mixture was then precipitated in acetonitrile and centrifuged at 7500 rpm for 5 min to remove unreacted gentamicin and salt formed. The supernatant was collected and the precipitation step in acetonitrile was repeated three times. Anhydrous magnesium sulfate was used as a drying agent to remove water from the mixture for further reaction with NO gas to introduce the NO-releasing NONOate moiety to the polymer–drug conjugates in acetonitrile.Attachment of NONOate to conjugated POEGMA-b-PVBA with gentamicin
The conjugated POEGMA-b-PVBA with gentamicin (0.3 g) was dissolved in acetonitrile (5 mL) and placed in a Parr apparatus and clamped. The apparatus was then purged and evacuated with nitrogen three times and pressurized to 5 atm NO at 25 °C for 48 h to form NONOate NO donors. Excess NO was then vented through purging with nitrogen gas. The NONOate polymer was then stored at 4 °C until required for further analysis.Determination of NO release by Griess assay and amperometric measurement
NO released from the polymer at specified time intervals was determined using a standard Griess reagent kit (G-7921, Molecular Probes), which is normally used for nitrite determination. NONOate readily releases NO upon contact with water at physiological pH. Typically, 10 mg gentamicin-NONOate containing polymer sample was dissolved in 2 mL of phosphate-buffered saline (PBS). The solution was enclosed in a sealed dialysis membrane (Cellu-Sep 3500 MWCO) that allows free diffusion of NO. The membrane was then immersed in a 6 mL PBS solution and incubated at 37 °C for up to 24 h. At various time points, a 100 μL aliquot from the PBS solution was taken for determining concentration of NO. Since NO readily oxidises to nitrite and nitrate upon contact with water, first the reduction of nitrate to nitrite was conducted through a nitrate reductase. For each 100 μL of sample, 12.5 μL of nitrate reductase and 12.5 μL of enzyme cofactor were added into the solution and incubated at room temperature for 30 min. Then, 120 μL of Griess reagents was added to the sample and left to incubate at room temperature for 30 min. The sample was then topped up with 395 μL of distilled water to make up a total volume of 640 μL. The preparation procedure was repeated for samples at different time points. The UV-Vis absorbance of the resulting solutions was determined at 548 nm and the total nitrite concentration in the sample solutions at different time points were calculated from a standard curve and converted to cumulative NO release.NO was detected amperometrically by using a TBR4100 free radical analyzer with Lab-Trax-4 digital recorder (World Precision Instruments, Sarasota, USA) and fitted with an NO specific sensor (ISO-NOP). The NO sensor, which was freshly calibrated using S-nitroso-N-acetylpenicillamine (SNAP) and copper sulfate according to the manufacturer's instructions, was immersed in a vial containing 10 mL PBS (pH 7.4) and continuously stirred at 37 °C. After the baseline had stabilized, 100 μL of 100 mM gentamicin-NONOate containing polymer solution was added into the vial and instantaneous NO levels were monitored over 3.5 h. After this time, 50 μL of a 50 mM solution of the free radical scavenger 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO) was injected into the vial in order to confirm that the amperometric signals being observed were due to NO.
Analytical instruments
OEGMA monomer conversion was determined via1H-NMR spectroscopy by the following equation: αOEGMA = 1 − (∫5.6 ppm/(∫4.1 ppm/2)), where ∫ is the peak integral of monomer (vinyl proton at 5.6 ppm, 1H) and the polymer (ester proton at 4.1 ppm, 2H).
The experimental Mn, NMR was calculated by using the dithiobenzoate end group peak (i.e.7.8 ppm) in the 1H-NMR as a reference, as follows:
M n, NMR = (∫4.1 ppm/2)/(∫7.8 ppm) × Mw, OEGA + Mw, CPADB. ∫4.1 ppm and ∫7.8 ppm represent the peak integral of OEGMA peak at 4.1 ppm (2H) and the dithiobenzoate peak (1H) at 7.8 ppm, respectively. Mw, OEGMA and Mw, CPADB represent the molar mass of OEGMA and CPADB, respectively.
VBA conversion was calculated from 1H NMR spectrum of the reaction mixture using the following equation: αOEGMA = ∫9.8 ppm/(∫10.0 ppm + ∫9.8 ppm), where ∫9.8 ppm and ∫10.0 ppm correspond to the integrals of aldehyde protons of poly(vinylbenzaldehyde) and vinyl benzaldehyde monomer, respectively.
NMR molecular weight was calculated according to Mn, NMR= ((∫9.8 ppm/(∫4.1 ppm/2)) × DPnOEGMA) × Mw, VBA + Mn, POEGMA macroRAFT, where Mw, VBA and Mn, POEGMA macroRAFT are the molecular weight of monomer and macro RAFT agent, respectively.
In addition, 1H NMR spectroscopy was used to demonstrate the conjugation of gentamicin drug to polymers as well as its release in acidic and neutral media, by monitoring changes in the signal at 9.8 ppm.
000000 g mol−1. Polymer solutions at 2–3 mg mL−1 were prepared in the eluent and filtered through 0.45 μm filters prior to injection.
Biofilm dispersal and killing assays
The laboratory strain P. aeruginosa PAO1 was used to characterize the effects of NO and/or antibiotic conjugated polymers on biofilm formation. Biofilms were grown as previously described40,58 with some modifications. Briefly, in all assays, overnight cultures in Luria Bertani medium were diluted to an OD600 of 0.005 in 1 mL M9 minimal medium (containing 48 mM Na2HPO4, 22 mM KH2PO4, 9 mM NaCl, 19 mM NH4Cl, 2 mM MgSO4, 20 mM glucose, 100 μM CaCl2, pH 7.0) in tissue-culture treated 24-well plates (Costar, Corning®). The plates were incubated at 37 °C with shaking at 180 rpm in an orbital shaker (model OM11, Ratek, Boronia, Australia) and the biofilms were allowed to grow for 6 h without any disruption.At this time, various treatments, including gentamicin-NONOate (GEN-NO) nanoparticles, free gentamicin or the NO donor N-[4-[1-(3-aminopropyl)-2-hydroxy-2-nitrosohydrazino]butyl]-1,3-propanediamine (spermine NONOate) (Cayman Chemical, USA), which has a half-life of ∼39 min at 37 °C,59,60 at different concentrations as indicated, were added to the wells. Each treatment was added from a 10 μL aliquot of a stock solution at the appropriate concentration of the compound dissolved in 10 mM NaOH and previously sterilized by passing through a 0.22 μm pore size filter. The plates were incubated for a further 1 h or 2.5 h before quantifying the biomass or viability of both planktonic and biofilm bacteria.
Biofilm biomass was determined by crystal violet staining. The biofilm on the well surfaces was first washed once with 1 mL of PBS, before adding 0.03% crystal violet stain made from a 1:10 dilution of Gram crystal violet (BD) in PBS. The plates were incubated on the bench for 20 min before washing the wells twice with PBS. Photographs of the stained biofilms were obtained using a digital camera. The amount of remaining crystal violet stained biofilm was quantified by adding 1 mL 100% ethanol and measuring OD550 of the homogenized suspension by using a microtitre plate reader (Wallac Victor2, Perkin-Elmer). OD measurements of control wells where no bacteria were added at the beginning of the experiment were subtracted from all values (i.e. OD550 = 0.10).
For viability measurements, the BacTiter-Glo Microbial Cell Viability Assay (Promega, Alexandria, Australia), which is based on quantitation of the ATP present in bacteria by using a thermostable luciferase and is known to correlate to viable cell counts, was used.61 After the final 1 h or 2.5 h incubation with various treatments, the planktonic solution was directly mixed with the BacTiter-Glo reagent following the manufacturer's instructions and after 5 min incubation, and the luminescence was measured by using a multimode microtitre plate reader (Wallac Victor2, Perkin-Elmer). In order to measure the viability of biofilm bacteria, biofilms on the interior surfaces of the wells were first washed twice with PBS before being re-suspended and homogenized in PBS by incubating in an ultrasonication bath (150 W, 40 kHz; Unisonics, Australia) for 20 min. This resuspension method is used similarly for analyzing colony-forming units (CFU) from biofilms.58 Re-suspended biofilm cells were then mixed with BacTiter-Glo reagent and their viability quantified by luminescence measurement as described above.
