
Evidence of methane adsorption over Mo2C involving single C–H bond dissociation instead of facile carbon exchange†
Adrianna
Brush
a,
Gregory M.
Mullen
a,
Robin
Dupré
b,
Shruti
Kota
a and
C. Buddie
Mullins
 *abcd
*abcd
aMcKetta Department of Chemical Engineering, University of Texas at Austin, 1 University Station, C0400 Austin, USA. E-mail: mullins@che.utexas.edu
bDepartment of Chemistry, University of Texas at Austin, 1 University Station, C0400 Austin, USA
cTexas Materials Institute and Center for Nano- and Molecular Science, University of Texas at Austin, 1 University Station, C0400 Austin, USA
dCenter for Electrochemistry, University of Texas at Austin, 1 University Station, C0400 Austin, USA
First published on 10th November 2016
Abstract
Mo2C catalysts have been widely studied for methane reforming reactions. One of the proposed mechanisms for Mo2C catalysts is a redox type mechanism that includes active participation of the carbide carbon in the reaction. While evidence for this mechanism has been provided by several studies, one of the most surprising results previously reported asserts that a stream of pure methane can undergo significant, facile carbon exchange with the Mo2C catalyst at temperatures above 550 °C. Using pulses of 13CH4, we have found no evidence of carbon exchange between methane and Mo2C, even at 800 °C, in contrast to these previous results. In addition, by using pulses of CD4, we have found evidence of a small degree of dissociative methane adsorption at 800 °C, involving the breaking and reforming of a single methane–hydrogen/deuterium bond. While the present study does not contradict the model of active carbide carbon participation via a redox mechanism in methane reforming reactions, it doesn't support the notion of significant and facile carbon exchange between methane and Mo2C without an oxidant.
Introduction
Transition metal carbides have been a focus of research since Levy and Boudart first demonstrated that tungsten carbide exhibited catalytic similarity to Pt.1 Further, molybdenum carbide (Mo2C) based catalysts have been an active area of research since Claridge et al. demonstrated the ability of Mo2C to catalyze methane reforming reactions with high activity, comparable to 5% Ir/Al2O3 or 5% Rh/Al2O3.2 Subsequent research has demonstrated Mo2C as an active catalyst for dry methane reforming,3–7 steam methane reforming,2,8 water gas shift,8,9 and several other reactions.10–12 Despite this interest, the mechanisms of reactions on Mo2C based catalysts, and carbide catalysts in general, are still somewhat speculative. Claridge et al. originally proposed two possible mechanisms for carbide catalysts.2 One is the redox mechanism, involving formation of a carbon vacancy (denoted by “[]”) by the oxidant, followed by carbon from methane decomposition filling the carbon vacancy:| CO2 → CO + O* | (1) |
| Mo − C − Mo(s) + O* → Mo − [] − Mo + CO | (2) |
| Mo − [] − Mo(s) + O* → Mo − O − Mo (oxide formation, beginning of catalyst deactivation) | (3) |
| CH4 → C* + 2H2 | (4) |
| Mo − [] − Mo(s) + C* → Mo − C − Mo | (5) |
This mechanism implies active participation of the carbide carbon in the reaction, similar to a Mars-van Krevelen type mechanism. The suggestion that the carbon in the carbide may participate in the reaction makes some intuitive sense. Mo2C is often synthesized from MoO3 using temperature programmed reduction in hydrogen and methane, indicating that methane can decompose to form carbide carbon.10,13,14 Additionally, most Mo2C catalysts deactivate via oxidation to MoO2, indicating that the oxidant (often CO2 or H2O) is able to react with a carbide carbon and remove it.2,4,7,8 In addition, reaction (3) also provides a potential a first step for catalyst deactivation when an oxygen atom fills the vacancy instead of carbon.
The second mechanism is a more traditional noble metal type mechanism, not directly involving the carbide carbon in the reaction mechanism:
| CH4 → C* + 2H2 | (6) |
| CO2 → CO + O* | (7) |
| O* + C* → CO | (8) |
In addition, a few isotopically labeled experiments have demonstrated that the carbide carbon can participate to some degree in various reactions. Shou and Davis performed a steady state isotopic transient kinetic analysis (SSITKA) experiment, where they switched from 12CO to 13CO during a reaction of H2 and CO to form CH4 and C2+ products.19 In addition to measured transient responses, they saw a persistent background of 12C in the CH4 and C2+ species, indicating some of the 12C from the Mo2C support was participating in the reaction even an hour after the switch to 13CO. Xiao et al. synthesized Mo213C, then performed several pulses of 12CH4 + O2 and monitored the amounts of 12C and 13C in CO and CO2 produced.20 The first pulse showed that the majority of the carbon in the produced CO and CO2 was 13C. The subsequent pulses showed declining amounts of 13CO/13CO2 and increasing amounts of 12CO/12CO2. Finally, Naito et al. flowed 13CH4 and 13CO2 over unsupported Mo212C and Mo212C supported on ZrO2 to produce CO and H2.21 They found that almost 50% of the CO produced initially was 12CO. Overall, these results show a definite participation of the carbide carbon from the Mo2C in these reactions.
The most surprising result from an isotopic study was reported by LaMont et al.22 They flowed pure 13CH4 over Mo212C while ramping from 100 °C to 800 °C at 25 °C per minute, taking samples at 5 points during the ramp and analyzing with a mass spectrometer. Their interpretation of their measurements indicated that, above 550 °C, a large portion of the effluent methane was 12CH4. Their data suggests that 50–75% of the 13CH4 underwent carbon exchange with the carbide carbon in Mo2C to form 12CH4. If true, this result suggests a rapid, facile decomposition and recombination of methane over the Mo2C catalyst. The results from this study have been used to support the primacy of the redox mechanism for dry methane reforming over Mo2C,5 and have been used to support the theory of active carbide carbon participation in methane reforming reactions.12
However, we question their interpretation of their reported results. Firstly, their results of 12CH4 and 13CH4 mole fractions in the effluent reported that the total methane mole fraction in the effluent was 220%, suspicious since, by definition, total mole fraction should be 100%, with any deviation from 100% likely due to experimental error. The evidence of significant carbon exchange between methane and Mo2C was given by the reported results that the effluent contained ∼50% mole fraction 13CH4 and ∼150% mole fraction 12CH4 at temperatures higher than 550 °C. Secondly, only masses 16 and 17 were monitored to detect 12CH4 and 13CH4, respectively. However, the CH3 fragment (mass 15 for 12CH3, mass 16 for 13CH3) is a major fragment of methane, giving a signal ∼80% of the parent mass signal.23 This means that the mass 16 signal would not change much between a stream of pure 12CH4 and a stream of pure 13CH4, complicating efforts to use mass 16 quantitatively for this experiment. While the researchers reported performing calibration curves, the fact that their calculated total mole fraction is 220% suggests experimental error significantly affected their quantitative measurements.
