
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cooperative polymeric platform for tumor-targeted drug delivery†
Wantong
Song
a,
Zhaohui
Tang
*a,
Dawei
Zhang
a,
Mingqiang
Li
a,
Jingkai
Gu
b and
Xuesi
Chen
*a
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, P. R. China. E-mail: ztang@ciac.ac.cn; xschen@ciac.ac.cn
bResearch Center for Drug Metabolism, College of Life Science, Jilin University, Changchun, 130012, P. R. China
First published on 26th October 2015
Abstract
In the pursuit of effective treatments for cancer, an emerging strategy is “active targeting”, where nanoparticles are decorated with targeting ligands able to recognize and bind specific receptors overexpressed by tumor cells or tumor vasculature so that a greater fraction of the administered drugs are selectively trafficked to tumor sites. However, the implementation of this strategy has faced a major obstacle. The interpatient, inter- and intra-tumoral heterogeneity in receptor expression can pose challenges for the design of clinical trials and result in the paucity of targetable receptors within a tumor, which limits the effectiveness of “active targeting” strategy in cancer treatment. Here we report a cooperative drug delivery platform that overcomes the heterogeneity barrier unique to solid tumors. The cooperative platform comprises a coagulation-inducing agent and coagulation-targeted polymeric nanoparticles. As a typical small-molecule vascular disrupting agent (VDA), DMXAA can create a unique artificial coagulation environment with additional binding sites in a solid tumor by locally activating a coagulation cascade. Coagulation-targeted cisplatin-loaded nanoparticles, which are surface-decorated with a substrate of activated blood coagulation factor XIII, can selectively accumulate in the solid tumor by homing to the VDA-induced artificial coagulation environment through transglutamination. In vivo studies show that the cooperative tumor-selective platform recruits up to 7.5-fold increases in therapeutic cargos to the tumors and decreases tumor burden with low systemic toxicity as compared with non-cooperative controls. These indicate that the use of coagulation-targeted nanoparticles, in conjunction with free small-molecule VDAs, may be a valuable strategy for improving standard chemotherapy.
Introduction
A key challenge of current chemotherapies is the enhancement of tumor targetability of drugs.1–4 Most chemotherapeutic agents attack both tumor and normal tissue, leading to limited therapeutic efficacy because of their severe side effects. Therefore, the development of highly effective delivery platforms that can selectively deliver chemotherapeutic agents to tumors while leaving healthy organs unharmed represents a key goal in modern drug delivery systems for cancer therapy.5–8“Active targeting” is an elegant concept for tumor-targeted drug delivery in cancer therapy.9,10 “Active targeting” drug delivery systems, utilizing targeting ligands such as sugars,11,12 lectins,13 vitamins,14–16 peptides,17–20 antibodies21,22 and aptamers,23–26 have attracted widespread attention. However, there are concerns about the implementation of current “active targeting” strategy.27–29 The expression of receptors within a tumor is not homogenous and is variable in density or structure over time.30 The type and level of expressed receptors within a tumor vary with patient, tumor type, subtype and stage.31–35 A great deal of interpatient, inter- and intra-tumor heterogeneity36–39 can pose challenges for the design of clinical trials and result in the paucity of targetable receptors available for nanoparticle binding within a tumor, which limits the application of “active targeting” nanomedicines.
Creation of a unique artificial environment with additional binding sites within a tumor by administering selective exogenous materials may overcome the heterogeneity barrier for the “active targeting” delivery of chemotherapeutic agents.40 Previously, SBY1 bacteria have been utilized to create a bacteria-accumulated tumor artificial environment for differential anticancer drug delivery, but this design may be limited by the potential immune response to the bacteria.41 Similarly, the coagulation cascade in tumors, activated by photothermal treatment or tissue factor proteins, has been exploited to amplify the delivery of diagnostic or therapeutic cargos to tumors. Very encouraging results have been obtained, but this approach is still limited by shallow tissue penetration for light or a potential immune response to the administered proteins.42 Alternatively, the small-molecule vascular disrupting agent (VDA), ombrabulin, can selectively target tumors, leading to a locally elevated presentation of protein p32. This strategy successfully amplified nanoparticle recruitment in vivo, suggesting that creation of a unique artificial environment in tumors may represent a significant advantage for targeted anticancer drug delivery.43 Specifically, small-molecule VDAs have been extensively studied to induce tumor collapse by selectively destroying existing tumor vessels, which is accompanied by hemorrhaging and coagulation within the tumor.44–49 However, to the best of our knowledge, small-molecule VDA-induced coagulation has never been used to target nanoparticles to tumors.
DMXAA (5,6-dimethylxanthenone-4-acetic acid, ASA404, Vadimezan), a small-molecule VDA currently in clinical trial,50 targets tumor vascular endothelium and activates tumor-associated macrophages to release high local levels of tumor necrosis factor-α (TNF-α). Release of the TNF-α disrupts established tumor blood vessels and results in the direct exposure of abnormal basement membrane to flowing blood, which activates the platelets and in turn induces the coagulation cascade.51–54 The effect of DMXAA is selective for tumor blood vessels,55–57 partly due to differences between tumor and normal tissue vasculatures58 and the pro-coagulant status of cancers.59 Thus, the administration of DMXAA creates a unique artificial coagulation environment in tumors, providing additional binding sites for tumor-targeted drug delivery.
The GNQEQVSPLTLLKXC peptide (A15) has been previously identified as a substrate of activated blood coagulation factor XIII (FXIIIa, a transglutaminase). A15 can specifically target coagulation enzyme activity by participating in the FXIIIa catalyzed fibrin cross-linking reaction and be covalently bound to fibrin clots through transglutamination.42 Because FXIIIa plays a key role in the final stages of blood coagulation,60 the specificity of A15 can be utilized to home drug-loaded nanoparticles to DMXAA-treated tumors with a unique coagulation environment.
In this report, we describe a novel cooperative tumor-targeted drug delivery platform that includes two main components (Fig. 1): (i) DMXAA (a coagulation-inducing agent), and (ii) A15 peptide-decorated poly(L-glutamic acid)-cisplatin conjugates (A15-PGA-CisPt, coagulation-targeted nanoparticles). By intravenous administration of DMXAA into tumor-bearing mice, the DMXAA specifically induces an artificial coagulation environment in tumors. Then, circulating A15-PGA-CisPt are selectively targeted to the coagulation site of DMXAA-treated tumors by covalently binding to fibrin clots through FXIIIa catalyzed transglutamination, leading to the selective eradication of tumor cells.