Confocal microscopy analysis
For microscopy analysis, P. aeruginosa biofilms were grown in glass-bottom, 24-well plates (MatTek Corporation, Ashland MA, USA) as described above. After 7 h incubation including 1 h treatment, biofilms were rinsed twice with PBS before being stained with LIVE/DEAD® BacLight™ bacterial viability kit reagents (L-7007, Molecular Probes) according to the manufacturer's procedure. One microliter of each of the two components were mixed thoroughly in 1 mL of PBS, then 0.3 mL of this solution was trapped between the sample and the glass microscopy slide and allowed to incubate at room temperature in the dark for 20 min. The samples were observed with an Olympus FV1000 Confocal Inverted Microscope, and imaged with a Leica DFC 480 camera. Cells that were stained green were considered to be viable, those that stained red and stained both green and red were considered to be non-viable.Statistical analysis
All assays included 2 replicates and were repeated in 2 independent experiments. Statistical analyses were performed with GraphPad Prism 6 (GraphPad Software) using one-way ANOVA followed by Dunnett's multiple comparison test comparing treated samples to the untreated control.Results and discussion
In this study, we designed polymeric nanoparticles for the co-delivery of nitric oxide and an antibiotic, gentamicin. To achieve a controlled release of gentamicin, we conjugated gentamicin to the polymer via a hydrolysable Schiff base linkage by reacting amino groups of gentamicin (GEN) with aldehyde groups. Aldehyde groups can react rapidly with primary amines to yield a hydrolysable linkage,36,62–66 that allows a slow release of antibiotic in the middle acidic microenvironment of biofilm.67,68 To confer water solubility to the polymers, we prepared an amphiphilic block copolymer, constituted of a hydrophilic block (POEGMA), which is closely related to polyethylene glycol leading to excellent biocompatibility, and a short hydrophobic block containing aldehyde groups (PVBA) for further conjugation with gentamicin.Synthesis of POEGMA-b-PVBA block copolymer
Block copolymer POEGMA-b-PVBA was synthesized using living polymerization (i.e. reversible addition fragmentation chain transfer (RAFT) polymerization (Scheme 1). Poly((oligoethylene glycol) methyl ether methacrylate) (POEGMA) macro-RAFT agent was prepared in toluene at 70 °C in the presence of 4-cyanopentanoic acid dithiobenzoate (CPADB) as a RAFT agent and oligo(ethylene glycol) methacrylate (OEGMA) as monomer. The monomer conversion was monitored via1H NMR spectroscopy by comparing the vinyl proton signals (at 6.1 and 5.6 ppm) with ester –OCH2 proton peaks (at 4.1 ppm). At ∼80% monomer conversion, the polymerization was stopped to avoid the formation of significant dead polymers; then, the polymer product was purified by several precipitations (three times) in petroleum spirits. The molecular weight obtained by SEC analysis is in good agreement with the theoretical value (Mn, theo = 10800 g mol−1, Mn, SEC = 11200 g mol−1, PDI = 1.08). Subsequently, POEGMA was successfully chain extended in the presence of 3-vinylbenzylaldehyde (VBA) to afford POEGMA-b-PVBA block copolymer. The conversion of VBA was determined to be around 50% using the vinyl signals at 5.0–6.0 ppm and aromatic signals at 6.5–7.5 ppm (ESI, Fig. S1†) to yield POEGMAx-b-VBAy, with x and y equal to 36 and 7. After purification, SEC analysis confirmed the successful chains extension by the molecular weight distribution shift to higher molecular weight (Mn, SEC = 13700 g mol−1) and a low polydispersity index (PDI = 1.13) was obtained (ESI, Fig. S2†). 1H NMR and FTIR spectroscopy confirmed VBA incorporation by the presence of characteristic signals at 9.8 ppm and at 1710 cm−1 attributed to aldehyde group, respectively. The final copolymer was constituted by a longer block of OEGMA (36 units) to confer good solubility in water, and a shorter block of VBA (7 units) for functionality. This composition appears ideal to afford well-defined nanoparticles after conjugation with gentamicin. Indeed, if a longer block of VBA were employed, the copolymer would not be soluble in water.
 | ||
| Scheme 1 Schematic approach for the preparation of gentamicin-NONOate nanoparticles via RAFT polymerization. | ||
Conjugation of POEGMA-b-PVBA to gentamicin
Gentamicin was conjugated via a hydrolysable bond (Schiff base/imine) by reaction of primary amines with the aldehyde group of the copolymer for 48 h in the presence of triethylamine in water (pH = 8.0). Due to the presence of several amine groups per gentamicin, the hydrophobic segments in the core of the nanoparticles were simultaneously conjugated and cross-linked (Scheme 1). DLS results showed the number-average size of 15 nm and the polydispersity index (PDI) of 0.1, which is in good agreement of TEM data (Fig. 1). Volume and intensity distributions (Fig. S3†) were similar to the number distribution, indicating the nanoparticles were not forming aggregates. The resultant cross-linked polymer was further characterized by DMAc SEC (ESI, Fig. S4†). We observed a substantial shift in molecular weight of block copolymer after conjugation with gentamicin, from 13700 g mol−1 to 110000 g mol−1, which demonstrates the successful formation of cross-linked nanoparticles with a narrow polydispersity (PDI = 1.34). It should be noted that the SEC system was calibrated with linear polystyrene standards and the number average molecular weight obtained normally underestimates the actual molecular weight of the polymer. Sumerlin and co-workers have described a similar method for the synthesis of core cross-linked star polymers using difunctional organic compounds.62,69–71 After purification, the polymer was dissolved in acetonitrile in the presence of magnesium sulfate to remove water for further reaction with NO gas.
