
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface water retardation around single-chain polymeric nanoparticles: critical for catalytic function?†
Patrick J. M.
Stals‡
 abc,
Chi-Yuan
Cheng‡
d,
Lotte
van Beek
ad,
Annelies C.
Wauters
ad,
Anja R. A.
Palmans
ab,
Songi
Han
*de and
E. W.
Meijer
*ab
abc,
Chi-Yuan
Cheng‡
d,
Lotte
van Beek
ad,
Annelies C.
Wauters
ad,
Anja R. A.
Palmans
ab,
Songi
Han
*de and
E. W.
Meijer
*ab
aLaboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P.O. Box 513, NL 5600 MB Eindhoven, The Netherlands. E-mail: e.w.meijer@tue.nl
bInstitute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, NL 5600 MB Eindhoven, The Netherlands
cDSM Coating Resins, Sluisweg 12, 5145 PE Waalwijk, The Netherlands
dDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106, USA. E-mail: songi@chem.ucsb.edu
eMaterials Research Laboratory, University of California, Santa Barbara, California 93106, USA
First published on 8th December 2015
Abstract
A library of water-soluble dynamic single-chain polymeric nanoparticles (SCPN) was prepared using a controlled radical polymerisation technique followed by the introduction of functional groups, including probes at targeted positions. The combined tools of electron paramagnetic resonance (EPR) and Overhauser dynamic nuclear polarization (ODNP) reveal that these SCPNs have structural and surface hydration properties resembling that of enzymes.
Dynamically folded single-chain polymeric nanoparticles (SCPNs) are a captivating class of polymer architectures.1 Intriguingly, these SCPNs – when appropriately functionalised – are effective catalysts in water, mimicking certain aspects of enzymes. In general, SCPNs consist of a conventional polymeric backbone, decorated with pendant functional groups capable of forming either dynamic covalent bonds or supramolecular non-covalent bonds. Upon exposure to a certain trigger in solution – e.g. (UV)-light,2 temperature2e,3 or a solvent-switch,3a – interactions are formed or broken between several pendant groups, leading to intramolecular crosslinked polymeric chains (Fig. 1). Although SCPNs are in general studied in organic solvents,1 several recent studies report on these particles in water.3a,b,4 In a strategy developed by our group, we generate water-soluble SCPNs consisting of a hydrophobic (methacrylate) backbone decorated with hydrophilic oligo(ethylene glycol) side-chains and hydrophobic benzene-1,3,5-tricarboxamides (BTA) pendants. These supramolecular BTA moieties form helical aggregates through strong, three-fold hydrogen bonding between the amides of adjacent BTAs, which serve as intramolecular crosslinks in the folded SCPN structure (Fig. 1).3,5
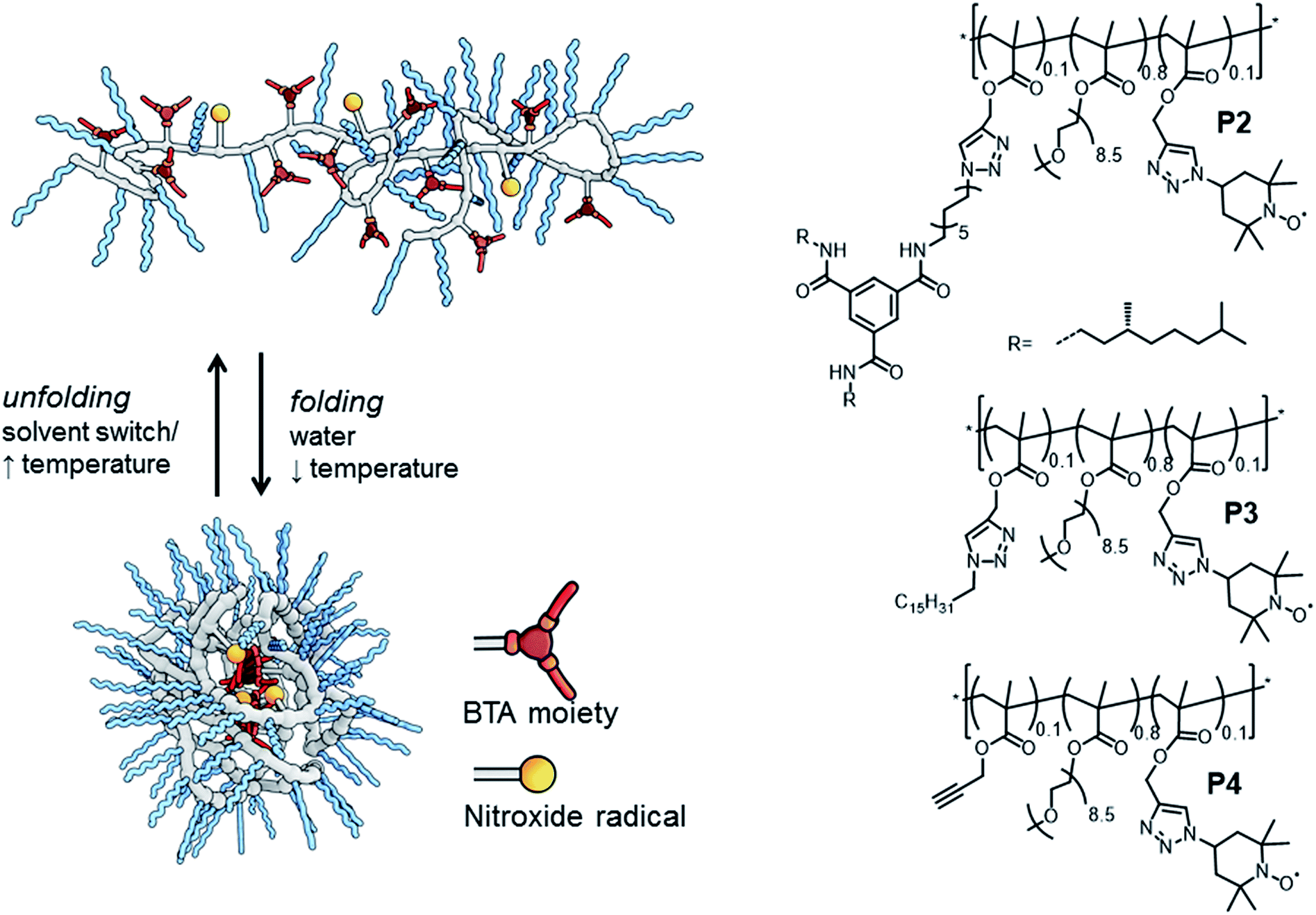 | ||
| Fig. 1 Folding and unfolding of a SCNP and chemical structures of the three polymers studied. | ||
The amphiphilic composition of these water-soluble polymers has been reported to result in the formation of a hydrophobic pocket inside the ellipsoidally shaped SCPNs.3b Crucially, it has been demonstrated that introducing a catalyst results in catalytically active SCPNs, but only after polymer folding in water has occurred. Both ruthenium-catalysed reductions6 and L-proline catalysed aldol reactions7 could be performed efficiently in water. In the latter, we found that there is a large dependence of the polymer architecture on the catalytic properties of the L-proline moieties in the SCPN. Remarkably, only an efficient aldol reaction occurred in polymers in which BTAs are present, while in derivatives where the BTA-moiety is replaced by a dodecyl-group no aldol reaction was observed.7 To further optimise the application of bio-inspired catalysts based on SCPNs, it is important to understand the role of the local polymer dynamics and hydration properties at the boundary between a hydrophilic and hydrophobic environment of SCPNs, where the catalyst centres are located.