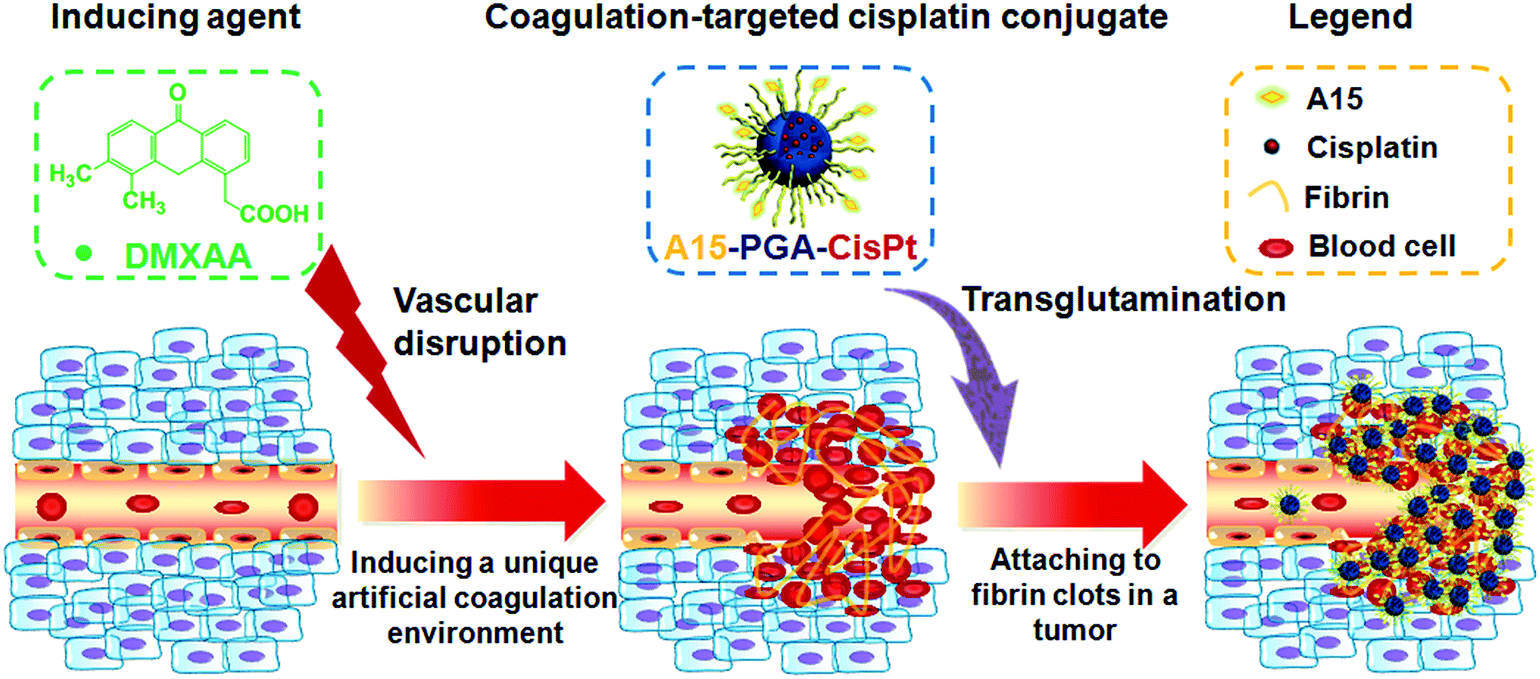 | ||
| Fig. 1 Illustration of a cooperative tumor-targeted drug delivery platform. DMXAA selectively disrupts the tumor vasculature and creates a unique artificial coagulation environment in a tumor. A15-PGA-CisPt are selectively recruited to the tumor by binding to fibrin clots via transglutamination. | ||
Results and discussion
Effect of DMXAA on the tumor environment
DMXAA is an analogue of flavone acetic acid and found to simultaneously target vascular endothelial cells and macrophages within the tumor microenvironment. This, in turn, induces an increase in the tumor concentration of TNF-α, leading to extensive hemorrhaging within treated tumors (Fig. 2a and S1†).61 Many hemorrhage sites in C26 tumors were observed 4 h following a single i.v. injection of DMXAA at 15.0 mg kg−1 (Fig. 2b), demonstrating the extent of coagulation that occurs following treatment. The induced hemorrhaging activates an extensive coagulation cascade within the tumors including: (i) cleavage of prothrombin to form thrombin, (ii) conversion of fibrinogen to fibrin and transglutaminase coagulation factor XIII to FXIIIa by thrombin and (iii) catalyzation of the cross-linking reaction between fibrins by FXIIIa, which leads to the formation of a stable fibrin-clot (Fig. 3a).62,63 To confirm these reactions, fluorescein isothiocyanate-labelled fibrinogen (fibrinogen-FITC) was co-injected with different doses of DMXAA into mice bearing C26 tumors. Abundant localization of fibrin (green) within tumors was observed 4 h following injection of DMXAA at 15.0 mg kg−1 (Fig. 2c and S2†), while no obvious hemorrhage or fibrin localization occurred in the vital organs at the same condition (Fig. S3†). These results further confirm that the administration of DMXAA will specifically induce an artificial coagulation environment in tumors.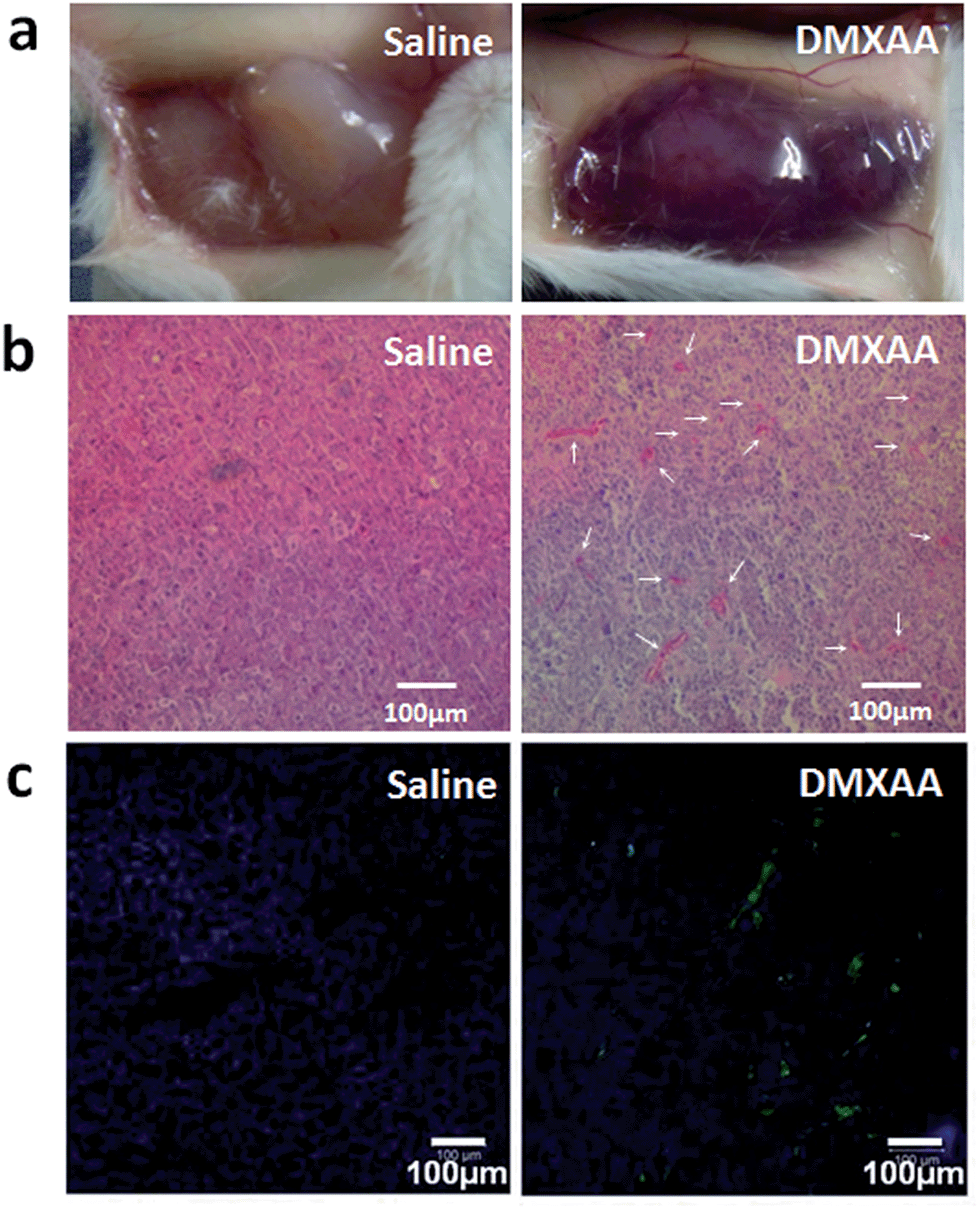 | ||
| Fig. 2 DMXAA induced C26 tumor environment changes at 4 h post injection (DMXAA dosage: 15.0 mg kg−1). (a) Photographs of tumors at 4 h after saline or DMXAA injection. (b) H&E staining of tumors harvested at 4 h following injection of saline or DMXAA. The white arrows in the right picture indicate the hemorrhage sites. (c) Histopathological analysis of fibrin localization in tumors at 4 h following injection of fibrinogen-FITC with saline or DMXAA. | ||
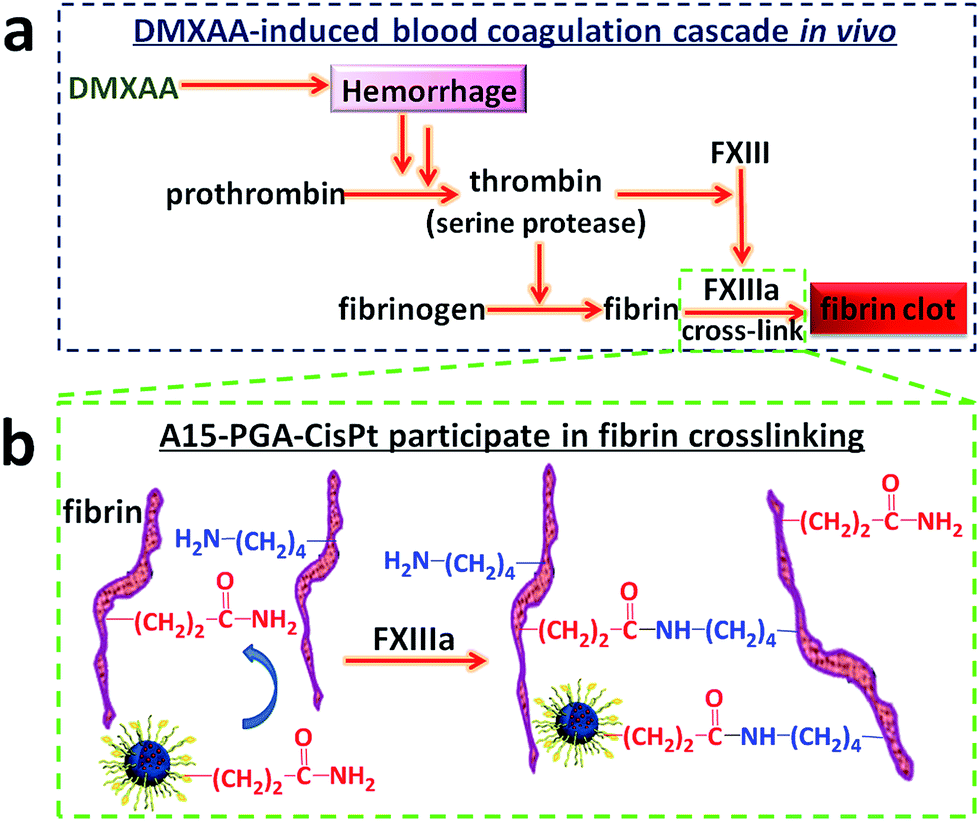 | ||
| Fig. 3 (a) Mechanism of DMXAA-induced coagulation cascade in tumors. (b) Mechanism of A15-PGA-CisPt participation in fibrin cross-linking reaction. | ||
Preparation of coagulation-targeted polymeric nanoparticles
Poly(L-glutamic acid)-g-poly(ethylene glycol), which has been optimized by our group previously for cisplatin loading, was synthesized here with slight modification.64,65 PGA-g-mPEG/MAL-PEG, was prepared by Steglich esterification between poly(L-glutamic acid) (PGA), mPEG-OH and MAL-PEG-OH (Scheme 1). The 1H NMR of PGA-g-mPEG/MAL-PEG was shown in Fig. S4.† The ratio of PGA/PEG was calculated based on the intensities ratio of signals at 1.96 and 1.83 ppm (>CHCH2–, d) and 3.63 ppm (–CH2CH2O–, f). The resultant molar ratio of glutamate unit/PEG monomer unit (1.0/5.8) was close to the feed ratio (1.0/6.0). The resultant mPEG/MAL-PEG ratio was 4.2/1.0, calculated according to the intensities ratio of peaks at 3.31 ppm (–OCH3, g) and 7.26 ppm (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CO–, h), which was consistent with the feed ratio (4.0/1.0). These indicated the esterification reaction was highly efficient. The GPC curves of mPEG-OH, MAL-PEG-OH, PGA and the obtained PGA-g-mPEG/MAL-PEG were shown in Fig. S5.† The polymers all exhibited unimodal peaks, and the peaks of PGA, mPEG-OH and MAL-PEG-OH did not appear in the GPC spectrum of the PGA-g-mPEG/MAL-PEG copolymer. The number-average molecular weight (Mn) of PGA-g-mPEG/MAL-PEG was 68.3 × 103 Da, with PDI = 1.28. These results demonstrated that the PGA-g-mPEG/MAL-PEG copolymer was obtained as desired.
CH–CO–, h), which was consistent with the feed ratio (4.0/1.0). These indicated the esterification reaction was highly efficient. The GPC curves of mPEG-OH, MAL-PEG-OH, PGA and the obtained PGA-g-mPEG/MAL-PEG were shown in Fig. S5.† The polymers all exhibited unimodal peaks, and the peaks of PGA, mPEG-OH and MAL-PEG-OH did not appear in the GPC spectrum of the PGA-g-mPEG/MAL-PEG copolymer. The number-average molecular weight (Mn) of PGA-g-mPEG/MAL-PEG was 68.3 × 103 Da, with PDI = 1.28. These results demonstrated that the PGA-g-mPEG/MAL-PEG copolymer was obtained as desired.
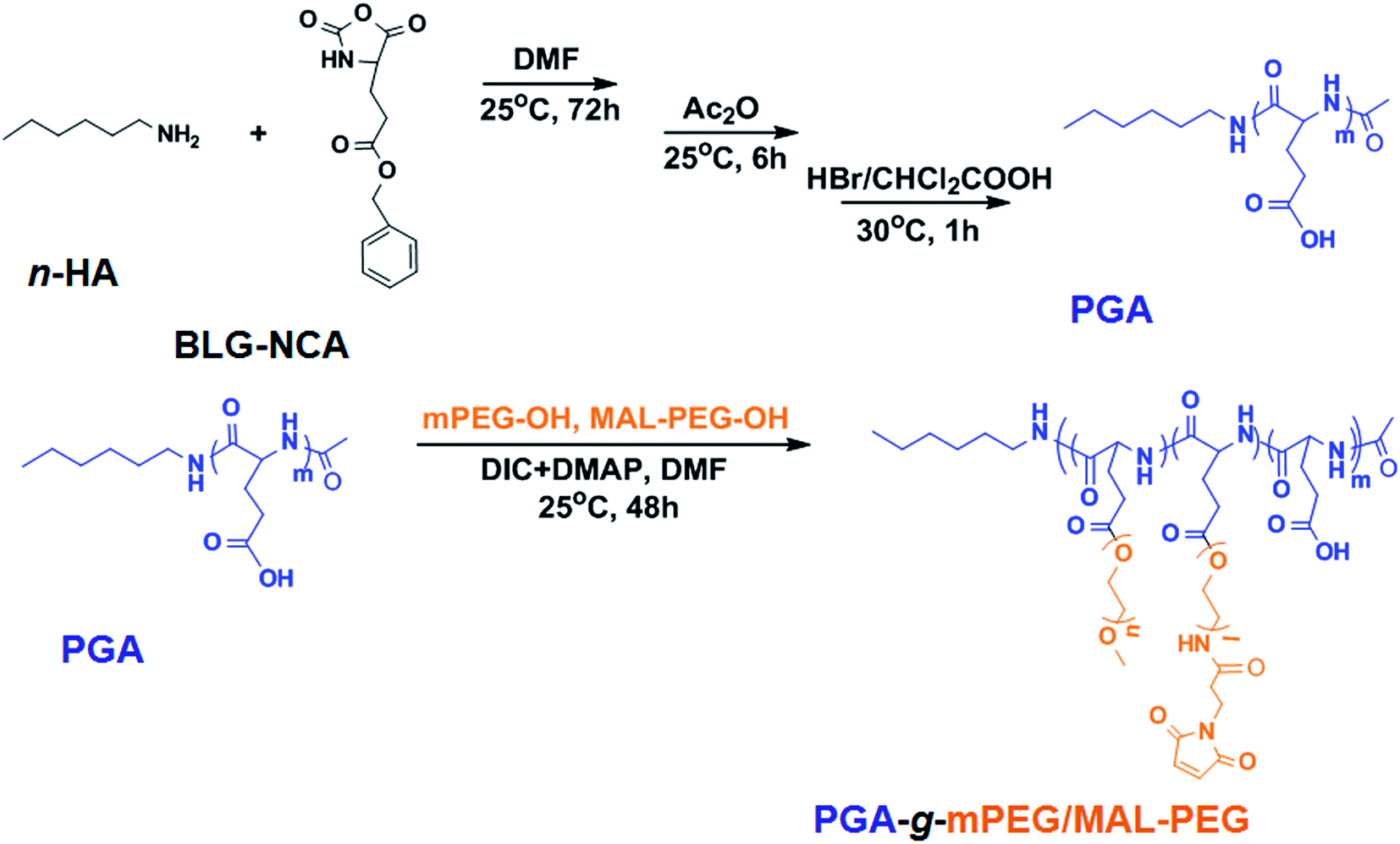 | ||
| Scheme 1 Preparation of PGA-g-mPEG/MAL-PEG. | ||
A15-decorated poly(L-glutamic acid)-cisplatin conjugates (A15-PGA-CisPt) were prepared by complexation of cisplatin with PGA-g-mPEG/MAL-PEG and subsequently decorating the surface of the cisplatin conjugates (PGA-CisPt) with the A15 peptide (Scheme 2). The obtained A15-PGA-CisPt are uniform spheres, with size of 42.7 ± 5.1 nm at dried condition and 63.5 ± 12.0 nm in aqueous solution (Fig. S6†). The surface zeta potential of the A15-PGA-CisPt is −9.8 ± 1.9 mV and the total cisplatin loading content is 15.3 wt% (Table S1†). The Mn of the A15-PGA-CisPt conjugates is 21.2 × 104 Da, with Mw/Mn of 1.48, as measured by GPC. The surface peptide of the A15-PGA-CisPt was characterized by X-ray photoelectron spectroscopy; C1s, N1s, O1s, S2p and Pt4f peaks of PGA-CisPt and A15-PGA-CisPt were taken, with S2p indicating the presence of the peptide GNQEQVSPLTLLKXC on the A15-PGA-CisPt (Fig. S7†). A15 has been shown to covalently crosslink to fibrin in clots catalyzed by FXIIIa in a highly specific manner.42,66 Therefore, we speculated that A15-PGA-CisPt could also bind to fibrin-clots through FXIIIa catalyzed transglutamination (Fig. 3b).