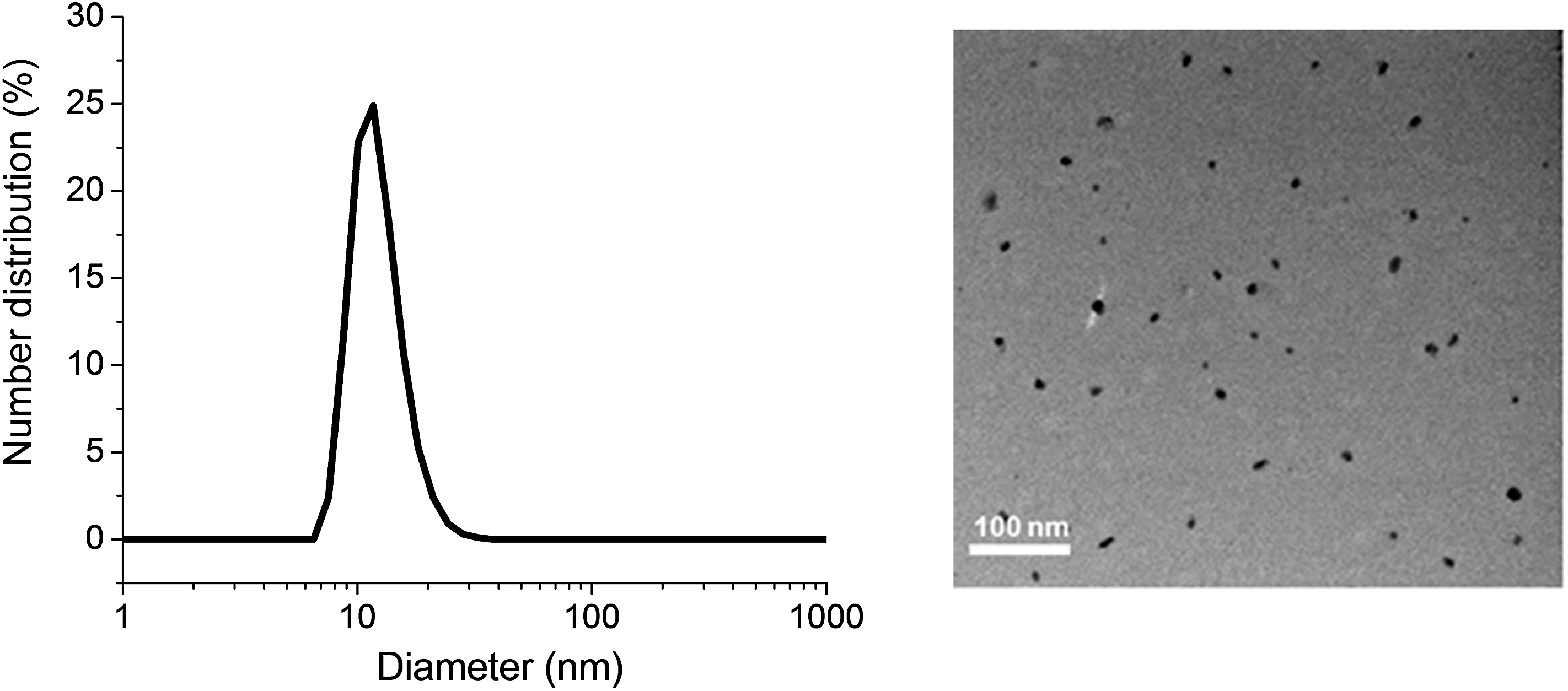 | ||
| Fig. 1 Dynamic light scattering (DLS) graph (left) and TEM image (right) depicting the size of POEGMA-b-PVBA-GEN nanoparticles. | ||
The successful attachment of gentamicin was also confirmed using 1H NMR, elemental analysis and ATR-FTIR analyses. The conversion of aldehyde was monitored by NMR that following the decrease in the signal at 9.8 ppm (Fig. 2 and 3), which corresponds to the aldehyde group. A conjugation efficiency of 72% was determined by comparing the intensity of aldehyde signal at 9.8 ppm with the –CH2O ester of OEGMA at 4.1 ppm before and after the reaction with gentamicin after 48 h. The reaction time was extended up to 72 h, but the conjugation efficiency was not improved. This result could be attributed to the steric hindrance of gentamicin limiting its reactivity with aldehyde. After purification by precipitation, the polymer was dissolved in D2O and analyzed by 1H NMR (600 MHz) (ESI, Fig. S5†). We exploited the characteristic signal at 5.8 ppm which corresponds to –CH of gentamicin to determine the amount of gentamicin in polymer by comparing with the –CH2O ester of OEGMA at 4.1 ppm. NMR results showed around 2.3 gentamicins per polymer chain. However, this number of gentamicin can be underestimated due to the encapsulation of gentamicin in the core of the nanoparticles, which could lead to the incomplete solvation. Such behavior has been observed in previous studies by us36,63 and others69,71 for various systems. We have further analyzed the composition of the polymer by elemental analysis using X-ray Photoelectron Spectroscopy (XPS). Gentamicin contains five nitrogen atoms (from amine groups), while the polymer is only constituted by oxygen and carbon. RAFT agent contains only one nitrogen atom. By comparing the nitrogen amount before and after conjugation with gentamicin, we were able to estimate that 2.8–3.2 gentamicins where incorporated in the polymer (ESI, Table S2†). This value is relatively close with NMR data. According to the values obtained by aldehyde conversion from 1H NMR (72%, corresponding to 5 reacted aldehyde groups) and the number of gentamicin (3 units) per polymer chain, we were able to calculate that, on average, three gentamicin molecules reacted with 3 aldehyde groups of the block copolymer and the other two aldehyde groups reacted with additional amine groups from these gentamicin molecules. This could explain the formation of cross-linked nanoparticles observed by SEC analysis.
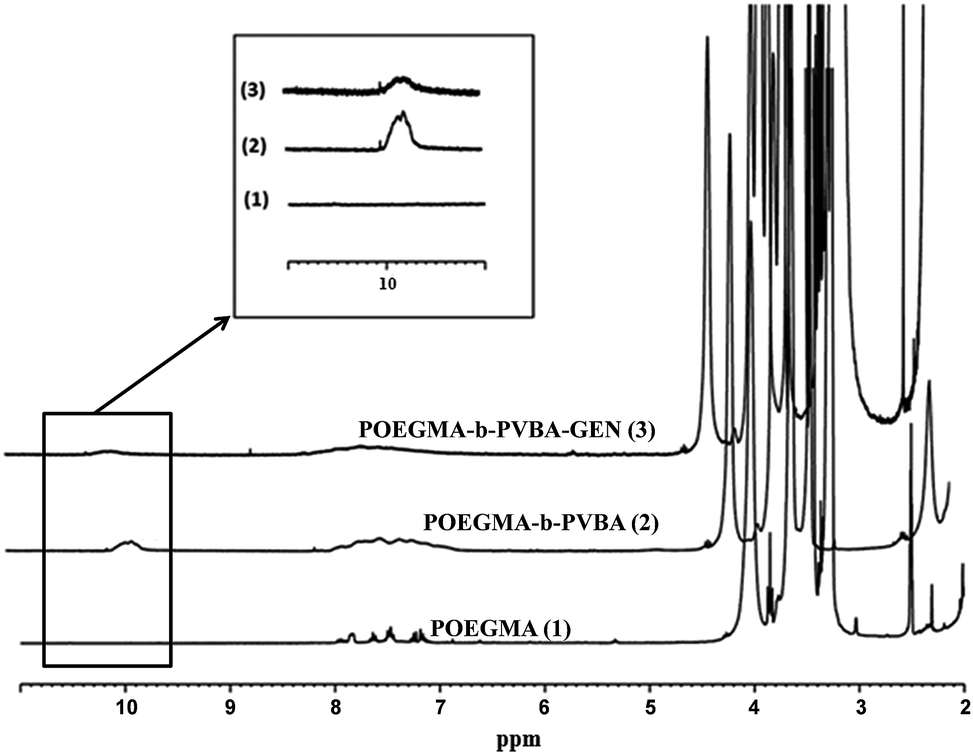 | ||
| Fig. 2 1H NMR spectra of purified POEGMA-b-PVBA-GEN overlaid with POEGMA-b-PVBA and POEGMA. | ||
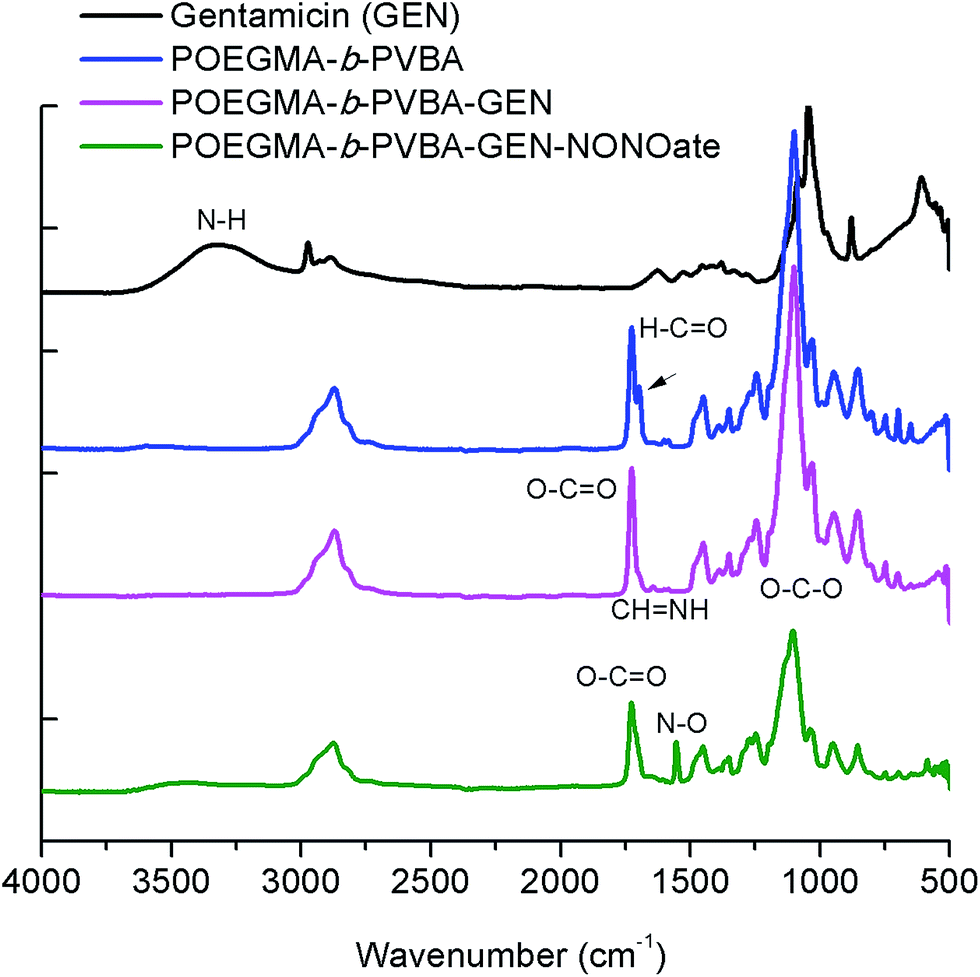 | ||
| Fig. 3 ATR-FTIR spectra of POEGMA-b-PVBA-GEN-NONOate compared with POEGMA-b-PVBA-GEN, POEGMA-b-PVBA and gentamicin. | ||
Finally, the conjugation of the copolymer to gentamicin was confirmed by ATR-FTIR spectroscopy. After conjugation with gentamicin, we noted a decrease in signals from the aldehyde bond at 1710 cm−1 and the presence of new absorption at ∼1620 cm−1 consistent with the formation of imine (Fig. 3).