An intriguing fundamental question that is asking to be answered is: “what is the molecular basis for the difference in catalytic function of the BTA/L-proline and the dodecyl/L-proline containing SCPNs in water?” More and more evidence is becoming available that the properties of hydration water – both its structure and dynamics – play a dominant role in enzyme catalysis.8 Therefore, we propose to determine whether these distinct properties of water around our catalytic site, or more specifically around the probe mimicking the catalytic site, are correlated with the higher degree of structure formed within the SCPNs. Hence, the questions are: how is the local environment of the catalyst centre at the BTA–PEG interface different (i) when BTA is replaced with a long carbon chain, such as a hexa- or dodecyl chain of a folded SCPN and (ii) when the BTA-based polymer is not folded up to an SCPN? For this, we employ nitroxide radical-based spin labels as analogues to tag the catalytically active L-proline SCPNs. This enables us to explore the local polymer backbone dynamics via tethered spin labels by electron paramagnetic resonance (EPR) line-shape analysis and the local water translational diffusion dynamics within 0.5–1 nm of tethered spin labels by solution-state Overhauser dynamic nuclear polarization (ODNP) NMR relaxometry9 under ambient solution conditions at the site where catalysis would occur.
We evaluate three polymers (Fig. 1), whose design closely resembles that of the earlier published catalytic SCPNs. We replaced the L-proline catalytic site in the SCPNs by the commonly used TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl) spin label for the current study. The polymer side-chains consist of 80% water-soluble oligo(ethylene glycol) moieties, 10% TEMPO moieties, and the remaining 10% of the polymer side-chains are BTAs (P2), hexadecyl chains (P3) or remain unfunctionalised (P4). Of these, only polymer P2 is able to fold into a SCPN with a structured, helical inner compartment. P3 likely forms small micelles due to a hydrophobic–hydrophilic phase separation, while P4 is thought to possess a random-coil polymer architecture.4b To prevent interference from the TEMPO radical during the polymerisation process – TEMPO is widely used as a radical-mediated polymerisation agent10 – we used a post-functionalisation strategy (ESI†) to introduce the functional moieties on the polymeric backbone. This post-functionalisation approach has the added benefit that the same prepolymer is used in all cases, eliminating batch-to-batch differences of polymers as a variable in our experiments. In the ligation strategy, we used the Huisgen 1,3-dipolar cycloaddition reaction since this reaction is known to be highly efficient for polymer analogous reactions.11 Furthermore, it was recently elucidated that the linker does not have a notable effect of the folding behaviour of BTA-containing SCPN.2e
We synthesized one random prepolymer (P1) from oligo(ethylene glycol)methacrylate (oEGMA with Mn = 475 and DP = 8.5–9) and 3-(trimethylsilyl)prop-2-yn-1-yl methacrylate in conventional reversible addition fragmentation chain-transfer (RAFT) polymerisation conditions (ESI†). To the resulting copolymer (Mn,NMR = 112 kDa, DPNMR = 267, Mn,SEC = 24.6 kDa, Đ = 1.90 (SEC in DMF relative to pEO standards)), we coupled the desired azides with an in situ silyl deprotection/click-reaction using sodium ascorbate and CuSO4 as the catalyst. For P2, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of BTA-azide and TEMPO-azide was coupled to the polymeric backbone. For P3, we coupled a 1:1 mixture of hexadecylazide and TEMPO-azide to the polymeric backbone, while for P4 we added an adequate amount of TEMPO-azide to functionalise 50% of the alkyne moieties on the backbone, leaving 50% as unreacted alkynes.
:1 mixture of BTA-azide and TEMPO-azide was coupled to the polymeric backbone. For P3, we coupled a 1:1 mixture of hexadecylazide and TEMPO-azide to the polymeric backbone, while for P4 we added an adequate amount of TEMPO-azide to functionalise 50% of the alkyne moieties on the backbone, leaving 50% as unreacted alkynes.