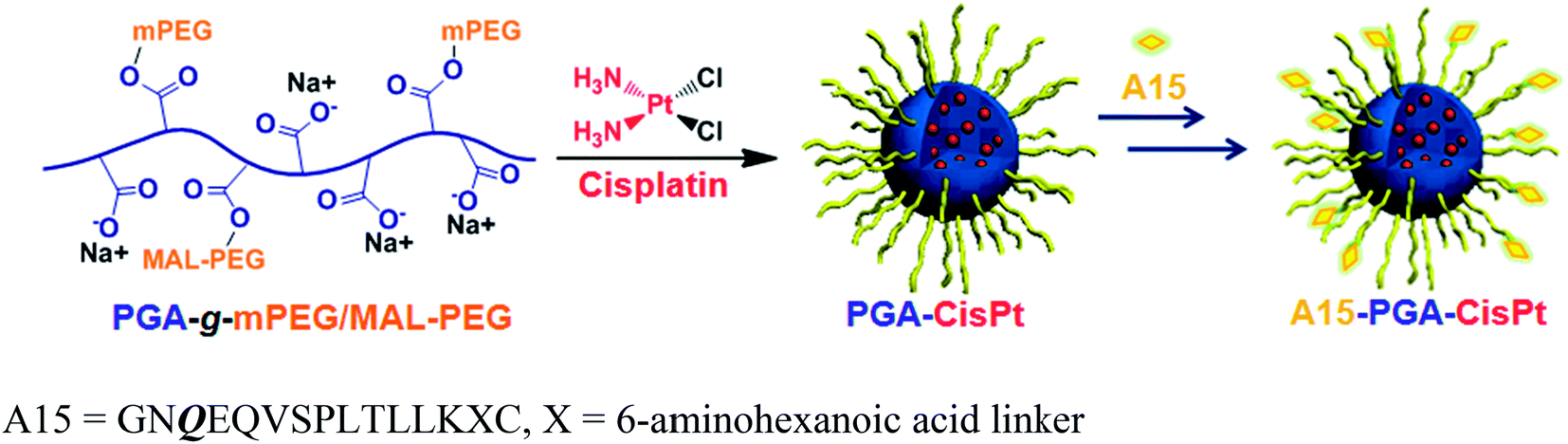 | ||
| Scheme 2 Preparation of coagulation-targeted poly(L-glutamic acid)-cisplatin conjugates (A15-PGA-CisPt). | ||
Solution behavior of A15-PGA-CisPt, in vitro cytotoxicity and pharmacokinetics
Similar to our previous report,67 cisplatin could be released from the A15-PGA-CisPt conjugates in the presence of saline, which was attributed to the exchange between chloride ions and the carboxyl groups (Fig. S8†). Besides, cathepsin B would increase the release rate due to the degradation of the polymer. pH does not have much effect on the release rate, as cisplatin was constantly released at similar rate at pH 7.4 and 5.5. The release rate affected the in vitro cytotoxicity of cisplatin. To both C26 tumor cells and HUVEC endothelial cells, A15-PGA-CisPt showed higher IC50 values than that of free cisplatin (Fig. S9, Table S2†). HUVEC cells were more resistant to A15-PGA-CisPt than C26 cells, with higher IC50 values and less cellular endocytosis after incubation with RhoB-labeled A15-PGA-CisPt (Fig. S10†), which suggested the selective toxicity of these conjugates to tumor cells.Before in vivo injection, it's important to evaluate the stability of the conjugates, especially in plasma. Here, the A15-PGA-CisPt conjugates were cultured in plasma for 30 min, 1 h, 3 h, 6 h and 24 h, and then separated by centrifugation and re-dispersed in water. The sizes of the separated conjugates were measured by DLS and the surface absorbed plasma proteins were measured by SDS-PAGE, following a method reported by D. Docter and co-workers.68 As shown in Fig. S11†, no obvious protein absorption was observed on the A15-PGA-CisPt conjugates until 24 h. Similar, the sizes did not change much during the observation period. These results confirmed the A15-PGA-CisPt conjugates can keep stable after getting contact with plasma and keep resistant to protein absorption due to the dense hydrophilic shell.
Then the pharmacokinetics of A15-PGA-CisPt was carried out on Wistar rats. After injection at a cisplatin dosage of 4.0 mg kg−1, A15-PGA-CisPt could keep high concentration in blood for quite a long time: the residue Pt concentration at 24 h was still 31.9% that of the maximum concentration at 0.05 h (Fig. S12, Table S3†). The terminal half-life (T1/2z) of cisplatin in A15-PGA-CisPt is 21.9 ± 1.4 h, and area under curve from 0 to 24 h (AUC0–t) 1125.5 ± 83.1 mg L−1 h−1, much higher than that of free cisplatin.69 The superior circulation ability guaranteed the A15-PGA-CisPt conjugates to accumulate and target to the coagulation sites in the DMXAA treated tumors.
Tumor-targeting ability of A15-NPs
To evaluate the targeting ability of A15-PGA-CisPt to the DMXAA-induced artificial coagulation environment in tumors, we labeled the A15-PGA-CisPt with RhoB (Scheme S1, Table S1†), and co-injected them with fibrinogen-FITC and DMXAA (15.0 mg kg−1) into Balb/C mice bearing C26 tumors. After 24 h, ex vivo fluorescent imaging of tumors revealed that the RhoB-labeled A15-PGA-CisPt (red) were primarily located around the coagulation regions (green) (Fig. 4). These data suggest that the peptide A15 directed the cisplatin conjugates to the coagulation sites induced by DMXAA inside tumors.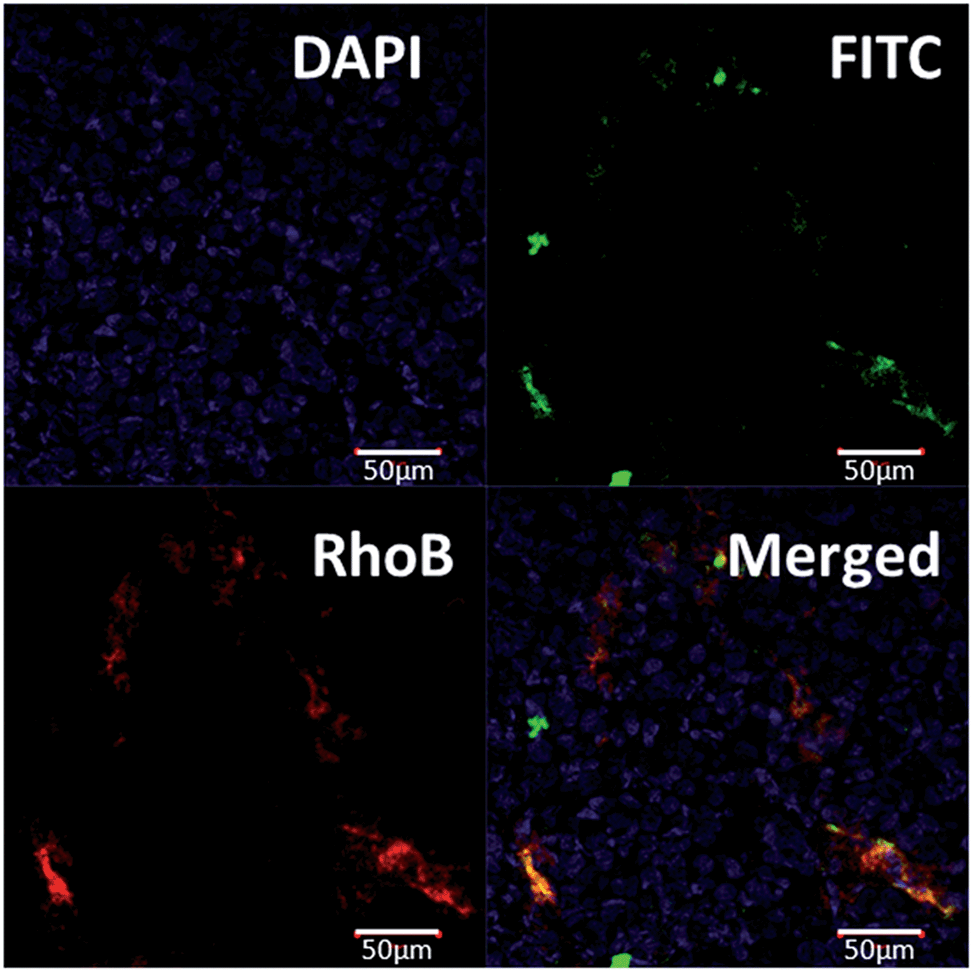 | ||
| Fig. 4 Histopathological analysis of the C26 tumor at 24 h following injection with DMXAA (dosage: 15.0 mg kg−1), fibrinogen-FITC and RhoB-labeled A15-PGA-CisPt. RhoB-labeled A15-PGA-CisPt (red) were primarily located around the coagulation regions (green), which suggests that peptide A15 directed the conjugates to the DMXAA-induced coagulation sites within the tumors. | ||
We subsequently labeled the A15-PGA-CisPt with IR830 (Scheme S2, Table S1†), a near-infrared probe with maximum absorbance at 817 nm in water (Fig. S13†), and used multispectral optoacoustic tomography (MSOT) in order to view the entire tumor. IR830-labeled A15-PGA-CisPt were injected into Balb/C nude mice bearing subcutaneous C26 tumors with or without injections of DMXAA (15.0 mg kg−1). After 24 h, images of the abdominal area were taken on a MSOT small animal scanner. The multispectrally processed images obtained from MSOT demonstrate the status of the exogenous absorbers, which indicates the location and amount of the IR830-labeled A15-PGA-CisPt.70 As shown in Fig. 5a, co-administration of DMXAA with the IR830-labeled A15-PGA-CisPt resulted in a much stronger signal of IR830 in the wider regions of the C26 tumors. This indicated that exposure to DMXAA improved the homing of IR830-labeled A15-PGA-CisPt to the C26 tumor site.
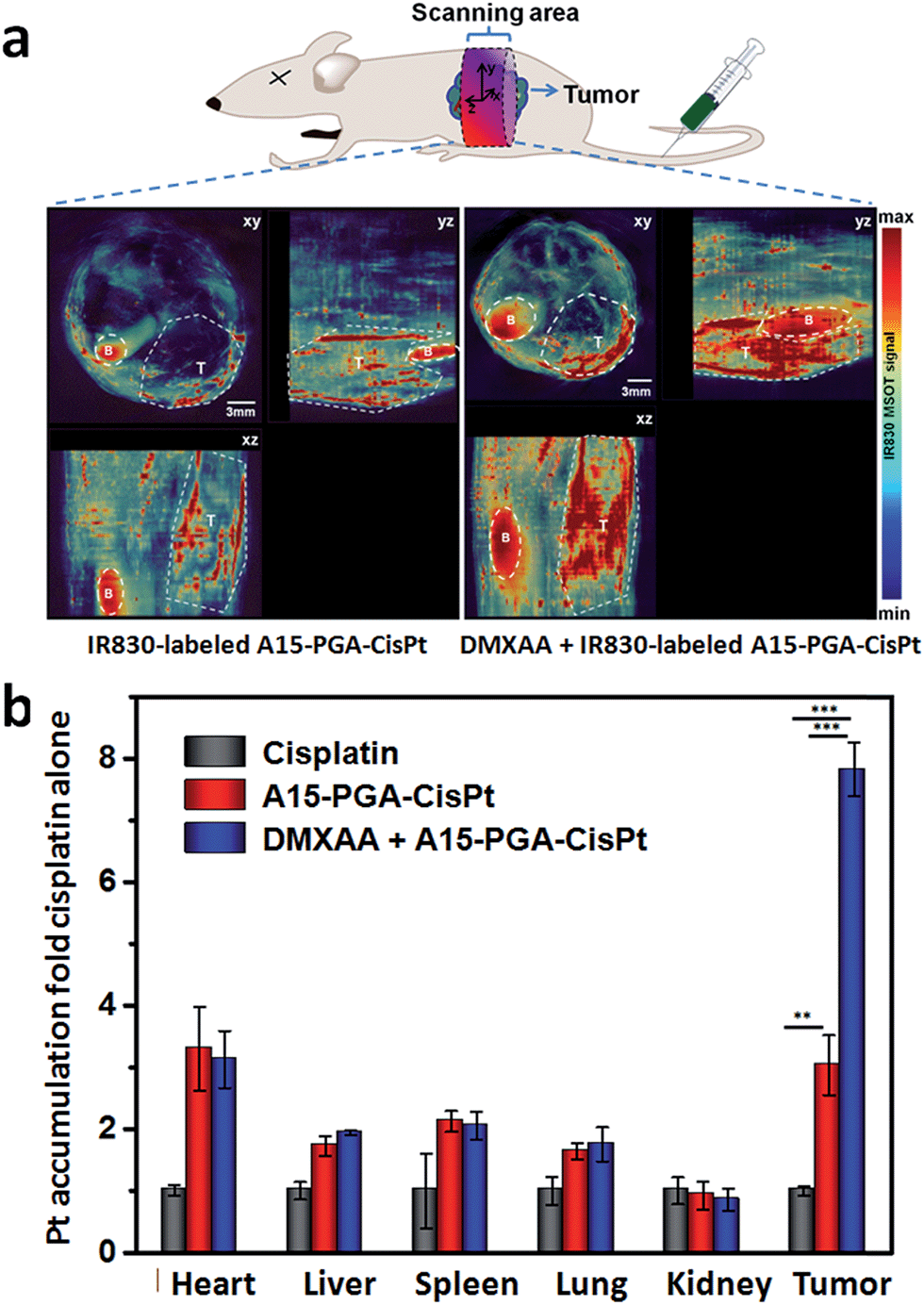 | ||
| Fig. 5 (a) Orthogonal views of MSOT images of C26 tumor-bearing mice at 24 h following injection of IR830-labeled A15-PGA-CisPt with or without DMXAA (DMXAA dosage: 15.0 mg kg−1). The 3D coordinate system defines the orientations and positions of the orthogonal views. B and T represent the bladder and tumor regions, respectively. (b) Pt accumulation in C26 tumors at 24 h following administration of free cisplatin (4.0 mg kg−1), A15-PGA-CisPt (4.0 mg cisplatin equivalent per kg), or DMXAA (15.0 mg kg−1) + A15-PGA-CisPt (4.0 mg cisplatin equivalent per kg) (n = 4, **: p < 0.01, ***: p < 0.001). | ||
We then examined the platinum (Pt) concentrations in tumors and other tissues to see the tumor targeting ability of the combination of DMXAA + A15-PGA-CisPt. Balb/C mice bearing C26 tumors were randomly divided into three groups and treated with one of the following: (i) cisplatin (4.0 mg kg−1), (ii) A15-PGA-CisPt (4.0 mg cisplatin equivalent per kg) or (iii) DMXAA (15.0 mg kg−1) + A15-PGA-CisPt (4.0 mg cisplatin equivalent per kg). After 24 h, the hearts, livers, spleens, lungs, kidneys and tumors were excised and the Pt concentrations were measured. It's noteworthy that the accumulation of Pt in the tumors of the DMXAA + A15-PGA-CisPt treated group was 2.5 fold that of the A15-PGA-CisPt group with no significant differences observed in the other organs between the two groups (Fig. 5b). The “passive targeting” of the A15-PGA-CisPt resulted in a 3.0 fold of Pt accumulation in tumors compared to free cisplatin. In contrast, the cooperative treatment of DMXAA + A15-PGA-CisPt resulted in a 7.5 fold of Pt accumulation in tumors compared to free cisplatin. Similar results were obtained in the biodistribution assay of platinum at 4 h, and the accumulation ratios between tumor and normal organs confirmed the selective accumulation of A15-PGA-CisPt in tumors when combined with DMXAA (Fig. S13, Table S4†). These results further confirm that DMXAA-induced coagulation in tumors could improve the tumor-targeted delivery of therapeutic agents that were loaded in coagulation-targeted nanoparticles.
In vivo therapeutic efficacy
Lastly, we compared the therapeutic efficacy of the cooperative tumor-targeted drug delivery platform with non-cooperative controls on Balb/C mice bearing C26 tumors. Saline, cisplatin, A15-PGA-CisPt, DMXAA or DMXAA + A15-PGA-CisPt was administered on the 1st, 3rd and 8th day. We found that the combination of DMXAA + A15-PGA-CisPt was significantly more effective at inhibiting tumor growth compared with all other treatment groups (Fig. 6). On the 14th day, cisplatin, A15-PGA-CisPt, DMXAA and DMXAA + A15-PGA-CisPt resulted in a tumor suppression rate of 79.6%, 57.9%, 72.9% and 95.9%, respectively. It is worth noting that the DMXAA + A15-PGA-CisPt group was the only therapeutic regimen tested able to completely inhibit tumor growth during the entire study period. Specifically, no tumor regrowth was observed in the DMXAA + A15-PGA-CisPt group on the 14th day, which was 6 days after the final treatment. In contrast, the tumor continued to grow in all other treatment groups by the 8th day after the last treatment. Furthermore, no obvious body weight loss was observed in the DMXAA + A15-PGA-CisPt group during the 14 days, indicating a low systemic toxicity of the cooperative tumor-targeted DDS. Collectively, these therapeutic studies demonstrate that the cooperative strategy of DMXAA + A15-PGA-CisPt possesses significantly enhanced anti-tumor efficacy with minimal systemic toxicity as compared to non-cooperative controls. This is the ultimate goal of modern drug delivery systems for cancer therapy.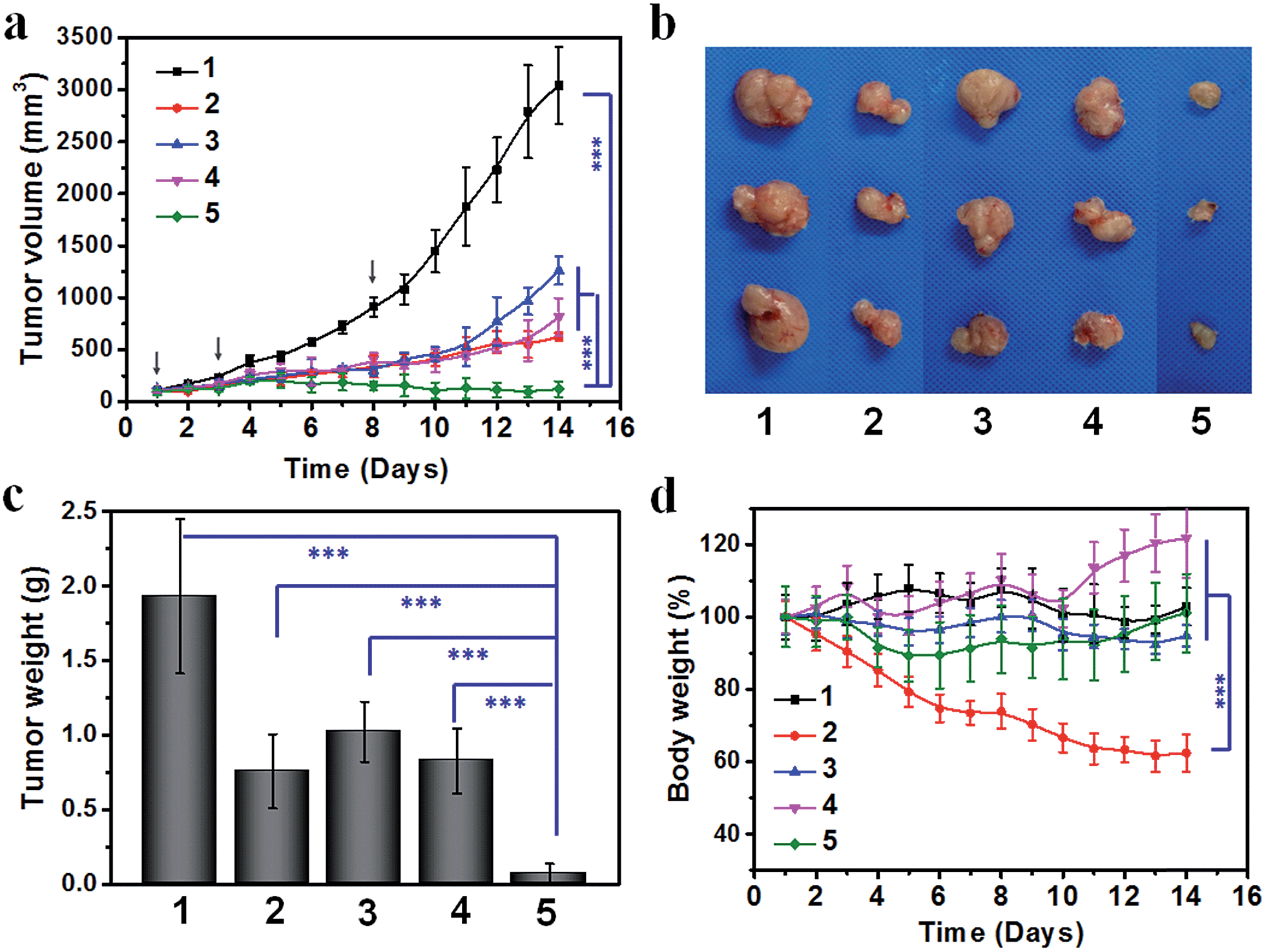 | ||
| Fig. 6 (a) Therapeutic efficacy of the cooperative drug delivery platform on C26 tumor-bearing Balb/C mice. (a) Tumor volumes (b) representative tumor images (c) average tumor weight after dissection on the 14th day (d) changes in body weight during the entire observation period. (1) Saline; (2) cisplatin, 4.0 mg kg−1; (3) A15-PGA-CisPt (4.0 mg cisplatin equivalent per kg); (4) DMXAA (15.0 mg kg−1); (5) DMXAA (15.0 mg kg−1) + A15-PGA-CisPt (4.0 mg cisplatin equivalent per kg). Drugs were administered on the 1st, 3rd and 8th day (n = 6, ***: p < 0.001). | ||
Conclusions
In summary, we developed a cooperative strategy for tumor-targeted delivery of an anticancer drug, using a small-molecule VDA as a coagulation-inducing agent along with a coagulation-targeted nanoparticle as a drug carrier. Using DMXAA as the model small-molecule VDA, we successfully fabricated a unique artificial coagulation environment in tumors. Under these conditions, we were able to selectively target tumors by administering coagulation-targeted cisplatin-conjugates (A15-PGA-CisPt), leading to tumor-targeted delivery of cisplatin and eradication of tumor cells. We demonstrated that the combination of DMXAA + A15-PGA-CisPt significantly enhanced the specificity and efficacy of treatment. Because a large number of small-molecule VDAs have proven effective in various murine and human tumor xenograft models and several small-molecule VDAs have entered clinical trials, the concept can be easily expanded to include other small-molecule VDAs and coagulation-targeted nanoparticles. This cooperative strategy is a promising method for the next generation of tumor-targeted nanomedicine development that could be applied to many other anticancer agents.Experimental
Cell culture and animal use
C26 murine colon carcinoma cells and HUVEC human umbilical vein endothelial cells were bought from Shanghai Bogoo Biotechnology Co. Ltd., China. Both cells were cultured in complete Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, supplemented with 50 U mL−1 penicillin and 50 U mL−1 streptomycin, and incubated at 37 °C in 5% CO2 atmosphere. Balb/C mice were obtained from the Laboratory Animal Center of Jilin University. Balb/C nude mice were obtained from Beijing Huafukang Biological Technology Co. Ltd. (HFK Bioscience, Beijing). The C26 xenograft tumor model was prepared by injecting 2.0 × 106 C26 cells into the right flank of Balb/C mice; the cells were then maintained by i.p. passage. All the animal experiments were conducted in accordance with the guidelines of Laboratory Protocol of Animal Care and Use Committee, Jilin University.Pathology and histopathological analysis of C26 tumor-bearing mice after injection with DMXAA and FITC-labeled fibrinogen
Balb/C mice bearing C26 tumors were injected with saline or DMXAA (15.0 mg kg−1), followed by 0.2 mL (2 nmol) of fibrinogen-FITC. After 4 h, the mice were sacrificed, tumor and heart, liver, spleen, lung, kidney were collected and photographed. Subsequently, these tumors and normal organs were submitted to H&E staining and frozen section respectively. The histological alterations were observed by microscopy (Nikon TI-S/L100), and the cryogenic slices were stained with 4′,6-diamidino-2-phenylindole (DAPI), and images were taken by confocal laser-scanning microscope (CLSM, Carl Zeiss LSM 710).Synthesis of PGA-g-mPEG/MAL-PEG
A graft copolymer PGA-g-mPEG/MAL-PEG was prepared by Steglich esterification between poly(L-glutamic acid) (PGA), mPEG-OH and MAL-PEG-OH in accordance with our previous work with slight modification.64 First, PGA was prepared by ring opening polymerization of BLG-NCA followed by deprotection of the benzyl group. Briefly, BLG-NCA (36.8 g, 140.0 mmol) was dissolved in 270.0 mL DMF, then 1.0 mL n-hexylamine (1.0 mM in DMF) was added and the mixture allowed to react for 72 h at 25 °C. Then, acetic anhydride (Ac2O, 2.0 g, 20.0 mmol) was added and the solution was stirred for another 6 h. The reaction solution was precipitated into excess diethyl ether/ethanol (2/1, v/v), and the resulting poly(γ-benzyl-L-glutamate) (PBLG) was dried under vacuum for 24 h. 5.0 g PBLG was dissolved in 50.0 mL dichloroacetic acid and 15.0 mL HBr/acetic acid (33 wt%) was added to remove the γ-benzyl group. After reacting for 1 h at 30 °C, the solution was precipitated in excess diethyl ether and the PGA was obtained (yield: 86.5%). In the second step, PGA (1.7 g, 13.2 mmol Glu monomer), mPEG-OH (2.8 g, 63.6 mmol EG monomer) and MAL-PEG-OH (0.7 g, 15.9 mmol EG monomer) were dissolved in 150.0 mL DMF, then DIC (178 mg, 1.4 mmol) and DMAP (196 mg, 1.6 mmol) were added into the reaction mixture. After 48 h at 25 °C, the solution was precipitated into excess diethyl ether and then dialyzed against distilled water for 3 days. The PGA-g-mPEG/MAL-PEG was obtained after freeze-drying (yield: 86%). The 1H NMR (Bruker AV 400 M NMR spectrometer) results in D2O were as follows: δ 7.26 ppm (d, –CHCH–CO–), 4.25 ppm (t, –CH<), 3.63 ppm (s, –CH2CH2O–), 3.31 ppm (s, –OCH3), 2.18 ppm (m, –CH2COO–), 1.96 and 1.83 ppm (m, >CHCH2–), 1.10–1.02 ppm (m, –CH2CH2–), 0.78 ppm (t, –CH2–CH3). Gel permeation chromatography (GPC) measurements of mPEG-OH, MAL-PEG-OH, PGA, and PGA-g-mPEG/MAL-PEG were conducted on a Waters GPC system (Waters Ultrahydrogel Linear column, 1515 HPLC pump with 2414 Refractive Index detector) using phosphate buffer (0.1 M, pH 7.4) as eluent (flow rate: 1 mL min−1, 25 °C, with polyethylene glycol as standards).
Preparation of poly(L-glutamic acid)-cisplatin conjugates decorated with A15 (A15-PGA-CisPt)
A15-PGA-CisPt were prepared by a two-step method. First, PGA-g-mPEG/MAL-PEG (80.0 mg) was dissolved in 15.0 mL water and the pH of the solution was adjusted to 7.5 by adding 0.1 mol L−1 NaOH. Then, cisplatin (23.0 mg, 76.7 μmol) was added and the mixture allowed to react at 37 °C for 72 h. The reaction mixture was purified by ultrafiltration. The obtained poly(L-glutamic acid)-cisplatin conjugates (PGA-CisPt) solution was added to A15 (4.8 mg, 3.0 μmol), reacted at 37 °C for 12 h, purified 5 times by ultrafiltration and preserved at 4 °C for use (A15-PGA-CisPt).Histopathological analysis with RhoB-labeled A15-PGA-CisPt
Balb/C mice bearing C26 tumors were prepared as described above. DMXAA (15.0 mg kg−1), fibrinogen-FITC (2 nmol per mouse) and RhoB-labeled A15-PGA-CisPt were injected via the tail vein. After 24 h, the mice were sacrificed, and the tumors were removed by dissection. The cryogenic slices of the tumor tissues were stained with 4′,6-diamidino-2-phenylindole (DAPI), and images were taken by CLSM.Multispectral optoacoustic tomography (MSOT) imaging
The multispectral optoacoustic tomographic equipment (inVision 128) was from iThera Medical (Munich, Germany). Balb/C nude mice bearing C26 tumors in the right abdomen were prepared by injecting 2.0 × 106 C26 cells suspended in 0.2 mL PBS into the right flank of the mice. IR830-labeled A15-PGA-CisPt with or without DMXAA (DMXAA dosage: 15.0 mg kg−1) were injected via the tail vein. After 24 h, the mice were anaesthetized with 2% isoflurane and placed into the MSOT system. The multispectral process (MSP) scanning was performed at 680 nm, 715 nm, 730 nm, 760 nm, 815 nm, 850 nm and 900 nm. The data were reconstructed using a model linear algorithm, and multispectral processing was performed by linear regression as implemented by the ViewMOST software (iThera Medical, Munich, Germany).Biodistribution of platinum
C26 tumor-bearing Balb/C mice were prepared similar to the methods described above. Mice were divided into 3 groups (4 mice per group), and cisplatin, A15-PGA-CisPt, or DMXAA + A15-PGA-CisPt were administered via tail vein (dosage: 4.0 mg cisplatin equivalent per kg body weight, 15.0 mg DMXAA per kg body weight; DMXAA and A15-PGA-CisPt were injected at the same time in the combination group). At 4 and 24 h, 4 mice in each group were sacrificed, and the hearts, livers, spleens, lungs, kidneys and tumors were excised. The organs were weighed, decomposed by heating in nitric acid, and ICP-MS was used to measure the Pt concentration.Tumor therapeutic efficacy
The tumor therapeutic efficacy was evaluated utilizing C26 tumor xenograft bearing Balb/C mice. Treatment started on the 4th day after cell implantation, which was designated as day 1. The mice were weighed and randomly divided into 5 groups (6 mice per group) to receive the following treatments: saline, cisplatin, A15-PGA-CisPt, DMXAA or DMXAA + A15-PGA-CisPt (dosage: 4.0 mg cisplatin equivalent per kg body weight, 15.0 mg DMXAA per kg body weight; DMXAA and A15-PGA-CisPt were injected at the same time in the combination group). The injections were carried out on the 1st, 3rd and 8th day via the tail vein. We assessed the treatment efficacy and conducted a safety evaluation by measuring the tumor volume and body weight, respectively. Calipers were used to measure tumor volume, which was then calculated as follows: tumor volume (V) = a × b2/2, where a is the major axis and b is the minor axis of the tumor. Similarly, we calculated the tumor suppression rate (TSR) as follows: TSR (%) = [(Vc − Vx)/Vc] × 100%, where c represents the control group and x represents the treatment group. The experiment was stopped on the 14th day and the tumors were removed by dissection then weighed and photographed.Statistical analysis
Data are expressed as the mean ± standard deviation. Statistical significance was determined using the Student's t-test.Acknowledgements
This research was financially supported by National Natural Science Foundation of China (Projects 51173184, 51233004, 51373168, 51390484, 81430087 and 51403204), Ministry of Science and Technology of China (International Cooperation and Communication Program 2011DFR51090), and the People's Government of Jilin Province (20130206058GX and 20130521011JH).References and notes
- H.-W. Yang, M.-Y. Hua, H.-L. Liu, R.-Y. Tsai, C.-K. Chuang, P.-C. Chu, P.-Y. Wu, Y.-H. Chang, H.-C. Chuang, K.-J. Yu and S.-T. Pang, ACS Nano, 2012, 6, 1795–1805 CrossRef CAS PubMed.
- S. Lee, H. Koo, J. H. Na, S. J. Han, H. S. Min, S. J. Lee, S. H. Kim, S. H. Yun, S. Y. Jeong, I. C. Kwon, K. Choi and K. Kim, ACS Nano, 2014, 8, 2048–2063 CrossRef CAS PubMed.
- Z. P. Zhen, W. Tang, Y. J. Chuang, T. Todd, W. Z. Zhang, X. Lin, G. Niu, G. Liu, L. C. Wang, Z. W. Pan, X. Y. Chen and J. Xie, ACS Nano, 2014, 8, 6004–6013 CrossRef CAS PubMed.
- K. Sano, T. Nakajima, P. L. Choyke and H. Kobayashi, ACS Nano, 2013, 7, 717–724 CrossRef CAS PubMed.
- P. M. Valencia, E. M. Pridgen, M. Rhee, R. Langer, O. C. Farokhzad and R. Karnik, ACS Nano, 2013, 7, 10671–10680 CrossRef CAS PubMed.
- R. Duncan and R. Gaspar, Mol. Pharmaceutics, 2011, 8, 2101–2141 CrossRef CAS PubMed.
- V. P. Chauhan and R. K. Jain, Nat. Mater., 2013, 12, 958–962 CrossRef CAS PubMed.
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- E. C. Dreaden, S. W. Morton, K. E. Shopsowitz, J.-H. Choi, Z. J. Deng, N.-J. Cho and P. T. Hammond, ACS Nano, 2014, 8, 8374–8382 CrossRef CAS.
- S. Dharap, Y. Wang, P. Chandna, J. Khandare, B. Qiu, S. Gunaseelan, P. Sinko, S. Stein, A. Farmanfarmaian and T. Minko, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12962–12967 CrossRef CAS PubMed.
- L. Yang, H. Xiao, L. Yan, R. Wang, Y. Huang, Z. Xie and X. Jing, J. Mater. Chem. B, 2014, 2, 2097–2106 RSC.
- S. Kawakami and M. Hashida, J. Controlled Release, 2014, 190, 542–555 CrossRef CAS PubMed.
- L. Neutsch, E. M. Wirth, S. Spijker, C. Pichl, H. Kählig, F. Gabor and M. Wirth, J. Controlled Release, 2013, 169, 62–72 CrossRef CAS PubMed.
- F. Porta, G. E. M. Lamers, J. Morrhayim, A. Chatzopoulou, M. Schaaf, H. den Dulk, C. Backendorf, J. I. Zink and A. Kros, Adv. Healthcare Mater., 2013, 2, 281–286 CrossRef CAS PubMed.
- Z. Yang, J. H. Lee, H. M. Jeon, J. H. Han, N. Park, Y. He, H. Lee, K. S. Hong, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 11657–11662 CrossRef CAS PubMed.
- W. Wang, D. Cheng, F. Gong, X. Miao and X. Shuai, Adv. Mater., 2012, 24, 115–120 CrossRef CAS.
- S. Kunjachan, R. Pola, F. Gremse, B. Theek, J. Ehling, D. Moeckel, B. Hermanns-Sachweh, M. Pechar, K. Ulbrich, W. E. Hennink, G. Storm, W. Lederle, F. Kiessling and T. Lammers, Nano Lett., 2014, 14, 972–981 CrossRef CAS PubMed.
- W. T. Song, Z. H. Tang, D. W. Zhang, Y. Zhang, H. Y. Yu, M. Q. Li, S. X. Lv, H. Sun, M. X. Deng and X. S. Chen, Biomaterials, 2014, 35, 3005–3014 CrossRef CAS PubMed.
- B. R. Smith, C. Zavaleta, J. Rosenberg, R. Tong, J. Ramunas, Z. Liu, H. J. Dai and S. S. Gambhir, Nano Today, 2013, 8, 126–137 CrossRef CAS PubMed.
- J. Ruiz-Rodriguez, M. Miguel, S. Preciado, G. A. Acosta, J. Adan, A. Bidon-Chanal, F. J. Luque, F. Mitjans, R. Lavilla and F. Albericio, Chem. Sci., 2014, 5, 3929–3935 RSC.
- F. Chen, H. Hong, Y. Zhang, H. F. Valdovinos, S. Shi, G. S. Kwon, C. P. Theuer, T. E. Barnhart and W. Cai, ACS Nano, 2013, 7, 9027–9039 CrossRef CAS PubMed.
- M. Ding, N. Song, X. He, J. Li, L. Zhou, H. Tan, Q. Fu and Q. Gu, ACS Nano, 2013, 7, 1918–1928 CrossRef CAS.
- W. Xu, I. A. Siddiqui, M. Nihal, S. Pilla, K. Rosenthal, H. Mukhtar and S. Gong, Biomaterials, 2013, 34, 5244–5253 CrossRef CAS PubMed.
- R. Wang, G. Zhu, L. Mei, Y. Xie, H. Ma, M. Ye, F.-L. Qing and W. Tan, J. Am. Chem. Soc., 2014, 136, 2731–2734 CrossRef CAS.
- Y.-H. Lao, K. K. L. Phua and K. W. Leong, ACS Nano, 2015, 9, 2235–2254 CrossRef CAS PubMed.
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS PubMed.
- Y. H. Bae, J. Controlled Release, 2009, 133, 2–3 CrossRef CAS PubMed.
- J. W. Nichols and Y. H. Bae, J. Controlled Release, 2014, 190, 451–464 CrossRef CAS PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- J. Li, Y.-C. Chen, Y.-C. Tseng, S. Mozumdar and L. Huang, J. Controlled Release, 2010, 142, 416–421 CrossRef CAS.