Post-modification of gentamicin conjugated POEGMA-b-PVBA nanoparticles with nitric oxide
As mentioned earlier, the aim of this study was to develop a nanocarrier system to disperse bacterial biofilms and kill bacteria in planktonic form. Our approach was to develop a compound that could disperse the biofilm, thereby making the bacteria more susceptible to antimicrobial agents, and then simultaneously treat the planktonic bacteria. In this study, gentamicin was chosen for this dual purpose owing to the presence of both primary and secondary amine groups. These functional groups allow for an easy conjugation of gentamicin to aldehyde functionalized polymers and the formation of gentamicin-N-diazeniumdiolate (gentamicin-NONOate) conjugated polymers by reaction of the secondary amine with nitric oxide (NO) gas. This is a novel approach, which combines the benefit of NO to the existing antibiotics. The NONOate group in gentamicin-NONOate complex can slowly release NO to re-generate native gentamicin. As gentamicin contains one secondary amine group, theoretically, one NONOate group could be attached per gentamicin. After purification, the polymeric nanoparticles were analyzed by three different techniques: UV-Vis spectroscopy, elemental analysis and ATR-FTIR. Firstly, UV-Vis was performed to quantify the amount of NONOate group by comparing the signal centered at 250 nm (ESI, Fig. S6†) before and after NO treatment using molar extinction coefficient for NONOate of 8500 M−1 cm−1.59,72 The UV-Vis after NO treatment shows an increased signal at around 250 nm, which demonstrates the successful attachment of NO. The amount of NONOate calculated by UV-Vis was close to one NONOate per gentamicin, which is in agreement with the expected values, i.e. 3 NONOate per polymer chain. Secondly, elemental analysis was carried out to quantify the amount of nitrogen. After NO treatment, we observed a significant increase of nitrogen (ESI, Table S2†), which corresponds to one NONOate per gentamicin, i.e. 3 NONOate per polymer chain. Both UV and elemental results are in good agreement. Finally, ATR-FTIR analysis confirmed the presence of the N–O band at 1510 cm−1 (Fig. 3), indicating the successful attachment of the NONOate group on gentamicin.Determination of gentamicin and nitric oxide release
The imine bond has previously been employed by our group and others for drug conjugation62–66,69,70 owing to its ability to slowly hydrolyze, which allows a sustainable release of therapeutic compounds. Bacterial biofilms and infected tissue by bacteria usually present a slight acidic pH (typically between 5.5–7.2), which should favor the release of gentamicin from the nanoparticles.68,73 Gentamicin conjugated nanoparticles were incubated in both pH 7.4 (phosphate buffer) and pH 5.5 (acetate buffer) (ESI, Fig. S7 and S8†). Nanoparticles were placed in a dialysis membrane with MWCO 3500 Da and the samples were taken at different time points for gentamicin and NO release. The gentamicin release kinetic from POEGMA-b-PVBA nanoparticles was monitored by comparing the aldehyde proton –CHO peak at 9.8 ppm and the –CH2O-proton peaks at 4.1 ppm using 1H NMR analysis. As expected, the intensity of the signal at 9.8 ppm increased over time, indicating the release of gentamicin. The release rate of gentamicin at pH 5.5 was slightly faster than at pH 7.4. After 17 h, around 50% of gentamicin had been released in both pH values (ESI, Fig. S9†). The slow release of gentamicin is desirable as it allows a prolonged action for a long treatment. Concurrently with the release of the gentamicin, the cross-linked structure disassembled into free block copolymer as shown by a decrease of molecular weight by SEC (ESI, Fig. S4 and Table S1†).NO release from GEN-NO nanoparticles was assessed by the Griess assay, which is commonly employed to monitor the cumulative release of NO by several groups24,74–81 (ESI, Fig. S10†) and by amperometric measurement (ESI, Fig. S11†) following a previous procedure established by us.24 Griess assay measures the accumulation of nitrite and nitrate in water due to the rapid oxidation of NO in aerobic conditions, while amperometric measurement measures the instantaneous release of NO. The media employed to determine the NO release can affect the measurement as demonstrated by Schoenfisch's group82 and Reynolds' group.77,82 For this reason, we decided to perform the release in phosphate buffer, which has been demonstrated to give more accurate results.
As indicated in Fig. 4 (and ESI, Fig. S10 and S11†), both tests showed a prolonged release of NO for several hours. Interestingly, NO released from GEN-NO nanoparticles followed first order kinetics that had a half-life of approximately 1 h at pH 7.4. After 5 h, over 75% of NO was released from the polymeric nanoparticles according to Griess assay (Fig. 4). Amperometric measurement (ESI, Fig. S11†) showed a rapid release of NO as the beginning of the experiment, which is consistent with Griess assay (i.e. approximately 10% of NO has been released after 10 min). More importantly, amperometric experiment showed a continuous release of NO for over 3.5 h. After 3.5 h, we added a free radical scavenger, i.e. 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO), into the vial in order to confirm that the amperometric signals being observed were due to NO. The signal of NO rapidly decreased after addition of PTIO. Interestingly, the encapsulation of NONOate in the core of nanoparticles appeared to enhance the stability of NONOate. Indeed, NONOate compounds such as diethylamine NONOate and spermine NONOate have very short half-lives (i.e. few minutes) as NONOates can spontaneously decompose to release NO in the presence of water.83 This relative slow release is desirable for our application to achieve a long dispersion of biofilms and avoid a rapid reformation of biofilm, which allows the gentamicin to kill bacteria.