The successful ligation of our functional moieties to the polymeric backbone was confirmed by a combination of techniques. With size exclusion chromatography (SEC), we showed an increase in molecular weight after ligation for all polymers, while IR (ESI†) clearly showed a vibration at 1350 cm−1, which is indicative for a nitroxide vibration.12 Peaks at 1640 cm−1 and 1450 cm−1 are indicative for triazole ring vibrations.13 Also, in P2, peaks at 1660 cm−1 and 1540 cm−1 are indicative for the carbonyl vibration and amide II vibration originating from the presence of the amides of benzene-1,3,5-tricarboxamides.14 Lastly, EPR analysis of the polymers after dialysis to remove free spin labels showed a comparable degree of functionalisation with TEMPO for all polymers (vide supra). With circular dichroism (CD) spectroscopy, we evaluated the incorporation of the BTA moieties, by comparing a solution of P2 (50 μM of BTAs based on theoretical incorporation of BTAs) with literature data for similar BTA containing SCPN at that concentration (P5, ESI†).3b The CD curves of P2 (with TEMPO) and P5 (without TEMPO) remain unaltered, indicating that (i) the TEMPO moiety is not interfering with the self-assembly of the BTAs into its helical superstructure and (ii) the build-in of BTAs in the polymer was indeed roughly 10%.
Dynamic light scattering (DLS) studies were used to verify the size of the architectures of P2–P4 in water. At a polymer concentration of 540 μM, the polymers showed a hydrodynamic diameter (Dh) of 23 ± 2 nm (P2) and 19 ± 2 nm (P3), consistent with sizes that were obtained for similar systems and indicating that single-chain aggregates are formed.3b Interestingly, polymer P4, which was expected to be ‘unfolded’ in water, did not show any scattering density, likely due to its excellent water solubility. P4 thus lacks any particle character and has no significant density difference with the surrounding water.
The local backbone dynamics around the TEMPO spin-probe in P2–P4 were studied by EPR, a well-known method to measure the local conformational freedom in synthetic systems.15 The EPR line-shapes of all three polymers in water were equal (ESI†), implying that the spin labels experience comparable mobility whether it comprises an unfolded polymer chain or a folded SCPN in water. We also found that the EPR line-shapes of these three polymers are the same upon the addition of the viscogen sucrose at 30 wt%, suggesting that the EPR line-shape is not dominated by the global tumbling of the polymer, and rather that the local spin-label mobility remains genuinely unhindered upon polymer folding. The rotational correlation time of the spin-labelled side-chain of the three polymers is ∼2.5 ns, a value that is consistent with that on the solvent-exposed surface of a globular protein.16 Critically, the spin labels of folded P2 and P3 are unhindered in their rotational motion and are solvent-exposed.
Next, we probed the local hydration dynamics near the TEMPO spin label of SCNPs in water, i.e. near the active site using ODNP. ODNP selectively amplifies the 1H-NMR signal of local hydration water within 0.5–1 nm of a specific spin label tethered near the molecular site of interest, by transferring polarization from electron to nearby water and relying on electron–nuclear dipolar relaxation, whose efficiency intimately depends on the translational diffusivity of local water at a 0.35 Tesla field.9 A detailed description of the ODNP measurements and analysis can be found in the ESI.† We derive the translational diffusion correlation time of water near the polymer-tethered spin label (ESI†), and display it as a retardation factor relative to that of bulk solvent, namely as τpolymer/τsolvent in Fig. 2a. We find a retardation factor of approximately 5 and 4 near the spin label of BTA-comprising P2 and hexadecyl-comprising P3, respectively. These values are on par with the 4–8-fold retarded translational water diffusion found on the surface of globular proteins,9a,16 which is in contrast to the typically smaller retardation factor of 2–3 found on unfolded proteins or polymers in solution.9b,16–18 The question is, what is the basis for and role of this strongly retarded water diffusion around the surface of P2 compared to P3, and yet smaller values found on P4? For unfolded P4, a retardation factor of ∼3 was found, which is consistent with that of an intrinsically disordered protein.17 Therefore, it seems evident that the stronger retardation of surface water observed on P2 is due to a more ordered packing of the folded core by BTA self-assembly in water. This ordered packing consequently displays a more ordered polymer surface and solvation structure.