- P. Völker, C. Gründker, O. Schmidt, K.-D. Schulz and G. Emons, Am. J. Obstet. Gynecol., 2002, 186, 171–179 CrossRef.
- A. K. Rajasekaran, G. Anilkumar and J. J. Christiansen, Am. J. Physiol., 2005, 288, C975–C981 CrossRef CAS PubMed.
- B. Felding-Habermann, B. M. Mueller, C. A. Romerdahl and D. A. Cheresh, J. Clin. Invest., 1992, 89, 2018–2022 CrossRef CAS PubMed.
- M. A. Salazar and M. Ratnam, Cancer Metastasis Rev., 2007, 26, 141–152 CrossRef CAS PubMed.
- M. A. Cobleigh, C. L. Vogel, D. Tripathy, N. J. Robert, S. Scholl, L. Fehrenbacher, J. M. Wolter, V. Paton, S. Shak, G. Lieberman and D. J. Slamon, J. Clin. Oncol., 1999, 17, 2639 CAS.
- P. L. Bedard, A. R. Hansen, M. J. Ratain and L. L. Siu, Nature, 2013, 501, 355–364 CrossRef CAS.
- M. R. Junttila and F. J. de Sauvage, Nature, 2013, 501, 346–354 CrossRef CAS PubMed.
- E. Ruoslahti, S. N. Bhatia and M. J. Sailor, J. Cell Biol., 2010, 188, 759–768 CrossRef CAS PubMed.
- A. Marusyk, V. Almendro and K. Polyak, Nat. Rev. Cancer, 2012, 12, 323–334 CrossRef CAS PubMed.
- L. Tang, Q. Yin, Y. Xu, Q. Zhou, K. Cai, J. Yen, L. W. Dobrucki and J. Cheng, Chem. Sci., 2015, 6, 2182–2186 RSC.
- M.-H. Xiong, Y. Bao, X.-J. Du, Z.-B. Tan, Q. Jiang, H.-X. Wang, Y.-H. Zhu and J. Wang, ACS Nano, 2013, 7, 10636–10645 CrossRef CAS.
- G. Von Maltzahn, J.-H. Park, K. Y. Lin, N. Singh, C. Schwöppe, R. Mesters, W. E. Berdel, E. Ruoslahti, M. J. Sailor and S. N. Bhatia, Nat. Mater., 2011, 10, 545–552 CrossRef CAS PubMed.
- K. Y. Lin, E. J. Kwon, J. H. Lo and S. N. Bhatia, Nano Today, 2014, 9, 550–559 CrossRef CAS.
- V. L. Heath and R. Bicknell, Nat. Rev. Clin. Oncol., 2009, 6, 395–404 CrossRef CAS PubMed.
- C. Sessa, P. Lorusso, A. Tolcher, F. Farace, N. Lassau, A. Delmonte, A. Braghetti, R. Bahleda, P. Cohen and M. Hospitel, Clin. Cancer Res., 2013, 19, 4832–4842 CrossRef CAS PubMed.
- A. K. Nowak, C. Brown, M. J. Millward, J. Creaney, M. J. Byrne, B. Hughes, G. Kremmidiotis, D. C. Bibby, A. F. Leske and P. L. Mitchell, Lung Cancer, 2013, 81, 422–427 CrossRef PubMed.
- M. Burge, A. B. Francesconi, D. Kotasek, R. Fida, G. Smith, A. Wilks, P. A. Vasey and J. D. Lickliter, Invest. New Drugs, 2013, 31, 126–135 CrossRef CAS PubMed.
- M. M. Shenoi, I. Iltis, J. Choi, N. A. Koonce, G. J. Metzger, R. J. Griffin and J. C. Bischof, Mol. Pharmaceutics, 2013, 10, 1683–1694 CrossRef CAS PubMed.
- S. Sengupta, D. Eavarone, I. Capila, G. L. Zhao, N. Watson, T. Kiziltepe and R. Sasisekharan, Nature, 2005, 436, 568–572 CrossRef CAS PubMed.
- M. Früh, R. Cathomas, M. Siano, G. Tscherry, A. Zippelius, C. Mamot, A. Erdmann, F. Krasniqi, D. Rauch, M. Simcock, E. Küttel, P. Fustier and M. Pless, Clin. Lung Cancer, 2013, 14, 34–39 CrossRef PubMed.
- M. Head and M. B. Jameson, Expert Opin. Invest. Drugs, 2010, 19, 295–304 CrossRef CAS PubMed.
- M. E. Watts, S. Arnold and D. J. Chaplin, Br. J. Cancer, Suppl., 1996, 27, S164–S167 CAS.
- L.-M. Ching, D. Goldsmith, W. R. Joseph, H. Körner, J. D. Sedgwick and B. C. Baguley, Cancer Res., 1999, 59, 3304–3307 CAS.
- A. S. Jassar, E. Suzuki, V. Kapoor, J. Sun, M. B. Silverberg, L. Cheung, M. D. Burdick, R. M. Strieter, L.-M. Ching, L. R. Kaiser and S. M. Albelda, Cancer Res., 2005, 65, 11752–11761 CrossRef CAS PubMed.
- S. F. Zhou, P. Kestell, B. C. Baguley and J. W. Paxton, Invest. New Drugs, 2002, 20, 281–295 CrossRef CAS PubMed.
- G. Rustin, C. Bradley, S. Galbraith, M. Stratford, P. Loadman, S. Waller, K. Bellenger, L. Gumbrell, L. Folkes and G. Halbert, Br. J. Cancer, 2003, 88, 1160–1167 CrossRef CAS PubMed.
- M. Jameson, P. Thompson, B. Baguley, B. Evans, V. Harvey, D. Porter, M. McCrystal, M. Small, K. Bellenger and L. Gumbrell, Br. J. Cancer, 2003, 88, 1844–1850 CrossRef CAS PubMed.
- B. C. Baguley, Lancet Oncol., 2003, 4, 141–148 CrossRef CAS PubMed.
- G. Y. Lip, B. S. Chin and A. D. Blann, Lancet Oncol., 2002, 3, 27–34 CrossRef CAS.
- C.-H. Tung, N.-H. Ho, Q. Zeng, Y. Tang, F. A. Jaffer, G. L. Reed and R. Weissleder, ChemBioChem, 2003, 4, 897–899 CrossRef CAS PubMed.
- B. C. Baguley and M. J. McKeage, Future Oncol., 2010, 6, 1537–1543 CrossRef CAS PubMed.
- L. Muszbek, V. C. Yee and Z. Hevessy, Thromb. Res., 1999, 94, 271–305 CrossRef CAS PubMed.
- R. L. C. Adams and R. J. Bird, Nephrology, 2009, 14, 462–470 CrossRef CAS PubMed.
- H. Yu, Z. Tang, D. Zhang, W. Song, Y. Zhang, Y. Yang, Z. Ahmad and X. Chen, J. Controlled Release, 2015, 205, 89–97 CrossRef CAS PubMed.
- C. S. Shi, H. Y. Yu, D. J. Sun, L. L. Ma, Z. H. Tang, Q. S. Xiao and X. S. Chen, Acta Biomater., 2015, 18, 68–76 CrossRef CAS PubMed.
- F. A. Jaffer, C.-H. Tung, J. J. Wykrzykowska, N.-H. Ho, A. K. Houng, G. L. Reed and R. Weissleder, Circulation, 2004, 110, 170–176 CrossRef CAS PubMed.
- W. Song, Z. Tang, D. Zhang, H. Yu and X. Chen, Small, 2015, 11, 3755–3761 CrossRef CAS PubMed.
- D. Docter, U. Distler, W. Storck, J. Kuharev, D. Wunsch, A. Hahlbrock, S. K. Knauer, S. Tenzer and R. H. Stauber, Nat. Protoc., 2014, 9, 2030–2044 CrossRef CAS PubMed.
- Z. H. Siddik, D. R. Newell, F. E. Boxall and K. R. Harrap, Biochem. Pharmacol., 1987, 36, 1925–1932 CrossRef CAS PubMed.
- W. Song, Z. Tang, D. Zhang, N. Burton, W. Driessen and X. Chen, RSC Adv., 2015, 5, 3807–3813 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental description of preparation of FITC-labeled fibrinogen, histopathological analyses at varied doses of DMXAA, preparation of RhoB- and IR830-labeled A15-PGA-CisPt, characterization of the prepared A15-PGA-CisPt conjugates, in vitro release, cytotoxicity, cellular uptake, stability test in plasma, and pharmacokinetic test. Schemes of syntheses of RhoB- or IR830-labeled PGA-g-mPEG/MAL-PEG, macroscopic observation of hemorrhages, histopathological analysis of fibrin localization, H&E and histopathological analysis of heart, liver, spleen, lung and kidney, 1H NMR spectrum and GPC curves of PGA-g-mPEG/MAL-PEG, DLS, TEM image, XPS spectra, in vitro release, cytotoxicity, CLSM images of cellular uptake, sizes and surface bound proteins measurement, pharmacokinetic results, UV-spectrum of IR830-labeled A15-PGA-CisPt in water, platinum biodistribution. See DOI: 10.1039/c5sc01698c |
| This journal is © The Royal Society of Chemistry 2016 |