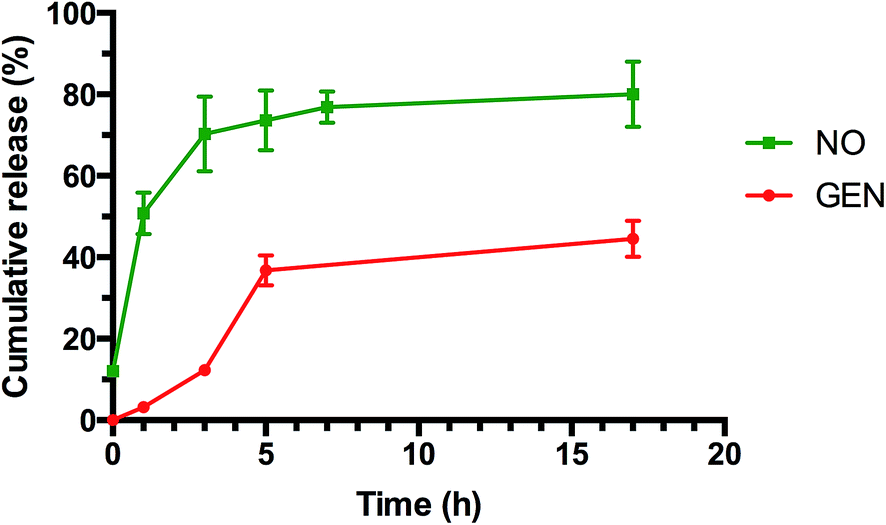 | ||
| Fig. 4 Cumulative release of NO and GEN from GEN-NO nanoparticles at pH 7.4, 37 °C. The concentration of GEN-NO nanoparticles was 5 mg mL−1; experiments were performed in triplicate (the points represent the average of three values). | ||
POEGMA-b-PVBA-gentamicin-NONOate eradicates P. aeruginosa biofilms
To evaluate the effect of the new POEGMA-b-PVBA-gentamicin-NONOate (GEN-NO nanoparticles) on biofilms, we first tested their ability to release NO and disperse biofilms. Pre-established biofilms of the opportunistic pathogen and model biofilm-forming organism P. aeruginosa that had been grown for 6 h in the absence of any treatment, were treated with various compounds: (i) NO donor, spermine NONOate (Sper-NO); (ii) free GEN; (iii) gentamicin-conjugated polymers (Poly-GEN) and (iv) GEN-NO nanoparticles (Poly-GEN-NO). After 1 h treatment, the GEN-NO nanoparticles at 5 μM (based on GEN, one mole of GEN-NO nanoparticles is equivalent to one mole of Sper-NO and gentamicin) were found to induce biofilm dispersal, leading to 83% reduction in biofilm biomass as determined by crystal violet (CV) staining, compared with untreated control biofilms (Fig. 5). Increasing the nanoparticle concentrations to 10–50 μM (based on GEN), while still clearly inducing biofilm dispersal, resulted in slightly higher levels of staining on the well surfaces, which was possibly due to a higher amount of cells that were killed but not dispersed and thus also stained with CV. The addition of the NO donor, Sper-NO, which was used at equimolar concentrations compared to GEN-NO nanoparticles, led to only 30% reduction in biomass at 50 μM (Fig. 5). This result is comparable to other NONOate-conjugated polymers that were previously shown to disperse biofilms.28,48 Treatment with the antibiotic gentamicin alone only induced a small decrease in biofilm biomass at high concentrations, with 50 μM free gentamicin leading to less than 14% reduction. Gentamicin-conjugated polymers (i.e. without NO) did not reduce the amount of cells attached on the surface at all concentrations tested (Fig. 5).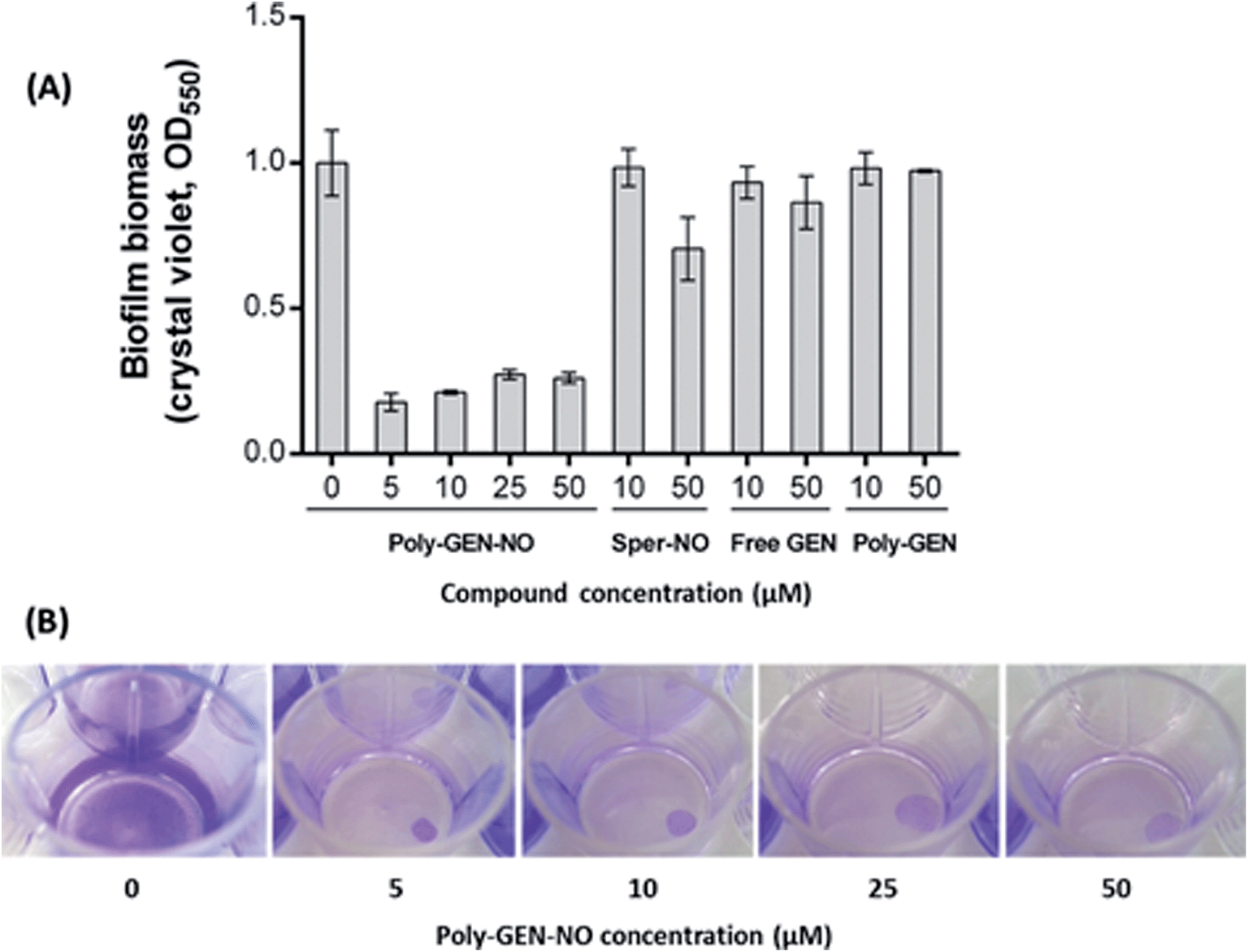 | ||
| Fig. 5 GEN-NO nanoparticles induced dispersal in P. aeruginosa biofilms. (A) Bacterial biofilms were grown in multi-well plates for 6 h in the absence of any treatment before being treated for a further 1 h with various concentrations (μM) of NO donor spermine NONOate (Sper-NO), free gentamicin or gentamicin-conjugated polymers (Poly-GEN) and GEN-NO nanoparticles (Poly-GEN-NO). Biofilm biomass was analyzed by crystal violet staining. Error bars represent standard error (n = 2). (B) Stained biofilms treated with the indicated concentrations of GEN-NO nanoparticles. Note: concentration based on GEN, one mole of GEN-NO nanoparticles is equivalent to one mole of Sper-NO and gentamicin. | ||
Furthermore, confocal microscopy was used to evaluate the ability of the GEN-NO nanoparticles to disperse biofilms. Biofilm cells were stained with LIVE/DEAD dyes, where live and dead cells appear green and red, respectively. Cultures treated with GEN-NO nanoparticles at 10 μM displayed greatly reduced biofilm biovolume and exhibited more dead cells, compared with untreated control biofilms or those inoculated with the NO donor or gentamicin alone (Fig. 6). Overall, the crystal violet and confocal microscopy results confirmed that GEN-NO nanoparticles were able to release NO, which was made available to biofilms, and consequently, induced dispersal of biofilm cells.
 | ||
| Fig. 6 Representative confocal images showing P. aeruginosa biofilms stained with LIVE/DEAD kit. Biofilms were grown for 6 h and then treated with NO donor spermine NONOate (Sper-NO), free gentamicin, GEN-NO nanoparticles or left untreated for a further 1 h before staining. Viable and non-viable bacteria appear green and red, as well as those stained both green/red, respectively. Scale bar = 50 μm. Note: concentration based on GEN, one mole of GEN-NO nanoparticles is equivalent to one mole of Sper-NO and gentamicin. | ||
Next, the bactericidal properties of GEN-NO nanoparticles were investigated. P. aeruginosa biofilms were grown in vitro for 6 h as described above before being exposed to various treatments, including the NO donor, free GEN and GEN-NO nanoparticles. Then instead of analyzing the biofilm cultures by crystal violet staining, which can only account for total biomass, the viability of the cultures was assessed by measuring the ATP content of both biofilm and planktonic cells (Fig. 7). After 1 h treatment, a strong killing effect was observed in cell cultures treated with GEN-NO nanoparticles at 5–50 μM, compared with the untreated control, free gentamicin or NO donor alone. At 10 μM, GEN-NO nanoparticles almost completely eradicated both biofilm and planktonic cells. The viability of bacteria decreased significantly by 90% and 94% (P < 0.0001) in the biofilm and planktonic phases, respectively, compared with untreated cultures (Fig. 7). In contrast, gentamicin alone at 10 μM induced only a 7% and 5% decrease in biofilm and planktonic viability, respectively. In the presence of higher concentrations (i.e. 25 or 50 μM), gentamicin showed a slight increase in the reduction of biofilm cells, by up to 20%, but gentamicin did not affect planktonic viability (Fig. 7).
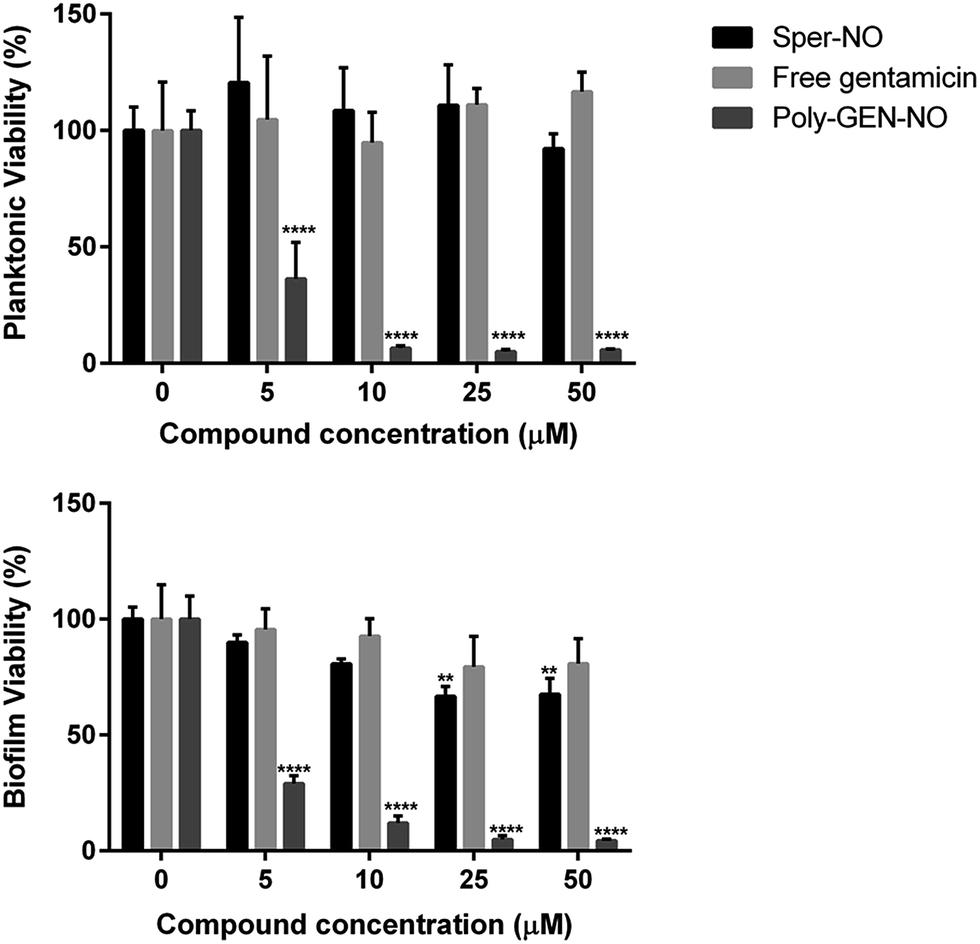 | ||
| Fig. 7 Effect of GEN-NO nanoparticles on P. aeruginosa viability after combined release of NO and gentamicin. P. aeruginosa biofilms were grown in multi-well plates for 6 h in the absence of any treatments and treated further for 1 h in the presence of 5–50 μM the NO donor spermine NONOate (Sper-NO), free gentamicin and GEN-NO nanoparticles (Poly-GEN-NO) before analyzing planktonic (top) and biofilm (bottom) viability by measuring the ATP content of bacteria. Error bars represent standard error (n = 4). Asterisks indicate statistically significant difference of treatments versus untreated culture (**, P < 0.01; ****, P < 0.0001). Note: concentration based on GEN, one mole of GEN-NO nanoparticles is equivalent to one mole of Sper-NO and gentamicin. | ||
These results appeared consistent with previously published data, where a minimum concentration of 100 μM gentamicin was needed to eradicate P. aeruginosa.84,85 Furthermore, the NO donor spermine NONOate alone at a concentration of 10 μM, releasing an equivalent amount of NO compared to GEN-NO nanoparticles, did not display any toxicity towards planktonic cells (Fig. 7). Indeed, spermine NONOate induced an increase in cell viability by approximately 10% in the planktonic phase and a concomitant 19% reduction in the biofilm phase, indicative of dispersal events. At higher concentrations (i.e. 50 μM), the NO donor only caused a small and non-significant decrease in the planktonic phase, which was less than 8%. Results from the spermine NONOate cell viability assay indicated that the amount of NO in the GEN-NO nanoparticles was not involved in the killing effect of the compound, which was mainly due to the bactericidal activity of gentamicin. Taken together, these results strongly suggest that the combination of NO and gentamicin into a single polymeric structure leads to synergistic effects of biofilm dispersal and enhanced bactericidal activity and represents a highly promising strategy for combatting biofilm-related infections.
Conclusions
In summary, we have synthesized a novel dual-action polymer based on an NO donor and the aminoglycoside gentamicin and demonstrated its potential for use in controlling P. aeruginosa biofilms. Combined and simultaneous delivery of NO and gentamicin is an attractive feature that would allow removing bacterial biofilms and killing the dispersed bacteria with one treatment. Encapsulated within the polymeric matrix the two agents are likely to have enhanced pharmacodynamic properties for systemic or local treatments. Furthermore, these compounds might be useful when applied as surface coating for the inhibition and prevention of biofilm formation on clinical surfaces or implants.Acknowledgements
C. B. acknowledges the Australian Research Council for his Future Fellowship (FT120100096) and UNSW – Australia. N. B. is supported by ARC grant DE120101604.References
- J. P. Guggenbichler, O. Assadian, M. Boeswald and A. Kramer, GMS Krankenhaushygiene interdisziplinar, 2011, 6, 18 Search PubMed.
- B. Spellberg, R. Guidos, D. Gilbert, J. Bradley, H. W. Boucher, W. M. Scheld, J. G. Bartlett and J. Edwards, Clin. Infect. Dis., 2008, 46, 155–164 CrossRef PubMed , t. I. D. S. o. America.
- M. B. Harney, Y. Zhang and L. R. Sita, Angew. Chem., Int. Ed., 2006, 45, 6140–6144 CrossRef CAS PubMed.
- N. Høiby, T. Bjarnsholt, M. Givskov, S. Molin and O. Ciofu, Int. J. Antimicrob. Agents, 2010, 35, 322–332 CrossRef PubMed.
- T. Bjarnsholt, P. Ø. Jensen, M. J. Fiandaca, J. Pedersen, C. R. Hansen, C. B. Andersen, T. Pressler, M. Givskov and N. Høiby, Pediatr. Pulmonol. 2009, vol. 44, pp. 547–558 Search PubMed.
- W. Costerton, R. Veeh, M. Shirtliff, M. Pasmore, C. Post and G. Ehrlich, J. Clin. Invest., 2003, 112, 1466–1477 CrossRef CAS PubMed.
- A. G. Gristina, M. Oga, L. X. Webb and C. D. Hobgood, Science, 1985, 228, 990–993 CAS.
- D. Lebeaux, J. M. Ghigo and C. Beloin, Microbiol. Mol. Biol. Rev., 2014, 78, 510–543 CrossRef CAS PubMed.
- A. Ito, A. Taniuchi, T. May, K. Kawata and S. Okabe, Appl. Environ. Microbiol., 2009, 75, 4093–4100 CrossRef CAS PubMed.
- J. S. Madsen, M. Burmolle, L. H. Hansen and S. J. Sorensen, FEMS Immunol. Med. Microbiol., 2012, 65, 183–195 CrossRef CAS PubMed.
- S. M. Moskowitz, J. M. Foster, J. Emerson and J. L. Burns, J. Clin. Microbiol., 2004, 42, 1915–1922 CrossRef CAS PubMed.
- I. Keren, N. Kaldalu, A. Spoering, Y. Wang and K. Lewis, FEMS Microbiol. Lett., 2004, 230, 13–18 CrossRef CAS PubMed.
- L. Yang, J. A. J. Haagensen, L. Jelsbak, H. K. Johansen, C. Sternberg, N. Høiby and S. Molin, J. Bacteriol., 2008, 190, 2767–2776 CrossRef CAS PubMed.
- M. Klausen, A. Heydorn, P. Ragas, L. Lambertsen, A. Aaes-Jørgensen, S. Molin and T. Tolker-Nielsen, Mol. Microbiol., 2003, 48, 1511–1524 CrossRef CAS PubMed.
- A. Heydorn, B. Ersbøll, J. Kato, M. Hentzer, M. R. Parsek, T. Tolker-Nielsen, M. Givskov and S. Molin, Appl. Environ. Microbiol., 2002, 68, 2008–2017 CrossRef CAS PubMed.
- S. Molin and T. Tolker-Nielsen, Curr. Opin. Biotechnol., 2003, 14, 255–261 CrossRef CAS PubMed.
- H. Anwar, M. K. Dasgupta and J. W. Costerton, Antimicrob. Agents Chemother., 1990, 34, 2043–2046 CrossRef CAS PubMed.
- D. McDougald, S. A. Rice, N. Barraud, P. D. Steinberg and S. Kjelleberg, Nat. Rev. Microbiol., 2012, 10, 39–50 CAS.
- N. Barraud, D. J. Hassett, S. H. Hwang, S. A. Rice, S. Kjelleberg and J. S. Webb, J. Bacteriol., 2006, 188, 7344–7353 CrossRef CAS PubMed.
- D. P. Arora, S. Hossain, Y. Xu and E. M. Boon, Biochemistry, 2015, 54, 3717–3728 CrossRef CAS PubMed.
- P. D. Lister, D. J. Wolter and N. D. Hanson, Clin. Microbiol. Rev., 2009, 22, 582–610 CrossRef CAS PubMed.
- N. Barraud, D. Schleheck, J. Klebensberger, J. S. Webb, D. J. Hassett, S. A. Rice and S. Kjelleberg, J. Bacteriol., 2009, 191, 7333–7342 CrossRef CAS PubMed.
- N. Liu, Y. Xu, S. Hossain, N. Huang, D. Coursolle, J. A. Gralnick and E. M. Boon, Biochemistry, 2012, 51, 2087–2099 CrossRef CAS PubMed.
- N. Barraud, B. G. Kardak, N. R. Yepuri, R. P. Howlin, J. S. Webb, S. N. Faust, S. Kjelleberg, S. A. Rice and M. J. Kelso, Angew. Chem., Int. Ed. Engl., 2012, 51, 9057–9060 CrossRef CAS PubMed.
- S. L. Chua, Y. Liu, J. K. Yam, Y. Chen, R. M. Vejborg, B. G. Tan, S. Kjelleberg, T. Tolker-Nielsen, M. Givskov and L. Yang, Nat. Commun., 2014, 5, 4462 CAS.
- L. D. Christensen, M. van Gennip, M. T. Rybtke, H. Wu, W. C. Chiang, M. Alhede, N. Hoiby, T. E. Nielsen, M. Givskov and T. Tolker-Nielsen, Infect. Immun., 2013, 81, 2705–2713 CrossRef CAS PubMed.
- N. Barraud, M. J. Kelso, S. A. Rice and S. Kjelleberg, Curr. Pharm. Des., 2015, 21, 31–42 CrossRef CAS PubMed.
- H. T. T. Duong, N. N. M. Adnan, N. Barraud, J. S. Basuki, S. K. Kutty, K. Jung, N. Kumar, T. P. Davis and C. Boyer, J. Mater. Chem. B, 2014, 2, 5003–5011 RSC.
- H. T. T. Duong, Z. M. Kamarudin, R. B. Erlich, Y. Li, M. W. Jones, M. Kavallaris, C. Boyer and T. P. Davis, Chem. Commun., 2013, 49, 4190–4192 RSC.
- B. Bonavida, S. Baritaki, S. Huerta-Yepez, M. I. Vega, D. Chatterjee and K. Yeung, Nitric Oxide, 2008, 19, 152–157 CrossRef CAS PubMed.
- A. Bratasz, N. M. Weir, N. L. Parinandi, J. L. Zweier, R. Sridhar, L. J. Ignarro and P. Kuppusamy, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3914–3919 CrossRef CAS PubMed.
- Y. Lu, D. L. Slomberg, A. Shah and M. H. Schoenfisch, Biomacromolecules, 2013, 14, 3589–3598 CrossRef CAS PubMed.
- Y. Lu, D. L. Slomberg and M. H. Schoenfisch, Biomaterials, 2014, 35, 1716–1724 CrossRef CAS PubMed.
- D. A. Riccio and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3731–3741 RSC.
- H. Zhang, G. M. Annich, J. Miskulin, K. Stankiewicz, K. Osterholzer, S. I. Merz, R. H. Bartlett and M. E. Meyerhoff, J. Am. Chem. Soc., 2003, 125, 5015–5024 CrossRef CAS PubMed.
- Y. Li, H. T. T. Duong, S. Laurent, A. MacMillan, R. M. Whan, L. V. Elst, R. N. Muller, J. Hu, A. Lowe, C. Boyer and T. P. Davis, Adv. Healthcare Mater., 2015, 4, 148–156 CrossRef CAS PubMed.
- S. Duan, S. Cai, Y. Xie, T. Bagby, S. Ren and M. L. Forrest, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2715–2724 CrossRef CAS.
- S. Duan, S. Cai, Q. Yang and M. L. Forrest, Biomaterials, 2012, 33, 3243–3253 CrossRef CAS PubMed.