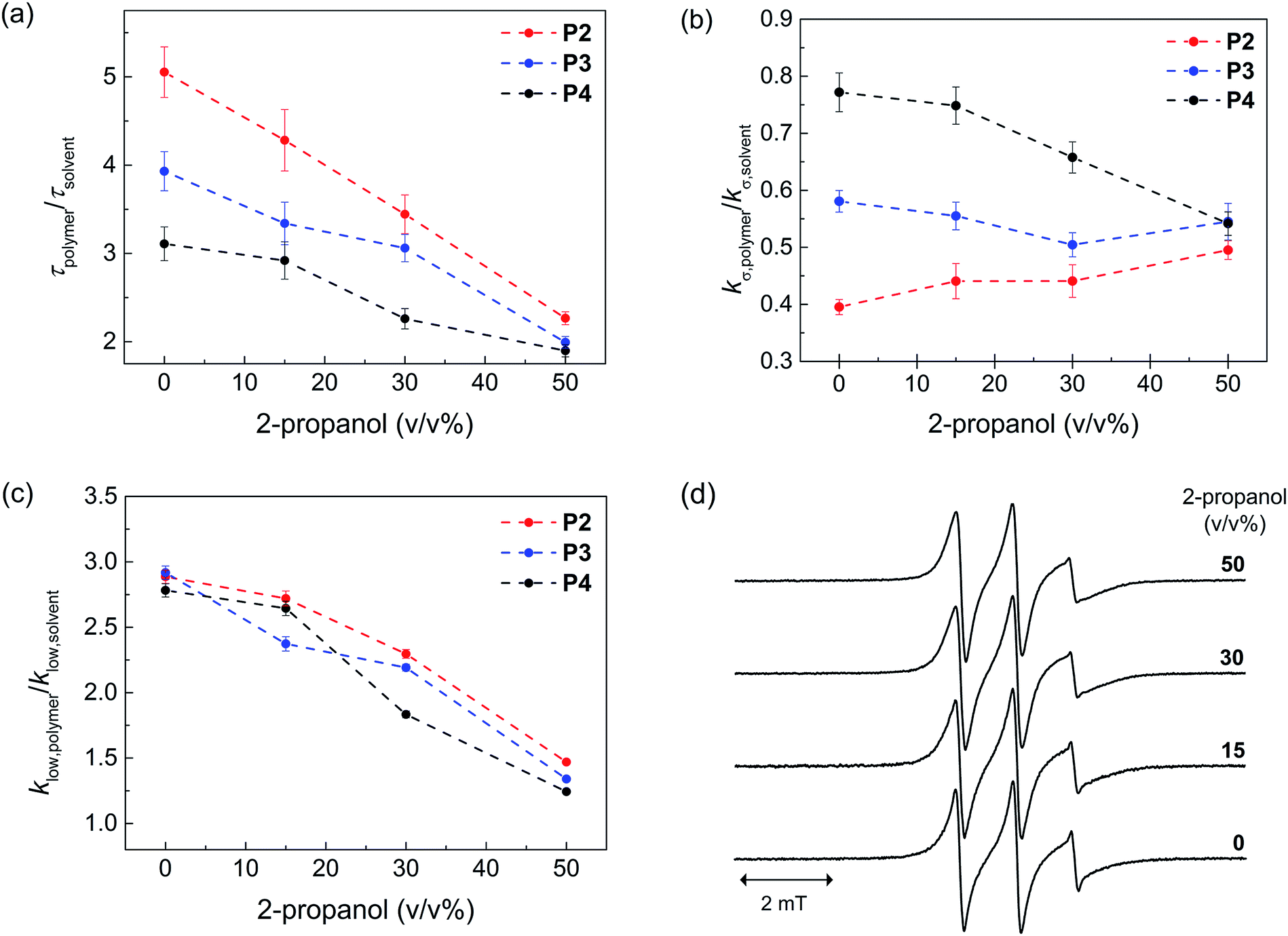 | ||
| Fig. 2 Local hydration dynamics of SCNPs by ODNP evaluated using the (a) translational correlation time, τpolymer (b) cross relaxivity kσ,polymer, (c) slow-motion component of the self-relaxivity klow,polymer. These values are compared with respect to those derived from the corresponding solvent condition (τsolvent, kσ,solvent, klow,solvent), in order to take into account the solvent viscosity. (d) EPR spectra of P2 at various 2-propanol concentrations. The τpolymer value represents the translational correlation time of water molecules within 5–10 Å of the nitroxide radical-based spin label tethered on the polymer surface. The value is inversely proportional to the local diffusion coefficient of water if the distances of closest approach between the spin label and water remains constant. kσ,polymer reflects on the contribution of freely diffusing, loosely bound water at picosecond to sub-nanosecond timescale on the polymer surface. klow,polymer reflects on the contribution of slow or bound water at nanosecond timescale on the polymer surface. | ||
In order to test this hypothesis, we measured the local spin label and solvation dynamics as a function of systematic unfolding of the polymers. Organic co-solvents, such as 2-propanol, have been shown to disturb any folded structure of SCPN by breaking apart the self-assembly of the BTA units.3a Here, we studied the hydration dynamics of P2–P4 SCNPs as a function of increasing 2-propanol concentration. The data (Fig. 2a) clearly illustrates that the retardation factor of local water diffusion gradually decreases with increasing 2-propanol concentration. Gratifyingly, the retardation factors of the three polymers merge to the same values of 2–2.5 at a 50 v/v% concentration of 2-propanol when the polymers are unfolded.
In order to understand the difference in the retardation factor between the folded polymers, we need to examine the role of surface water dynamics at different timescales. Either an increase in surface water diffusivity (fast, tens of picoseconds) or a decrease in bound water (slow, nanoseconds) population can result in the decrease in the retardation factor. Thus, we separated the contribution of water into a different motional timescale of local water dynamics around the surface of SCPNs by means of 1H NMR relaxivity at different frequencies as derived from ODNP data.9b,19 Here, the kσ value reports on 1H NMR relaxivity at ∼10 GHz, modulated by fast diffusive water on the picosecond timescale, where the higher the value the faster the water diffusivity and vice versa. In contrast, klow reports on 1H-NMR relaxivity at ∼15 MHz, modulated by