In an attempt to further explore the possibility of carbon exchange between pure methane and Mo2C, as well as gain some kinetic and mechanistic insights into it, we performed several pulse steady state isotopic transient kinetic analysis (SSITKA) experiments with 13CH4 and CD4 over Mo2C. The SSITKA technique was used due to its ability to measure various kinetic and mechanistic properties of a catalytic system, including number of active surface species, turnover frequency (TOF), and qualitative insights into the reaction mechanism.24,25 Using the same amount of Alfa Aesar Mo2C and the same flowrates as LaMont et al., but continuously monitoring additional masses, we find no evidence of carbon exchange between the 13CH4 and the Mo2C, even at 800 °C. Our experiments with CD4 corroborated this result, but also gave evidence for a small degree of methane dissociation and recombination involving a single C–H bond, seen primarily at 800 °C (and likely higher temperatures). Additionally, experiments using temperature ramping similar to the LaMont et al. study confirm the lack of carbon exchange, even at 800 °C. These results are not consistent with a model involving facile carbon exchange between pure CH4 and Mo2C at temperatures exceeding 550 °C. Instead, it appears that dissociative methane adsorption involves only one C–H bond, and does not occur to a significant degree even at 800 °C. However, these results are not inconsistent with the previously proposed redox mechanism, where the carbide carbon can participate in reactions involving oxidants over Mo2C. The isotopically labeled studies performed by Shou and Davis,19 Xiao et al.,20 and Naito et al.21 have given evidence of this phenomenon for the CO hydrogenation, methane partial oxidation, and methane dry reforming reactions, respectively.
Experimental
The reactor and apparatus for delivering the pulses of isotopically labeled gas have been described in more detail in a separate publication,26 and a basic schematic of the reactor system can be seen in Fig. 1. Briefly, a straight quartz tube reactor (7 mm ID) with a restriction in the middle was packed with 0.03 g of quartz wool, and 1 g of commercial Mo2C (Alfa Aesar, 99.5% [metals basis], 325 mesh, lot # C03W025) was placed on top of the quartz wool. The vertical quartz tube reactor was heated by an external furnace (Applied Test Systems, Series 3210 Furnace) controlled by a proportional-integral-derivative (PID) controller. Four gas cylinders feed reactants into the system via electronic mass flow controllers (MFC). The four streams combine into one stream, leading to the quartz reactor tube. In this study, only 3 streams were used: one for 95% CH4/5% Ar, one for H2, and one for an inert gas (He or N2).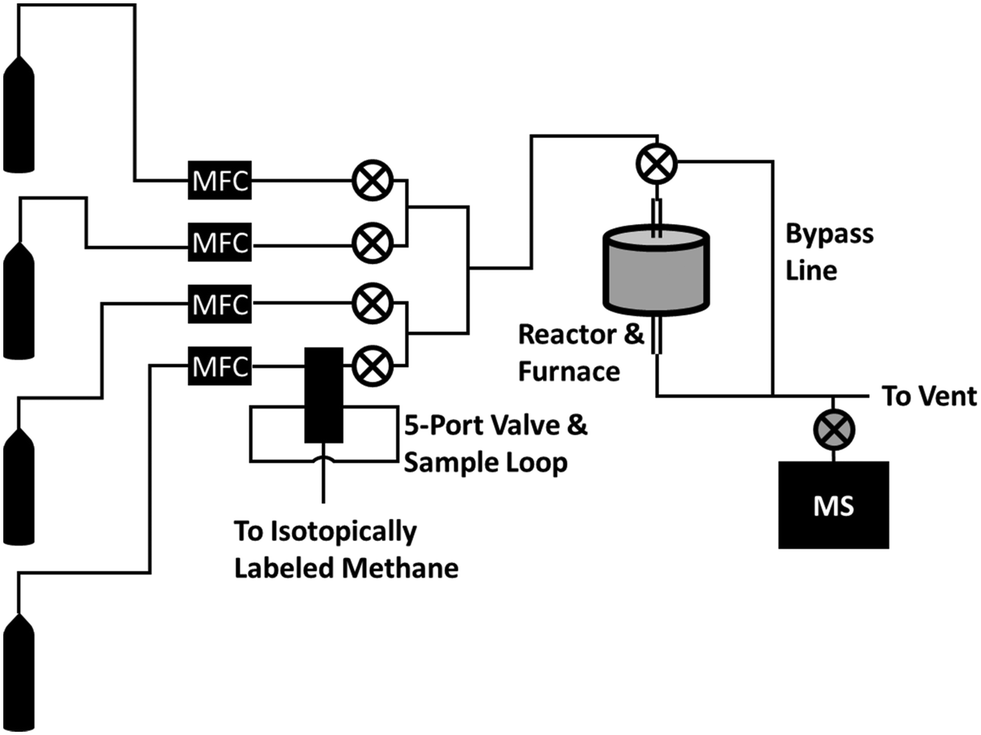 | ||
| Fig. 1 Basic schematic of the reactor used. From left to right, gas cylinders containing the reactants connect to mass flow controllers (“MFC”) which regulate flow of each reactant. The bottom most gas line represents the methane reactant, which passes through a 5-port valve with a sample loop of isotopically labeled methane. All of the gas lines are then fed to the reactor, and the effluent is analyzed with an on-line mass spectrometer. | ||
Isotopically labeled methane was introduced to the system via a 5-port valve with a 50 mL sample loop filled with the isotopically labeled methane. When the 5-port valve switch was flipped, the methane/argon stream was diverted first to the sample loop before being fed to the reactor, similar to the operation of a common GC 6-port valve. This causes a pulse of isotopically labeled methane (13CH4 or CD4) to be delivered to the reactor, pushed by the diverted 95% CH4/5% Ar stream. A flow diagram of the 5-port valve and isotope sample loop used to deliver the pulse of isotopically labeled methane can be seen in Fig. S1.† The resulting effluent was continuously analyzed with an on-line mass spectrometer. Our on-line mass spectrometer consisted of a 6-way ultra-high vacuum (UHV) cross connecting together an Extorr brand XT100 residual gas analyzer equipped with a quadrupole mass spectrometer operating with an electron bombardment energy of 70 eV, a turbomolecular pump backed by a mechanical pump, an ion-gauge, and finally a leak valve connected on-line to the reactor effluent stream for continuous sampling. The dwell time for each mass measured by the residual gas analyzer was 21 ms, and each mass was measured approximately once every 1.6 seconds.
95% CH4/5% Ar was used instead of pure methane in order to use Ar as an inert tracer for the transient studies. In SSITKA experiments and other transient studies, a small amount of inert gas is often incorporated into the non-isotopically labeled reactant in order to determine the gas phase hold-up of the system. This essentially gives the mass transport function of the isotopic pulse through the reactor system without any influence from reaction kinetics. For the isotopically labeled atom(s) in the pulse, any entrance into a kinetic pathway, such as a reaction or dissociative adsorption, causes a time delay in emergence from the reactor compared with the gas phase holdup function. This delay can be detected by normalizing and, if necessary, inverting the mass spectrometer curves so that all curves span from 0 to 1 to 0 during the pulse. The time-delays indicative of entrance into a kinetic pathway manifest as deviations of the reactant and product pulse functions from the inert pulse function. Although much more information about the catalytic system can be extracted from comparing these pulse functions,24,25 for the purposes of this study it is sufficient to understand that deviation of a species' pulse function from the inert pulse function indicates the presence of a kinetic pathway. In our results, we will present both the mass spectrometer data (smoothed using a 5-point FFT smoothing function in Origin 9.1 software and scaled using the inert tracer's baseline) as well as these normalized curves.
For the SSITKA experiments, the Mo2C was heated to 600 °C at 10 °C min−1 under 50 standard cubic centimeters per minute (SCCM) flowing inert gas (He for 13CH4 experiments, N2 for CD4 experiments). The catalyst was subjected to a reductive pretreatment by holding at 600 °C for 3 hours under 50 SCCM flowing H2. We used this pretreatment, since we found the 1 hour H2 at 600 °C pretreatment used by LaMont et al.22 to be insufficient for removing oxygen and adversely affected our results, as will be discussed in more detail below. After the reductive pretreatment, the catalyst was flushed with inert for one hour while cooling to 200 °C. 5.5 SCCM of 95% CH4/5% Ar was then flowed through the system until the system appeared to reach steady state, as determined by the mass spectrometer signals. 5.5 SCCM of 95% CH4/5% Ar was used in order to be similar to the 5.2 SCCM 13CH4 flowrate used by LaMont et al.22 Once at steady state, a pulse of 50 mL 13CH4 or CD4 was injected into the stream, corresponding to a pulse of approximately 9 minutes, and the responses were monitored with the mass spectrometer. One hour after the pulse injection, the reactor was heated to 600 °C at 10 °C min−1, still while flowing 95% CH4/5% Ar, and allowed to reach steady state. Once at steady state, a 50 mL pulse of 13CH4 or CD4 was injected into the stream. Similarly, one hour after the injection at 600 °C, the reactor was heated to 800 °C at 10 °C min−1, and allowed to reach steady state before injecting a 50 mL pulse of 13CH4 or CD4. One hour after the pulse injection, the reactor was cooled in a stream of inert. A control experiment was performed for each isotopically labeled methane species by injecting a pulse while flowing 5.5 SCCM of 95% CH4/5% Ar through the catalyst at room temperature.
A second set of experiments was performed in order to more closely resemble LaMont et al.'s experiment, where the Mo2C was pretreated with 1 hour of H2, cooled to ∼100 °C, then heated to 800 °C at 25 °C min−1 while flowing 5.3 SCCM 13CH4. In our experiments, the Mo2C was heated to 600 °C in 50 SCCM inert, subjected to a 3 hour, 50 SCCM H2 pretreatment, then cooled to 200 °C in 50 SCCM inert, similar to the SSITKA experiments. Once at 200 °C, 18 SCCM inert (He for 13CH4 experiments, N2 for CD4 experiments) and 2 SCCM 95% CH4/5% Ar was flowed over the Mo2C until steady state was achieved. Then, a 50 mL pulse of 13CH4 or CD4 was injected, and the temperature was ramped from 200 °C to 800 °C at 25 °C min−1, corresponding to a pulse of approximately 25 minutes. Our flowrates and ramp timing were chosen so that the entirety of the ramp occurred during the pulse of isotopically labeled methane.
Finally, the commercial Mo2C was characterized by XRD and BET. XRD was performed with a Rigatu R-Axis SPIDER with a Cu X-ray source operating at 40 kV and 40 mA, and rotating the sample at 10 degrees per s for 10 minutes. BET surface area analysis was performed using a Quantachrome Instruments NOVA 2000 high-speed surface area BET analyzer at a temperature of 77 K. The data was analyzed by Quantachrome Autosorb1 software, using multipoint BET analysis from P/P0 = 0.1 to 0.3 and achieving a correlation coefficient of 0.9999 and a C value of 43.
Results and discussion
Catalyst characterization
The XRD spectrum of the Alfa Aesar commercial Mo2C is shown in Fig. S2† along with the reference spectra for Mo2C. This spectrum confirms the sample identity to be Mo2C without any measurable crystalline oxide phase. In addition, the specific surface area of the commercial Mo2C was measured to be 0.336 m2 g−1. Both of these are in agreement with the LaMont et al.22 study, which found their catalyst to be pure Mo2C, with a specific surface area of 0.36 m2 g−1.Pulse SSITKA experiments
In order to probe the possibility of carbon exchange between CH4 and Mo2C, we first performed 50 mL 13CH4 pulses while flowing 5.5 SCCM 95% CH4/5% Ar over Mo2C. Fig. 2 shows the mass spectrometry signals (Fig. 2a–d), and normalized transient responses (Fig. 2e–h) for these pulses as performed at room temperature, 200 °C, 600 °C, and 800 °C, respectively. Fig. 2a–d depict the mass spectrometry signals for masses 15, 17, and 40 for all four conditions tested (room temperature, 200 °C, 600 °C, and 800 °C). Mass 40 corresponds to the Ar tracer and mass 17 corresponds to 13CH4. Mass 15 was chosen as the best representative for 12CH4, since it is a major fragment of 12CH4 (i.e.12CH3, ∼80% of parent mass signal) but a much smaller fragment of 13CH4 (i.e.13CH2, ∼23% of parent mass signal), corresponding to the most significant change in signal during the 13CH4 pulse.23 If carbon exchange is occurring between methane and Mo2C, the 13CH4 (represented by mass 17) signal should reach a lower maximum and the 12CH4 (represented by mass 15) signal should reach a higher minimum than is observed at room temperature, due to 13C incorporation into the bulk and formation of 12CH4 from the Mo2C. However, as can be seen in Fig. 2a–d, the relative magnitude of all masses are extremely consistent across all temperatures, even at 600 °C and 800 °C, where carbon exchange has been postulated to occur.22 While this does not disprove the possibility of a very small degree of carbon exchange between methane and Mo2C, it casts serious doubt on the assertion that a significant amount of methane in the stream undergoes carbon exchange with Mo2C, and certainly not the 50–75% as suggested previously.22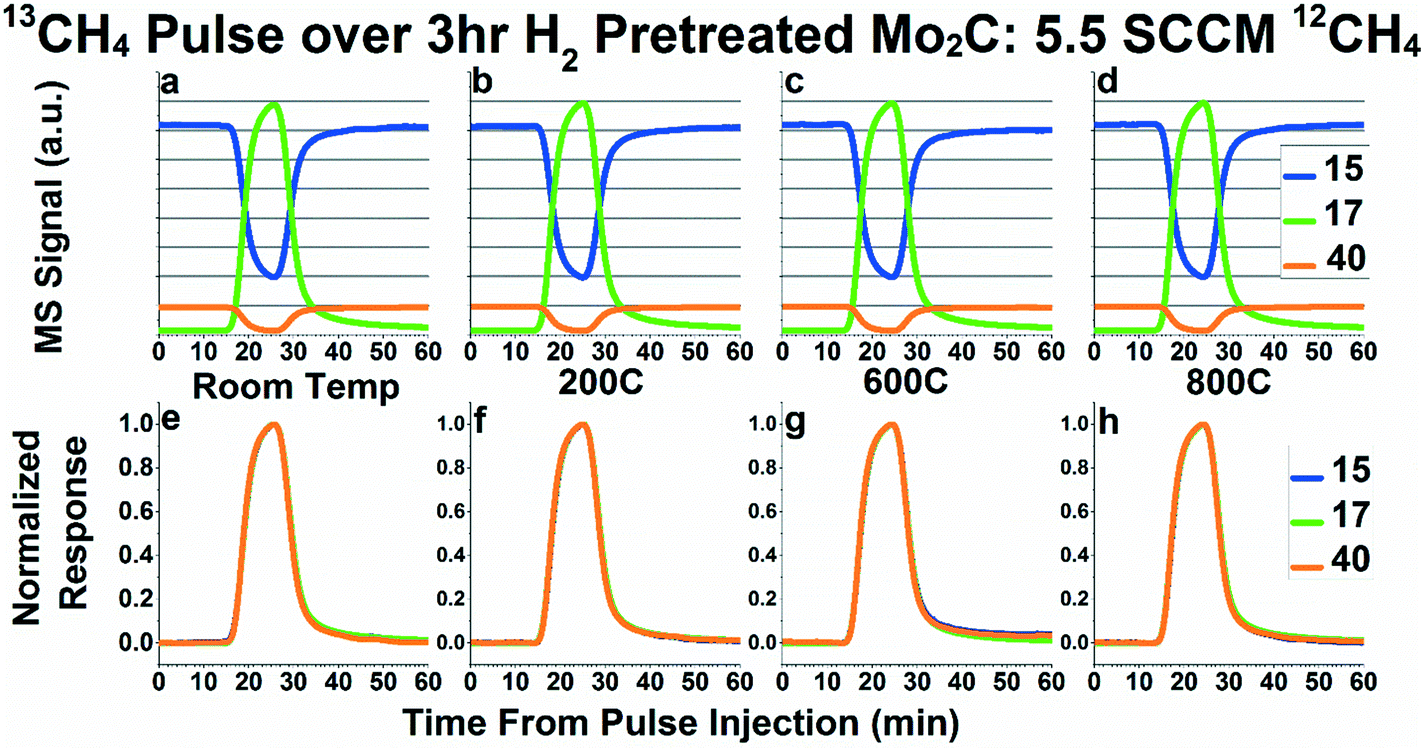 | ||
| Fig. 2 Raw mass spectrometry signals (a–d) and normalized responses (e–h) of masses 15 (representing 12CH4), 17 (representing 13CH4), and 40 (representing Ar inert tracer) for 13CH4 pulses while flowing 5.5 SCCM 95% CH4/5% Ar over Mo2C at room temperature (a and e), 200 °C (b and f), 600 °C (c and g), and 800 °C (d and h). In Fig. 2e–h, mass 15 has been inverted in addition to normalized, in order to overlay the pulse response functions. Pretreatment was 50 SCCM flowing H2 at 600 °C for 3 hours. | ||
In order to probe for any measurable kinetic steps for carbon exchange, the normalized transient response curves of masses 15, 17, and 40 during the 13CH4 pulses at the four temperatures (room temperature, 200 °C, 600 °C, and 800 °C) are plotted in Fig. 2e–h. In these plots, the curves of masses 15 and 17 line up almost perfectly behind the Ar inert tracer curve. The lack of any noticeable deviations from the inert tracer during the pulse experiments suggest that there is no kinetic step, giving more evidence against carbon exchange occurring between methane and the molybdenum carbide. While Fig. 2e–h depict only masses 15 and 17, we monitored all possible methane fragments (masses 12–17). As can be seen in Fig. S3e–h,† the pattern seen with masses 15 and 17 is consistent with all the mass fragments measured.
Fig. S3a–d† depict the same plots as Fig. 2a–d, but with the addition of mass 16 to demonstrate that the mass 16 curve does not decrease very much during the 13CH4 pulse. In addition, the normalized response curves in Fig. S3e–h† show that the baseline for mass 16 does not appear as steady as the rest of the masses monitored. This somewhat unstable baseline, combined with the small change in signal for mass 16 during the pulse, demonstrates the potential risk in using mass 16 to quantify 12CH4versus13CH4.
In order to further study the possible dissociation and recombination mechanism of methane over Mo2C, the same transient experiments discussed above involving 13CH4 were performed with CD4. Carbon exchange between methane and the carbide support would inherently require full dissociation of the four carbon–hydrogen/deuterium bonds, and then combination of four hydrogen/deuterium atoms with a carbon atom from the support. If this mechanism is correct, one would expect to see a mixture of CH4 (mass 16), CH3D (mass 17), CH2D2 (mass 18), CHD3 (mass 19), and CD4 (mass 20) with the distribution determined by the relative coverages of H and D atoms and the results of any kinetic isotope effect. We would expect a mixture of CH3D (mass 17), CD2H2 (mass 18) and CH3D (mass 19) even if the dissociation/recombination mechanism involved the breaking and reforming of 2 or more carbon–hydrogen/deuterium bonds on the methane. Alternatively, if only one methane–hydrogen/deuterium bond was broken and reformed in the mechanism, only CH4 (mass 16), CH3D (mass 17), CHD3 (mass 19), and CD4 (mass 20) should appear in any significant amount.
Fig. 3a–d shows the raw signals of 15, 17, 19, 20, and 40 during the CD4 pulse experiements at room temperature, 200 °C, 600 °C, and 800 °C, respectively. Mass 15, corresponding primarily to a CH3 fragment, represents CH4, since mass 16 is a major fragment of CD4. Mass 17 represents CH3D, mass 19 represents CD3H, mass 20 represents CD4, and mass 40 represents the inert Ar tracer. In Fig. 3a–d, the signals for masses 17 and 19 have all been multiplied 20× in order to depict them on the same plot as masses 20 and 40. In the room temperature plot (Fig. 3a), we can see an increase in 19 during the pulse, since there are some CD3H impurities in the CD4. Similarly, we see a decrease in mass 17 during the pulse, since there is some naturally occurring CH3D and 13CH4 in the CH4 reactant. As can be seen in Fig. 3b and c, the plots for 200 °C and 600 °C appear similar to room temperature, giving no indication of methane dissociation and recombination. However, at 800 °C, we see the appearance of peaks in mass 17 and mass 19. The presence of these peaks in mass 17 (CH3D) and 19 (CD3H) indicate the presence of a small amount of dissociative methane adsorption accompanied by recombinative desorption. This dissociation is small enough that there is no noticeable difference in the mass 15 (representing CH4) and mass 20 (representing CD4) signals between the pulse performed at 800 °C (Fig. 3d), and the rest of the temperatures (Fig. 3a–c).
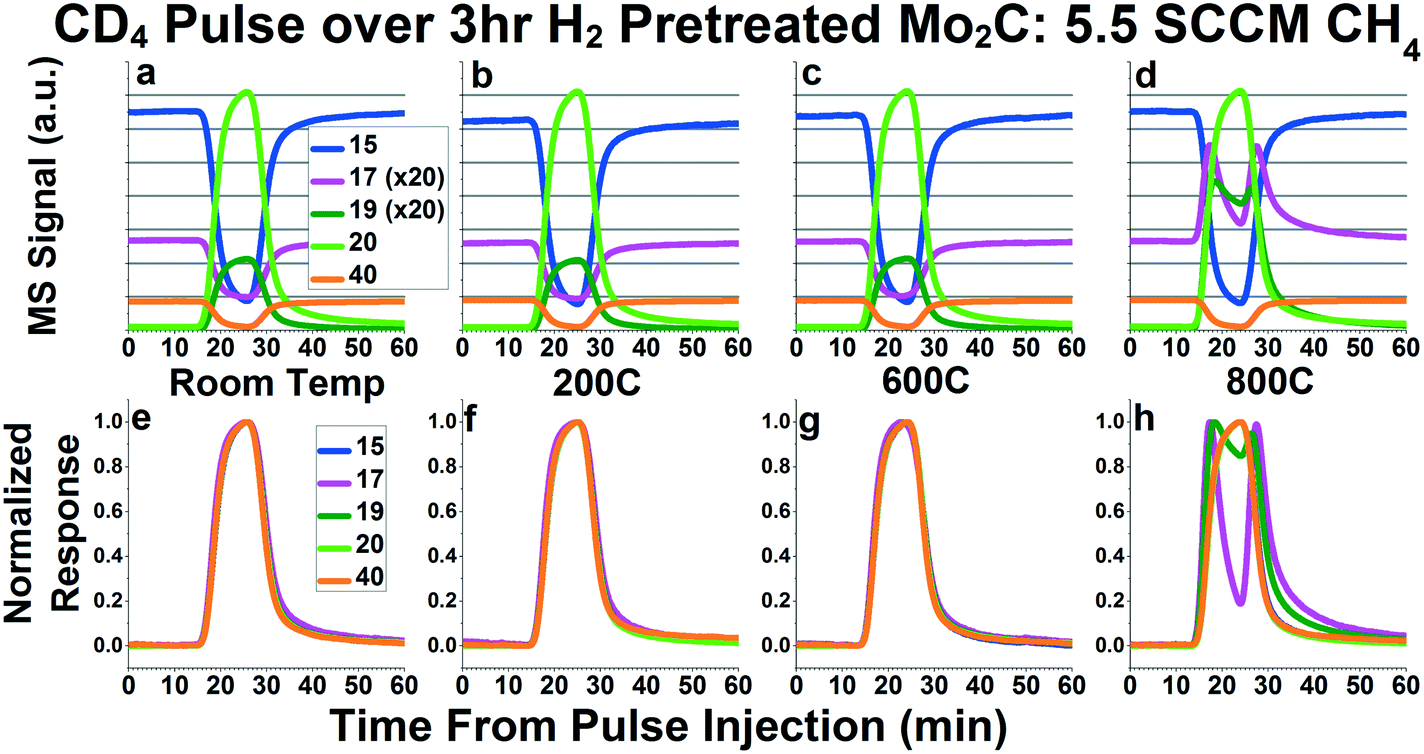 | ||
| Fig. 3 Mass spectrometry signals (a–d) and normalized responses (e–h) of masses 15 (representative of CH4), 17 (CH3D), 19 (CD3H), 20 (CD4) and 40 (Ar inert tracer) for CD4 pulses while flowing 5.5 SCCM 95% CH4/5% Ar over Mo2C in the bypass (a and e), at 200 °C (b and f), at 600 °C (c and g), and at 800 °C (d and h). In Fig. 3a–d, masses 17 and 19 have been multiplied by 20 in order to display them on the same plot as masses 15, 20, and 40. In Fig. 3e–g, masses 15 and 17 have been inverted in addition to normalized in order to overlay the pulse response functions. In Fig. 2h, only masses 15 has been inverted. | ||
As mentioned above, the mechanism involving carbon exchange between methane and Mo2C would inherently produce CD2H2, which has a mass of 18, in addition to CH3D and CD3H. However, mass 18 is a major fragment of CD4, requiring a signal subtraction of 0.78 × (mass 20 signal) in order to properly interpret the data. Plots similar to Fig. 3a–d, but including the corrected mass 18 signal, can be found in Fig. S4a–d.† This correction is not perfect, however, as can be seen in the room temperature plot, where mass 18 should theoretically be 0 in the whole plot. While there are features in mass 18 at 800 °C that appear similar to the peaks in masses 17 and 19 at 800 °C (Fig. S4d†), it is unclear whether this actually corresponds to the CD2H2 species. The features could be a result of imperfect signal subtraction, similar to the effect seen in the 200 °C plot. Since 18 would represent the CD3 fragment of CD3H, it is also possible that some or all of the features in the mass 18 signal could be attributed to CD3H fragmentation. Even if the entire signal for mass 18 in Fig. S4d† was attributable to CD2H2, the peaks in mass 18 are less than the ones in mass 17 or mass 19. This suggests that, if any CH2D2 is produced, it is significantly less than the CH3D and CD3H produced at 800 °C.
The normalized response curves for masses 15, 17, 19, 20 and 40 are depicted in Fig. 3e–h. Similar plots containing all masses monitored (masses 12–20 and 40) can be seen in Fig. S4e–h.† In the room temperature, 200 °C, and 600 °C plots, no noticeable deviations from the Ar tracer (mass 40) can be seen. However, at 800 °C (Fig. 3h), the peaks in mass 17 and 19 are clearly detectable. From these plots, we can see that the peaks in mass 17 and 19 occur when both CH4 and CD4 are in the stream, suggesting that this dissociation/recombination mechanism occurs relatively rapidly. Overall, the results from these CD4 pulse experiments do not provide evidence for significant and complete methane dissociation and recombination, as would be required for carbon exchange between methane and Mo2C. Further, the fact that the small degree of methane dissociative adsorption that can be detected at 800 °C results primarily in CH3D and CD3H species indicates that this dissociative adsorption involves the breaking and reforming of just one carbon–hydrogen/deuterium bond.
Ramp experiments
In order to more closely match the experimental conditions (i.e. exposure time and temperature profile) used in the LaMont et al. study, pulse experiments were performed while flowing 18 SCCM inert and 2 SCCM 95% CH4/5% Ar and ramping from 200 °C to 800 °C at 25 °C min−1. Fig. 4 depicts the results of 13CH4 pulses at room temperature (Fig. 4a), and while ramping from 200 °C to 800 °C at 25 °C min−1 (Fig. 4b). Fig. 4 compares mass 15 (best mass for 12CH4), 17 (13CH4), and 40 (the inert Ar tracer). It should be noted that the spikes seen in the curves around minute 4 are due to imperfect matching of the pressure of the 95% CH4/5% Ar reactant stream and the pressure in the sample loop filled with isotopically labeled methane, causing a temporary spike of extra methane or extra inert. Based on these results and other, unpublished results, we have not found this spike to affect our results. During the ramp, the curves for 13CH4 and 12CH4 appear identical to the room temperature pulse curves. If significant and facile carbon exchange occurred at temperatures above 550 °C, there should have been a significant decrease in the mass 17 and significant increase in the mass 15 curves around the middle of the pulse function. Even at 800 °C, no signs of carbon exchange can be seen.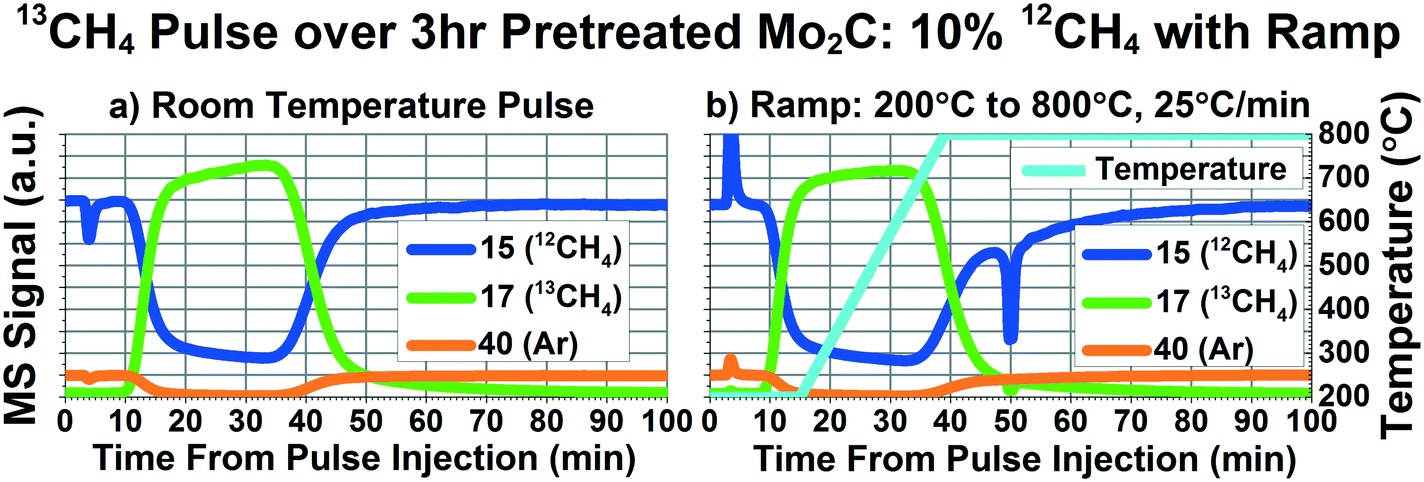 | ||
| Fig. 4 Raw mass spectrometry curves of masses 15 (representing 12CH4), 17 (representing 13CH4), and 40 (representing Ar inert tracer) for 13CH4 pulses delivered while flowing 18 SCCM He, and 2 SCCM 95% CH4/5% Ar after 3 hours of H2 pretreatment at 600 °C. a) Pulse performed at room temperature; b) pulse performed while ramping from 200 °C to 800 °C at 25 °C min−1, as indicated by the cyan temperature curve overlaid on the mass spectrometer curves. | ||
There is an interesting feature of note at minute 50 in Fig. 4b, where there is a significant decrease in both mass 17 and 15. Since carbon exchange would result in one of these masses increasing and the other decreasing, this feature is likely the result of some degree of oxidation of the methane. Fig. S5† depicts more masses measured during the same experiment described in Fig. 4. As can be seen in Fig. S5d,† the decrease in mass 15 and 17 is accompanied by increases in 28, 29, and a small amount of 18. This is indicative of some kind of oxygenated species being reduced by the methane. During our study, we found evidence of a couple different types of oxygenated species present in the Mo2C. The first type belongs to oxygen that can be removed with the 600 °C H2 pretreatment. As stated earlier, we found a one hour pretreatment to be insufficient for removing this type of oxygen species. Fig. S6† depicts the same ramp experiment as seen in Fig. 4, but with only 1 hour of hydrogen pretreatment, showing a significant decrease in masses 17 and 15 during the ramp, accompanied by increases in masses 28, 29, and 18. Since we do not see an increase in mass 15 corresponding to the decrease in 17, as well as seeing features for CO and H2O, this phenomena is likely due to methane consumption, not carbon exchange between the methane and Mo2C. This finding is supported by the additional H2O that is removed from the Mo2C during the second and third hour of the H2 pretreatment, as can be seen in Fig. S7.†
The spikes seen at minute 50 seem to represent a different type of oxygenated species than that removed by the H2 pretreatment. We see consumption of methane in the SSITKA studies as well, during the ramp to 600 °C and hold at 600 °C until steady state was achieved, as can be seen in Fig. S8.† Since this species is reduced at 600 °C with methane, but was not reduced during the H2 pretreatment at 600 °C, this species likely requires methane for reduction. We see a smaller, similar phenomenon during the ramp to 800 °C, as can be seen in Fig. S9,† but we cannot say whether or not this species required methane for reduction, or if a H2 pretreatment at a higher temperature would remove it. A SSITKA experiment, identical to that described in Fig. 2 was also performed with only 1 hour of H2 pretreatment at 600 °C, and we found no difference in the results, as can be seen in Fig. S10.†
We performed a similar ramping experiment with a CD4 pulse, and the results are depicted in Fig. 5. The ramping pulse (Fig. 5b) generally appears similar to the room temperature pulse (Fig. 5a), giving no indication of significant methane dissociation during the temperature ramp. There seems to be a slight dip in mass 20 at minute 22, which is likely due to the same oxidation phenomenon seen in the 13CH4 experiments. Fig. S11† depicts more of the masses measured during the pulse experiment depicted in Fig. 5. As can be seen in Fig. S11d,† the dip in mass 20 is accompanied by a peak in 44, likely indicative of the reduction of some oxygenated species. In addition, there may be a slight decrease in 20 starting at minute 30, accompanied by slight increases in masses 17, 19, and 44, before a spike of methane consumption at minute 40. In this experiment, it is difficult to determine whether the increases in masses 17 and 19 are due to H2O and HDO production or dissociative methane adsorption to form CH3D and CD3H. What is clear, however, is that we do not see evidence of significant, complete, methane dissociation and recombination, as would be required for methane–carbide carbon exchange, especially not on the order of 50–75%, as has been previously claimed.22
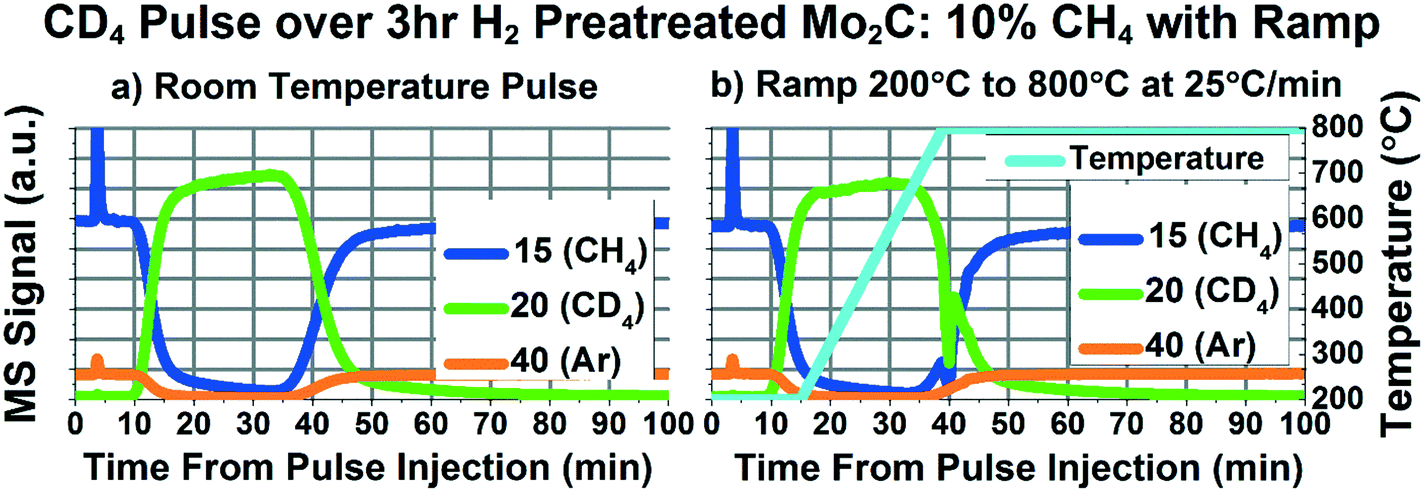 | ||
| Fig. 5 Raw mass spectrometry curves of masses 15 (representing CH4), 20 (representing CD4), and 40 (representing Ar inert tracer) for CD4 pulses delivered while flowing 18 SCCM He, and 2 SCCM 95% CH4/5% Ar after 3 hours of H2 pretreatment at 600 °C. a) Pulse performed at room temperature; b) pulse performed while ramping from 200 °C to 800 °C at 25 °C min−1, as indicated by the cyan temperature curve overlaid on the mass spectrometer curves. | ||
Conclusions
Both the SSITKA and ramping 13CH4 exchange studies do not exhibit significant carbon exchange between methane and the Mo2C carbide carbon, in contradiction to previous literature reports suggesting 50–75% of the methane stream undergoes carbon exchange at temperatures above 550 °C with the conditions used in this study.22 Further, no kinetic step could be measured at any condition or temperature tested using the SSITKA technique. This suggests that carbon exchange between a stream of methane and Mo2C, if any does occur, is extremely small and undetectable with these experiments.In addition, the CD4 exchange experiments seem to suggest a different methane adsorption mechanism. The appearance of mass 17 (CH3D) and 19 (CD3H) peaks at 800 °C in the SSITKA studies, indicates that methane likely does dissociate on Mo2C at 800 °C. However, since little to no amount of mass 18 could be attributed to CD2H2, our results suggest that methane adsorption involves the cleavage of only one carbon–hydrogen/deuterium bond to form CH3 or CD3 and H or D on the surface. In addition, the extremely small amount of CD3H and CH3D formed from the recombination suggests that this dissociation/recombination mechanism is still relatively minor, even at 800 °C. This implies that reactions involving methane over Mo2C require additional species, such as oxidants, to involve the carbide carbon and complete the redox mechanism.
Acknowledgements
We would like to thank the following people and organizations for their assistance in this work: Shallaco McDonald, Butch Cunningham, and Jim Smitherman for their help in designing, fabricating, and installing the reactor system and isotope delivery system. Kevin Haynes for helping manage the isotopically labeled gases. Edward Evans for his suggestions and insight. Prof. Keith Johnston for use of the Quantachrome NOVA 2000 BET surface analyzer. The X-ray Diffraction Lab of the Chemistry Department, University of Texas at Austin. We are thankful for the generous financial support of the Department of Energy [Grant DE-FG02-04ER15587] and the Welch Foundation [Grant F-1436].References
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CAS.
- J. B. Claridge, A. P. E. York, A. J. Brungs, C. Marquez-Alvarez, J. Sloan, S. C. Tsang and M. L. H. Green, J. Catal., 1998, 100, 85–100 CrossRef.
- A. R. S. Darujati and W. J. Thomson, Appl. Catal., A, 2005, 296, 139–147 CrossRef CAS.
- D. C. Lamont and W. J. Thomson, Appl. Catal., A, 2004, 274, 173–178 CrossRef CAS.
- D. C. LaMont and W. J. Thomson, Chem. Eng. Sci., 2005, 60, 3553–3559 CrossRef CAS.
- A. J. Brungs, A. P. E. York, J. B. Claridge, C. Marquez-Alvarez and M. L. H. Green, Catal. Lett., 2000, 70, 117–122 CrossRef CAS.
- J. Sehested, C. J. H. Jacobsen, S. Rokni and J. R. Rostrup-Nielsen, J. Catal., 2001, 201, 206–212 CrossRef CAS.
- A. R. S. Darujati, D. C. LaMont and W. J. Thomson, Appl. Catal., A, 2003, 253, 397–407 CrossRef CAS.
- J. Patt, D. J. Moon, C. Phillips and L. Thompson, Catal. Lett., 2000, 65, 193–195 CrossRef CAS.
- T. Xiao, H. Wang, J. Da, K. S. Coleman and M. L. H. Green, J. Catal., 2002, 211, 183–191 CAS.
- A.-M. Alexander and J. S. J. Hargreaves, Chem. Soc. Rev., 2010, 39, 4388–4401 RSC.
- A. P. E. York, T. Xiao, M. L. H. Green and J. B. Claridge, Catal. Rev.: Sci. Eng., 2007, 49, 511–560 CAS.
- A. Hanif, T. Xiao, A. P. E. York, J. Sloan and M. L. H. Green, Chem. Mater., 2002, 14, 1009–1015 CrossRef CAS.
- T. Xiao, A. P. E. York, K. S. Coleman, J. B. Claridge, J. Sloan, J. Charnock and M. L. H. Green, J. Mater. Chem., 2001, 11, 3094–3098 RSC.
- A. J. Medford, A. Vojvodic, F. Studt, F. Abild-Pedersen and J. K. Nørskov, J. Catal., 2012, 290, 108–117 CrossRef CAS.
- H. Tominaga and M. Nagai, Appl. Catal., A, 2007, 328, 35–42 CrossRef CAS.
- P. Liu and J. A. Rodriguez, J. Phys. Chem. B, 2006, 110, 19418–19425 CrossRef CAS PubMed.
- X. R. Shi, S. G. Wang and J. Wang, J. Mol. Catal. A: Chem., 2016, 417, 53–63 CrossRef CAS.
- H. Shou and R. J. Davis, J. Catal., 2013, 306, 91–99 CrossRef CAS.
- T. Xiao, A. Hanif, A. P. E. York, Y. Nishizaka and M. L. H. Green, Phys. Chem. Chem. Phys., 2002, 4, 4549–4554 RSC.
- S. Naito, A. Takada, S. Tokizawa and T. Miyao, Appl. Catal., A, 2005, 289, 22–27 CrossRef CAS.
- D. C. LaMont, A. J. Gilligan, A. R. Darujati, A. S. Chellappa and W. J. Thomson, Appl. Catal., A, 2003, 255, 239–253 CrossRef CAS.
- NIST Mass Spec Data Center and S. E. Stein, Mass Spectra, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, http://webbook.nist.gov, (retrieved July 26, 2016) Search PubMed.
- S. Shannon and J. G. J. Goodwin, Chem. Rev., 1995, 95, 677–695 CrossRef CAS.
- C. Ledesma, J. Yang, D. Chen and A. Holmen, ACS Catal., 2014, 4, 4527–4547 CrossRef CAS.
- A. Brush, S. McDonald, R. DuPré, S. Kota, G. M. Mullen and C. B. Mullins, In Preparation, 2017.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6re00141f |
| This journal is © The Royal Society of Chemistry 2016 |