- H. T. T. Duong, A. Ho, T. P. Davis and C. Boyer, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2099–2103 CrossRef CAS.
- N. R. Yepuri, N. Barraud, N. Shah Mohammadi, B. G. Kardak, S. Kjelleberg, S. A. Rice and M. J. Kelso, Chem. Commun., 2013, 49, 4791–4793 RSC.
- S. K. Kutty, N. Barraud, A. Pham, G. Iskander, S. A. Rice, D. S. Black and N. Kumar, J. Med. Chem., 2013, 56, 9517–9529 CrossRef CAS PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- G. M. Whitesides, Nat. Biotechnol., 2003, 21, 1161–1165 CrossRef CAS PubMed.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- Y. Lu, A. Shah, R. A. Hunter, R. J. Soto and M. H. Schoenfisch, Acta Biomater., 2015, 12, 62–69 CrossRef CAS PubMed.
- E. M. Hetrick and M. H. Schoenfisch, Chem. Soc. Rev., 2006, 35, 780–789 RSC.
- H. T. Duong, K. Jung, S. K. Kutty, S. Agustina, N. N. Adnan, J. S. Basuki, N. Kumar, T. P. Davis, N. Barraud and C. Boyer, Biomacromolecules, 2014, 15, 2583–2589 CrossRef CAS PubMed.
- D. L. Slomberg, Y. Lu, A. D. Broadnax, R. A. Hunter, A. W. Carpenter and M. H. Schoenfisch, ACS Appl. Mater. Interfaces, 2013, 5, 9322–9329 CAS.
- E. M. Hetrick, J. H. Shin, N. A. Stasko, C. B. Johnson, D. A. Wespe, E. Holmuhamedov and M. H. Schoenfisch, ACS Nano, 2008, 2, 235–246 CrossRef CAS PubMed.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- A. W. Carpenter, D. L. Slomberg, K. S. Rao and M. H. Schoenfisch, ACS Nano, 2011, 5, 7235–7244 CrossRef CAS PubMed.
- Y. Lu, D. L. Slomberg, B. Sun and M. H. Schoenfisch, Small, 2013, 9, 2189–2198 CrossRef CAS PubMed.
- P. N. Coneski and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3753–3758 RSC.
- B. V. Worley, K. M. Schilly and M. H. Schoenfisch, Mol. Pharmaceutics, 2015, 12, 1573–1583 CrossRef CAS PubMed.
- B. V. Worley, D. L. Slomberg and M. H. Schoenfisch, Bioconjugate Chem., 2014, 25, 918–927 CrossRef CAS PubMed.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- N. Barraud, J. A. Moscoso, J. M. Ghigo and A. Filloux, in Pseudomonas Methods and Protocols, ed. A. Filloux and J. L. Ramos, Humana Press, New York, NY, 2014, vol. 1149 Search PubMed.
- C. M. Maragos, D. Morley, D. A. Wink, T. M. Dunams, J. E. Saavedra, A. Hoffman, A. A. Bove, L. Isaac, J. A. Hrabie and L. K. Keefer, J. Med. Chem., 1991, 34, 3242–3247 CrossRef CAS PubMed.
- L. K. Keefer, R. W. Nims, K. M. Davies and D. A. Wink, in Methods Enzymol., ed. P. Lester, Academic Press: 1996, vol. 268, pp. 281–293 Search PubMed.
- X. Chen, H. Hirt, Y. Li, S.-U. Gorr and C. Aparicio, PLoS One, 2014, 9, e111579 Search PubMed.
- S. Mukherjee, A. P. Bapat, M. R. Hill and B. S. Sumerlin, Polym. Chem., 2014, 5, 6923–6931 RSC.
- J. Liu, H. Duong, M. R. Whittaker, T. P. Davis and C. Boyer, Macromol. Rapid Commun., 2012, 33, 760–766 CrossRef CAS PubMed.
- A. W. Jackson and D. A. Fulton, Chem. Commun., 2011, 47, 6807–6809 RSC.
- A. W. Jackson, C. Stakes and D. A. Fulton, Polym. Chem., 2011, 2, 2500–2511 RSC.
- A. W. Jackson and D. A. Fulton, Polym. Chem., 2013, 4, 31–45 RSC.
- H.-S. Lee, S. S. Dastgheyb, N. J. Hickok, D. M. Eckmann and R. J. Composto, Biomacromolecules, 2015, 16, 650–659 CrossRef CAS PubMed.
- J. M. Vroom, K. J. de Grauw, H. C. Gerritsen, D. J. Bradshaw, P. D. Marsh, G. K. Watson, J. J. Birmingham and C. Allison, Appl. Environ. Microbiol., 1999, 65, 3502–3511 CAS.
- A. P. Bapat, D. Roy, J. G. Ray, D. A. Savin and B. S. Sumerlin, J. Am. Chem. Soc., 2011, 133, 19832–19838 CrossRef CAS PubMed.
- A. P. Bapat, J. G. Ray, D. A. Savin, E. A. Hoff, D. L. Patton and B. S. Sumerlin, Polym. Chem., 2012, 3, 3112–3120 RSC.
- A. P. Bapat, J. G. Ray, D. A. Savin and B. S. Sumerlin, Macromolecules, 2013, 46, 2188–2198 CrossRef CAS.
- R. Ferrero, F. Rodríguez-Pascual, M. T. Miras-Portugal and M. Torres, Br. J. Pharmacol., 1999, 127, 779–787 CrossRef CAS PubMed.
- A. Hoštacká, I. Čižnár and M. Štefkovičová, Folia Microbiol., 2010, 55, 75–78 CrossRef PubMed.
- Y. Li and P. I. Lee, Mol. Pharmaceutics, 2010, 7, 254–266 CrossRef CAS PubMed.
- B. J. Heilman, J. St. John, S. R. J. Oliver and P. K. Mascharak, J. Am. Chem. Soc., 2012, 134, 11573–11582 CrossRef CAS PubMed.
- J. Sun, X. Zhang, M. Broderick and H. Fein, Sensors, 2003, 3, 276 CrossRef CAS.
- J. L. Harding and M. M. Reynolds, Anal. Chem., 2014, 86, 2025–2032 CrossRef CAS PubMed.
- C. Vogt, Q. Xing, W. He, B. Li, M. C. Frost and F. Zhao, Biomacromolecules, 2013, 14, 2521–2530 CrossRef CAS PubMed.
- K. S. Bohl and J. L. West, Biomaterials, 2000, 21, 2273–2278 CrossRef CAS PubMed.
- K. A. Mowery, M. H. Schoenfisch, J. E. Saavedra, L. K. Keefer and M. E. Meyerhoff, Biomaterials, 2000, 21, 9–21 CrossRef CAS PubMed.
- A. B. Seabra, R. da Silva and M. G. de Oliveira, Biomacromolecules, 2005, 6, 2512–2520 CrossRef CAS PubMed.
- R. A. Hunter, W. L. Storm, P. N. Coneski and M. H. Schoenfisch, Anal. Chem., 2013, 85, 1957–1963 CrossRef CAS PubMed.
- L. K. Keefer, ACS Chem. Biol., 2011, 6, 1147–1155 CrossRef CAS PubMed.
- T.-F. Mah, B. Pitts, B. Pellock, G. C. Walker, P. S. Stewart and G. A. O'Toole, Nature, 2003, 426, 306–310 CrossRef CAS PubMed.
- S. M. Abdelghany, D. J. Quinn, R. J. Ingram, B. F. Gilmore, R. F. Donnelly, C. C. Taggart and C. J. Scott, Int. J. Nanomed., 2012, 7, 4053–4063 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, SEC traces, FTIR, UV-Vis spectra, NO release using amperometric measurement (Fig. S1–S11) are available free of charge via the internet. See DOI: 10.1039/c5sc02769a |
| ‡ Present address: School of Chemistry, University of Sydney, Sydney, NSW 2052, Australia. |
| This journal is © The Royal Society of Chemistry 2016 |