bound water moving on the order of the nanosecond timescale, where the higher the value the stronger the bound water contribution compared to the bulk solvent (ESI†). Crucially, we find that the difference between P2, P3 and P4 lies in the trend in kσ,polymer/kσ,solvent (Fig. 2b) rather than in klow,polymer/klow,solvent (Fig. 2c). Hence, the difference in the retardation factor for the diffusion correlation time of water for the three polymers (Fig. 2a) is the result of the differences in the contribution of the fast diffusive dynamics of hydration water. Fig. 2b illustrates that kσ,polymer is lowest in P2 folded in water, hence it is clear that indeed the solvent water is most retarded at the active site of P2 – the catalyst model – folded in water.
As expected, the ratio klow,polymer/klow,solvent, which represents the contribution from the slow timescale of bound water, decreases with increasing 2-propanol concentration and eventually reaches approximately 1 for all polymers (Fig. 2c). Interestingly, there is no difference between the three polymers. This result demonstrates that 2-propanol successively dehydrates the polymer surface, while no bound hydration water remains at the SCNP surfaces in 50% 2-propanol. Finally, the EPR lineshapes (Fig. 2d) do not show any measurable changes at various 2-propanol concentrations, suggesting that differences in polymer mobility or packing, per se, is not the deciding factor for mimicking an enzyme surface.
Taken together, the local surface water diffusion at the interface between the hydrophobic pocket and hydrophilic PEG, i.e. the location of the probe – being the model for the L-proline catalyst7 – within the SCPN, is most strongly retarded in P2 when folded in water. At the same time, there is significant contribution of bound water on the surfaces of P2–P4 in water, whereas the spin label mobility remains high and unaltered. Retarded surface water diffusivity and the bound water population on solvent-exposed macromolecular surfaces are hallmarks of a folded protein surface.16–19 This study finds that surface water diffusivity at the surface of SCPNs is the only experimental signature that reliably follows the structural transition from intramolecular structural/folded to unfolded SCNPs and differentiates between the surfaces of a structured (P2) versus non-structured (P3) SCPN. Thus, hydration retardation is the only clearly different physical parameter identified between the polymer structures that have dramatically different catalytic activities. Whether the signature of a retarded hydration shell is canonically critical to catalysis, and if so why, still remains to be answered with future studies.
Acknowledgements
P. J. M. S., A. R. A. P. and E. W. M. acknowledge financial support from the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035) and the European Research Council (FP7/2007-2013, ERC Grant Agreement 246829). C.Y.C. and S.H. acknowledge financial support from the 2011 NIH Director New Innovator Award. This work made use of Materials Research Laboratory Central Facilities supported by NSF through MRSEC (DMR 111053). The MRL is a member of the NSF-funded Material Research Facilities Network (http://www.mrfn.org). This work was funded by the UCSB-CISEI program through the NSF Research Experiences for Undergraduates (NSF DMR 0843934) to L. V. B. and A. C. W. A. J. H. Spiering is acknowledged for synthetic support. The ICMS Animation Studio (Eindhoven University of Technology) is acknowledged for providing the artwork.Notes and references
-
(a) C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181–197 RSC
; (b) M. Artar, E. Huerta, E. W. Meijer and A. R. A. Palmans, Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties, ACS Symposium Series, ed. J.-F. Lutz, T. Y. Meyer, M. Ouchi and M. Sawamoto, American Chemical Society, Washington, DC, 2014, vol. 1170, ch. 21, pp. 313–325 Search PubMed
-
(a) E. J. Foster, E. B. Berda and E. W. Meijer, J. Am. Chem. Soc., 2009, 131, 6964–6966 CrossRef CAS PubMed
-
(a) M. A. J. Gillissen, T. Terashima, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Macromolecules, 2013, 46, 4120–4125 CrossRef CAS
-
(a) E. A. Appel, J. Dyson, J. del Barrio, Z. Walsh and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 4185–4189 CrossRef CAS PubMed
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC
-
(a) M. Artar, T. Terashima, M. Sawamoto, E. W. Meijer and A. R. A. Palmans, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 12–20 CrossRef CAS
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2013, 52, 2906–2910 CrossRef CAS PubMed
-
(a) P. W. Snyder, J. Mecinović, D. T. Moustakasa, S. W. Thomas III, M. Harder, E. T. Mack, M. R. Lockett, A. Héroux, W. Sherman and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17889–17894 CrossRef CAS PubMed
-
(a) B. D. Armstrong and S. Han, J. Am. Chem. Soc., 2009, 131, 4641–4647 CrossRef CAS PubMed
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed
- L. Rintoul, A. S. Micallef and S. E. Bottle, Spectrochim. Acta, Part A, 2008, 70, 713–717 CrossRef CAS PubMed
- H. Li, Q. Zheng and C. Han, Analyst, 2010, 135, 1360–1364 RSC
- P. J. M. Stals, J. F. Haveman, R. Martín-Rapún, C. F. C. Fitié, A. R. A. Palmans and E. W. Meijer, J. Mater. Chem., 2009, 19, 124–130 RSC
-
(a) D. Hinderberger, H. W. Spiess and G. Jeschke, J. Phys. Chem. B, 2004, 108, 3698–3704 CrossRef CAS
- B. D. Armstrong, J. Choi, C. Lopez, D. A. Wesener, W. Hubbell, S. Cavagnero and S. Han, J. Am. Chem. Soc., 2011, 133, 5987–5995 CrossRef CAS PubMed
- C.-Y. Cheng, J. Varkey, M. R. Ambroso, R. Langen and S. Han, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16838–16843 CrossRef CAS PubMed
- A. Pavlova, E. R. McCarney, D. W. Peterson, F. W. Dahlquist, J. Lew and S. Han, Phys. Chem. Chem. Phys., 2009, 11, 6833–6839 RSC
- S. Hussain, J. M. Franck and S. Han, Angew. Chem., Int. Ed., 2013, 52, 1953–1958 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, CD- and IR spectra, additional information on ODNP and EPR spectra. See DOI: 10.1039/c5sc02319j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |